

Abstract
Compared to adults, immature metallothionein I & II knockout (MT−/−) mice incur greater neuronal loss and a more rapid rate of microglia accumulation following target deprivation-induced injury. Since minocycline has been proposed to inhibit microglial activation and associated production of neuroinflammatory factors, we investigated its ability to promote neuronal survival in the immature, metallothionein-deficient brain. Following ablation of the visual cortex, 10-day-old MT−/− mice were treated with minocycline or saline and sacrificed 24 or 48 hours after injury. Using stereological methods, the number of microglia and neurons were estimated in the ipsilateral dorsal lateral geniculate nucleus (dLGN) by an investigator blinded to the treatment. No effect on neuronal survival was observed at 24 hours, but 48 hours after injury an unanticipated but significant minocycline-mediated increase in neuronal loss was detected. Further, while failing to inhibit microglial accumulation, minocycline treatment increased the proportion of amoeboid microglia in the ipsilateral dLGN. To understand the molecular mechanisms underlying this neurotoxic response, we identified minocycline-mediated changes in the expression of three potentially pro-apoptotic/ inflammatory genes: growth arrest- and DNA damage-inducible gene 45γ (GADD45γ); interferon-inducible protein 1 (IFI1) and cytokine induced growth factor (CTGF). We also observed increased mitogen-activated protein kinase (MAPK) p38 phosphorylation with minocycline treatment. Although minocycline inhibited calpain activity at 12 hours post-injury, this effect was not sustained at 24 hours. Together, these results help to explain how minocycline has a deleterious effect on neuronal survival in this injury model.
Keywords: microglia, minocycline, metallothionein, traumatic brain injury
Introduction
The functional deficits that follow traumatic brain injury (TBI) result from both the primary “mechanical” injury and later secondary insults that include elements of inflammation, excitotoxicity, neurotrophin withdrawal and oxidative damage (Potts et al. 2006; Ray et al. 2002; Robertson 2004). The immature brain appears particularly vulnerable to such insults (Natale et al. 2002; Potts et al. 2006; Repici et al. 2003; Robertson et al. 2006) and an effective therapy to inhibit neuronal loss in pediatric patients would likely target multiple degenerative pathways.
Minocycline is a second generation tetracycline with anti-inflammatory and anti-apoptotic properties as well as associated potential neuroprotective properties (Domercq and Matute 2004; Zemke and Majid 2004). Through inhibition of microglia activation, minocycline has been shown to reduce transcription of pro-inflammatory mediators including inducible nitric oxide synthase, nitric oxide, cyclooxygenase 2 and interleukin 1β (Tikka et al. 2001; Yrjanheikki et al. 1998; Yrjanheikki et al. 1999). Inhibition of MAPK p38 is another well described property of minocycline (Lin et al. 2001; Suk 2004). Phosphorylation of MAPK p38 is not only central to microglial activation and increased production of inflammatory mediators (Saccani et al. 2002; Suk 2004), but also leads to direct activation of cell death pathways (Du et al. 2001; Lin et al. 2001). In vitro, minocycline inhibits MAPK p38 phosphorylation and promotes neuronal survival (Lin et al. 2001; Pi et al. 2004; Wei et al. 2005). Other demonstrated in vitro effects of minocycline include caspase-1 inhibition, up-regulation of anti-apoptotic Bcl-2, and blockade of pro-apoptotic cytochrome c and SMAC/Diablo release from the mitochondria (Wang et al. 2004; Wang et al. 2003b; Zhu et al. 2002).
Through these and other less well described mechanisms, systemic administration of minocycline has been neuroprotective in a number of animal models of CNS injuries including ischemia (Morimoto et al. 2005; Wang et al. 2003a; Yrjanheikki et al. 1998), excitotoxicity (Tikka et al. 2001; Tikka and Koistinaho 2001) and spinal cord injury (Stirling et al. 2004; Teng et al. 2004). Particular to TBI, Sanchez et al. (Sanchez Mejia et al. 2001) found minocycline to be neuroprotective in a mouse experimental model. However, a second study by Bye et al. found minocycline’s protective effects to be only transient (Bye et al. 2006). In the setting of injury in the immature brain, minocycline’s neuroprotective effects have been similarly inconsistent. Although the majority of studies report favorable outcomes (Arvin et al. 2002; Dommergues et al. 2003; Fan et al. 2005a; Fan et al. 2005b), minocycline conferred only transient protection in the immature rat brain following focal cerebral ischemia-reperfusion (Fox et al. 2005), and worsened outcome in a neonatal mouse model of hypoxia-ischemia (Tsuji et al. 2004). In the face of such variable results, further investigation is required to understand the conditions under which minocycline provides neuroprotection, and those where it may be detrimental.
In the immature mouse brain, loss of thalamocortical neurons proceeds more rapidly following in vivo target deprivation than in the adult brain. This is partly due to an age-dependent deficiency in the anti-oxidant and anti-inflammatory factors metallothionein I & II (MT I&II) (Natale et al. 2004). Deficient expression of MT I&II has also been associated with an enhanced inflammatory cell response (Giralt et al. 2002; Penkowa et al. 2005; Potter et al. 2007) that contributes to increased generation of pro-inflammatory cytokines and further neuronal injury. Since minocycline has been shown to limit the inflammatory response and promote neuronal survival, we hypothesized that it would overcome the developmental deficiency in MT I&II and limit neuronal loss in the immature brain following in vivo target deprivation. We chose to first test this hypothesis in the most extreme case of metallothionein deficiency: immature mice that completely lack MT I&II.
In this study we report that minocycline not only failed to inhibit microglia accumulation following in vivo target deprivation, but enhanced loss of thalamocortical neurons. Since these results add to the already controversial body of literature on minocycline’s effects in the CNS, we carefully investigated the mechanisms underlying its neurotoxicity to better describe minocycline’s detrimental effects in this model of injury. To do so, we characterized minocycline-mediated changes in gene expression, levels of phospho-p38, and both caspase-3 and calpain activity. At the doses used in this study of MT−/− mice subjected to visual cortical ablation, minocycline promoted pro-inflammatory and pro–apoptotic events that may account for enhanced loss of thalamocortical neurons. Interestingly, in wild-type mice, despite its ability to inhibit the microglial response to cortical injury, minocycline was also found to promote neuronal loss from the dLGN.
Materials and Methods
Mice
MT knock out and wild-type (MT−/−, 129S7/SvEvBrd-Mt1tmBriMt2tm1Bri) mice purchased from Jackson Laboratories (Bar Harbor, ME) were provided care in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal experiments were conducted on immature post-natal day (P) 10 mice. All procedures were approved by the Institutional Animal Care and Use Committee of the Children’s National Medical Center and every effort was made to minimize both the suffering and number of animals used.
Target deprivation-induced thalamic injury and treatment
P10 MT−/− or wild-type mice were subjected to visual cortex ablation as previously described (Natale et al. 2002; Natale et al. 2004). Briefly, mice were anesthetized with isofluorane and nitrous oxide in oxygen. With the skull immobilized in a mouse stereotaxic apparatus (Stoelting, Wood Dale, IL), a 4 × 4-mm cranial flap exposed the right visual cortex. A 20-gauge metal cannula connected to low-grade suction was used to aspirate the right visual cortex. The cortical defect was filled with an absorbable gelatin mesh (Gelfoam: Pharmacia and Upjohn, Kalamazoo, MI), the bone flap replaced, and the skin incision sutured closed. In sham procedures, mice received a craniotomy only. Based on initially promising preliminary data obtained in our lab and effective dosing regimens reported in the literature (Tikka et al. 2001; Yrjanheikki et al. 1998; Yrjanheikki et al. 1999), we chose the following scheme for dose and timing of minocycline administration. Beginning 30 minutes after surgery, mice received 90 mg/kg of minocycline (dissolved in saline, pH corrected to 7.4; Sigma, St. Louis, MO) or an equi-volume of saline via intraperitoneal injection. Additional doses were given at 12 and 24 hours post-injury. Mice were sacrificed 6, 12, 24 or 48 hours post-injury.
Immunohistochemistry
Under deep anesthesia, MT−/− and wild type mice were perfused transcardially with ~100 mls of phosphate buffered saline followed by an equal volume of 4% paraformaldehyde. Brains were postfixed overnight, cryoprotected and sectioned at 40 μm on a sliding microtome. Microglia were identified with monoclonal rabbit anti-ionized calcium-binding adaptor molecule 1 (Iba1, 1:250; Wako Chemicals USA Inc, Richmond, VA) as previously described (Potter et al. 2007). Immunoreactivity was visualized using horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody (1:500; Bio-Rad, Hercules, CA) was used in conjunction with the chromogen 3,3′-diaminobenzidine-4HCl (DAB; Vector Laboratories, Burlingame, CA).
Stereology
We used non-biased stereological methods to quantify microglia and neurons in the entire dLGN ipsilateral to the cortical injury in MT−/− and wild type mice. Typically the LGN extended through approximately twenty-five 40 μm-thick sections. Alternate sections were stained for the microglial protein Iba1, mounted onto gelatin-coated slides and stained with Cresyl Violet (Sigma, St Louis, MO) to yield a section sampling fraction (ssf) of ½. For quantification, the computer-assisted stereological toolbox (CAST)-Grid software (Olympus, Denmark) was used on a PC connected to an Olympus BX50 microscope. A motorized automatic stage was used to guide movement in the x, y plane (Proscan, Prior Scientific, MA, USA) while movement in the vertical (Z) direction was monitored using a microcator (MT12, Heidenhain, Germany). Microglial and neuronal numbers were estimated using a counting frame area (a(frame)) of 2817 μm2 with a step length of 100 μm in both the x- and y-plane between fames. The resultant area sampling fraction (asf = a(frame)/ a(step)) was 2817 μm2/ 10000 μm2 = 0.2817. Multiple measurements of the section thickness (t) were made throughout the LGN and averaged. A dissector of height 15 μm (h) with a guard of 3 μm was employed and the thickness sampling fraction (tsf) estimated as t/h. Neurons and microglia were counted under a 100X oil-immersion objective with a numerical aperture of 1.4. Cells were counted only when their nuclei came into focus within the inclusion area of the counting frame. Cell counts were made by an investigator blinded to the treatment condition.
The optical fractionator method, as outlined by West et al. (West et al. 1996; West et al. 1991), was used to estimate the total number of neurons (N) in the dLGN. This method calculates the total number of cells within the reference space (dLGN) as a sum of the objects counted (Q) multiplied by the reciprocal of the ssf, asf and tsf:
Assessment of microglial morphology
In MT−/− mice, microglia were classified into three categories; resting, activated and amoeboid by an investigator masked to the treatment condition. Resting microglia were recognized by a small cell body and numerous, highly ramified processes. Activated microglia were distinguished by an enlarged cell body and short, stout processes. Spheroid microglia with few or no processes were classified as amoeboid. Such morphological features are considered indicative of the stereotypical microglial response to neuronal injury and are described in detail by multiple investigators (Dailey and Waite 1999; Ito et al. 2001; Kreutzberg 1996).
RNA extraction, amplification and hybridization to microarrays
At 6 and 12 hours post-surgery, sham (N=4) and injured (N=3 / treatment group at each time-point) MT−/− mice were euthanized with carbon dioxide. Following brain extraction, the right (ipsilateral) LGN was rapidly dissected from the thalamus and immediately frozen in chilled isopentane.
Approximately 15 mg of LGN tissue from each brain was homogenized in guanidine thiocyanate buffer (Buffer “RLT”, Qiagen Inc., Valencia, CA) using a Polytron homogenizer (Brinkmann Instruments, West bury, NY). Samples were not pooled; mRNA derived from a single brain was used to generate each array profile. Total RNA was isolated using an RNeasy Mini kit (Qiagen Inc., Valencia, CA). Six micrograms of total RNA from each sample was converted to into double-stranded cDNA using an oligo-dT primer containing the T7 RNA polymerase promoter (Invitrogen, Carlsbad, CA). Using a MEGAscript T7 polymerase (GeneChip® Expression 3′-Amplification Reagents for IVT labeling, Affymetrix, Santa Clara, CA), purified double-stranded cDNA was then converted into biotin-labeled cRNA. Following fragmentation to approximately 200 bp, 15 micrograms of biotin-labeled cRNA was hybridized to mouse genome 430 2.0 oligonucleotide microarrays (Affymetrix, Santa Clara, CA) for 16 hours at 60 rpm, 45 °C. Each microarray was stained on the Affymetrix Fluidics Station 450 using instructions and reagents provided by Affymetrix. cRNA hybridization was detected using a streptavidin conjugated fluorophore and confocal laser scanning according to the manufacturers recommendations (Affymetrix). Raw intensity data were captured, and the signal intensity calculated using both dCHIP difference model (Diff) and PLIER (Affymetrix) probe set algorithms.
Microarray quality control
Stringent quality control methods were employed as previously described (2004). In brief, all 16 arrays included in the study demonstrated a present call rate above 45%, a linear scaling factor below 4 and a signal ratio of 3′ to 5′ human glyceraldehydes-4-phosphate dehydrogenase below 3.
Statistical methods and selection of differentially expressed genes
In parallel, data from the dCHIP Diff and PLIER (Seo et al. 2006) algorithms were filtered using GeneSpring GX v.7.3 (Agilent Technologies, Santa Clara, CA). Genes that were not “present” in at least one of the chips were eliminated. Probe set signal intensities at each post-injury time point were normalized to the mean signal intensities at time zero (sham injured). Genes differentially expressed between saline and minocycline at 6 and 12 hours were identified using ANOVA (one-way, variances not assumed equal (Welch t-test)) analyses with no multiple testing corrections and a p value cutoff of 0.01. The subsequent gene lists generated by dCHIP and PLIER were filtered by Venn diagram to identify only those genes found to be differentially regulated by both algorithms. This yielded a list of 116 genes which were then subjected to supervised hierarchical (gene tree; Pearson correlation with average linkage clustering algorithm) clustering.
Quantitative real-time RT-PCR
In a separate experiment with 8 MT−/− mice treated in the same manner as described for the microarray experiment, total RNA was isolated from the LGN of saline and minocycline treated mice 6 hours post-injury as described above. 2 μg of total RNA was used to synthesize a single-stranded cDNA template using an oligo-dT primer containing the T7 RNA polymerase promoter. Each sample was diluted 1: 5. Taqman PCR primers were designed using the Invitrogen D-LUX™ (Light upon eXtension) Fluorogenic designer software and labeled with 6-carboxy-fluorescein (FAM). An additional primer set for protein-tyrosine phosphatase, receptor type N (Ptprn) was designed and labeled with 6-carboxy-4′, 5′-dichloro-2 (JOE) to serve as an internal control. Primers sequences were as follows; Ptprn (forward) 5′-GAC CAA TAC CCA TCT GCC ACC TTG G [JOE] C-3′ and (reverse) 5′-CCT TCC TTC CCT CCC GAC TC-3′; NFκB1A (forward) 5′-GAT TCG TTC ACA AAG ACA ACA GCC GAA [FAM] C-3′ and (reverse) 5′-ACA CCA CAC AGC GCC TAG ACC-3′; FosL2 (forward) 5′-CGC ACC AAT GAC TCT GGA AGA AGT C [FAM] G-3′ and (reverse) 5′-AGA AGT TCG TGC AGT GCT TTC C-3″. Pre-designed primers (D-LUX™ Pre-Designed Gene Expression Assay, Invitrogen) were purchased for GADD45γ (MLUX3314382_FAM_Gadd45g and MLUX3314382_FAM_Gadd45g), CTGF (MLUX3304218_FAM_Ctgf and MLUX3304218_Ctgf) and IFI1 (MLUX3306145_FAM_Igrm and MLUX3306145_Igrm). 5 μl of cDNA template was added to a reaction mixture containing 2X Platinum Quantitative PCR SuperMix-UDG with 55, 6-carboxy-x-rhodamine (ROX) reference dye [60u/ml Platinum Taq polymerase, 40 mM Tris-HCl (pH 8.4), 100 mM KCl, 6 mM MgCl-2, 400 μM dGTP, 400 μM dATP, 400 μM dCTP, 800 μM dUTP, 40 U/ml UDG, 1 μM ROX], forward FAM labeled LUX primer (10 μM) and reverse unlabeled primer (10 μM). Estimation of mRNA levels were quantified relative to Ptprn to account for variations in sample quantity and reverse transcriptase efficiency. mRNA levels were calculated using the ΔΔCT method as described in the ABI Prism 7700 Sequence Detection System (SDS) manual (Applied Biosystems, Foster City, CA).
Immunoblots
Protein lysates were prepared from fresh-frozen, MT−/− LGN tissue. Samples were homogenized in ~ 10 μl/mg of cold lysis buffer (20 mM Tris HCl, pH 7.4; 1 mM EDTA; 5 mM EGTA; 10 mM benzamide, protease inhibitor cocktail (Roche Diagnostics Corporation, Indianapolis, IN)) using a handheld motorized pestle. To ensure hydrolysis, samples were incubated on ice for 30 min before centrifugation at 14,000 rpm for 5 min at 4 °C. The total protein concentration was quantified using the Bradford method (Bradford 1976).
For detection of αII-spectrin, 20 μg of protein was separated in a 4 % tris-glycine gel (Invitrogen). Protein was transferred to a nitrocellulose membrane before blocking in 0.05% Tween-20/ Phosphate buffered saline (PBS) /5% nonfat milk for 1 hour at room temperature, followed by incubation with mouse monoclonal anti- αII-spectrin antibody (Biomol, Plymouth Meeting, PA) at a concentration of 1:4000 in blocking buffer at 4 °C overnight. After washing in 0.05% Tween-20/PBS, membranes were incubated with a 1:10,000 dilution of goat-anti-mouse HRP-conjugated secondary antibody (Bio-Rad Laboratories, Hercules, CA) for 1 hour at room temperature. After further washing, protein bands were detected using enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Pierce, Rockford, IL) followed by exposure to Kodak X-OMAT film. To avoid signal saturation, a shorter exposure time (~ 10 seconds) was used for quantification of the more abundant 145 kDa product while and a longer exposure (~ 1 minute) was required for the 120 kDa product. Membranes were incubated in stripping buffer (0.1 M ß-mercaptoethanol, 2% Sodium dodecyl sulphate, 62.5mM Tris HCl pH 6.8) prior to re-probing with monoclonal mouse anti-vinculin antibody (Sigma, St Louis, MO) to assess uniformity of protein transfer to the blot.
For detection of phospho-p38, 40 μg of protein was separated in a 12 % bis-tris gel (Invitrogen) for 1 hour at a constant 200 V. Following protein transfer, membranes were blocked in 0.05% Tween-20/ Tris buffered saline (TBS) /5% bovine serum albumin (BSA) for 1 hour at room temperature, prior to incubation with rabbit polyclonal anti- Phospho-p38 MAP Kinase (Thr180/Tyr182) antibody (1:3000; Cell Signaling, Danvers, MA) at 4 °C overnight. After washing, a 1:50,000 dilution of goat anti-rabbit HRP-conjugated secondary antibody (Bio-Rad) was applied to the membrane for 1 hr at room temperature. Protein bands were detected using enhanced chemiluminescence (SuperSignal West Dura Extended Duration Substrate; Pierce). Membranes were stripped and re-probed with rabbit polyclonal anti-p38 antibody (1:800; Cell Signaling). Total extracts from C-6 glioma cells prepared with and without anisomycin treatment served respectively as the positive and negative controls for P-p38 (Cell Signaling, Danvers, MA).
Statistics
Data are expressed as mean ± standard error of the mean (SEM). Two-tailed Mann-Whitney U-tests were used to compare microglial and neuronal numbers in saline and minocycline treated mice. These statistical tests were conducted using GraphPad Prism 4.00 (GraphPad Prism Software, San Diego, CA). P values < 0.05 were considered significant.
Results
Minocycline fails to inhibit microglial accumulation within the dLGN of MT−/− mice after cortical ablation
Consistent with previous reports (Milligan et al. 1991; Muessel et al. 2002; Potter et al. 2007), we found target deprivation to invoke a robust microglial response. By 48 hours after ablation of the visual cortex, the microglial population had increased by ~2.5 fold within the ipsilateral dLGN in both minocycline and saline treatment groups (Fig. 1A). However, minocycline administration did not alter the number of microglia accumulating within the ipsilateral dLGN either 24 or 48 hours after injury compared to saline treatment. Within the contralateral dLGN 48 hours following injury, the number of microglia remained relatively low (~1000), and was again unaffected by minocycline treatment.
Fig. 1. Minocycline does not inhibit accumulation of microglia but is associated with an increased frequency of amoeboid microglia in the ipsilateral dLGN 48 hours after cortical ablation in P10, MT−/− mice.
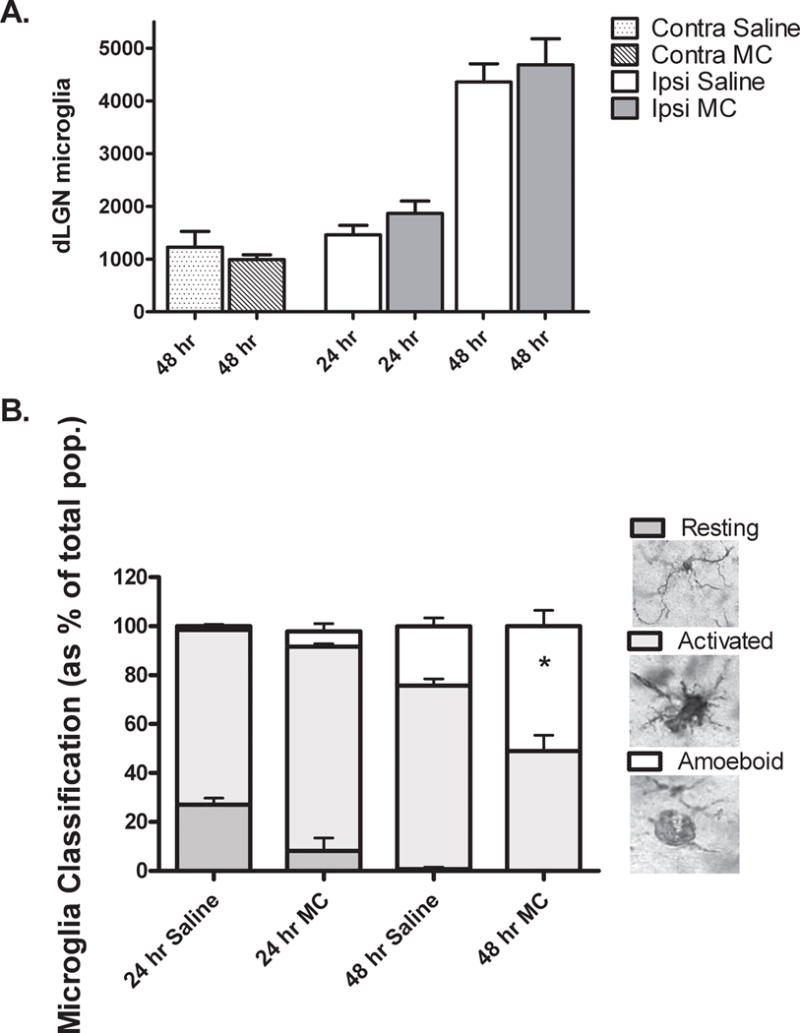
A. Using unbiased stereological methods, the number of Iba1 positive microglia in the contralateral (contra) and ipsilateral (ipsi) dLGN were estimated by an investigator blinded to the treatment condition. No significant difference was found between numbers of microglia in saline (Sal) vs. minocycline (MC) treated mice at any time point after injury. *P=0.03, saline vs. minocycline, Mann-Whitney U, N= 3 /contra group, N= 5 /ipsi group. B. An investigator blinded to treatment condition classified Iba1 positive microglia as resting, activated or amoeboid according to morphological criteria (Dailey and Waite, 1999; Ito, et al., 2001; Kreutzberg, 1996). Resting microglia were recognized by a small cell body and numerous, highly ramified processes. Activated microglia were distinguished by an enlarged cell body and short, stout processes. Spheroid microglia with few or no processes, were classified as amoeboid. Stacked columns show each morphology as a percentage of total population with time after injury and treatment. No significant difference was determined between treatment groups at 24 hrs. Percentage of amoeboid microglia was increased in minocycline (MC) vs. saline (Sal) treated mice at 48 hrs, *P= 0.03 Mann-Whitney U, N = 4/ group.
Since minocycline did not influence the overall number of microglia that accumulated within the ipsilateral dLGN, we next asked whether it affected the microglial activation state. Regardless of treatment, the percentage of resting microglia declined from 24 to 48 hours after injury as the population became almost wholly activated (Fig. 1B). However 48 hours after injury, minocycline treatment significantly increased the proportion of more highly activated amoeboid microglia compared to saline.
Minocycline promotes neuronal loss in MT−/− mice after cortical ablation
While we were unable to detect an effect of minocycline on the number of neurons within the contralateral dLGN 48 hours after cortical ablation, on the ipsilateral side treatment with minocycline was associated with greater neuronal loss (Fig. 2). Minocycline had no effect on neuronal numbers 24 hours after cortical injury.
Fig. 2. Minocycline promotes neuronal loss from the ipsilateral dLGN 48 hours after cortical ablation in P10, MT−/− mice.
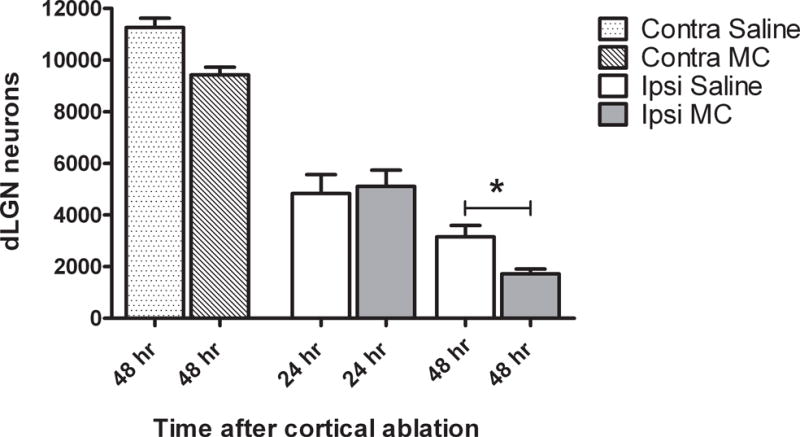
Using unbiased stereological methods, the number of cresyl violet stained neurons in the contralateral (contra) and ipsilateral (ipsi) dLGN were estimated by an investigator blinded to the treatment condition. At 24 hours post-injury, the number of neurons was unchanged, but by 48 hours post-injury minocycline (MC) treated mice had fewer neurons remaining in the dLGN compared to their saline (Sal) treated counterparts. *P=0.03, saline vs. minocycline, Mann-Whitney U, N= 3 /contra group, N= 5 /ipsi group.
Minocycline increases expression of factors associated with inflammation and apoptosis in the LGN after cortical ablation in MT−/− mice
116 genes were found to be differentially expressed between saline and minocycline at 6 and 12 hours post-injury (using criteria described in the methods section). Hierarchical clustering with these genes revealed a group of 26 genes up-regulated by minocycline treatment at the 6 hour time-point (Fig. 3). This group included 3 genes with function associated to immune response, 5 genes involved in control of gene expression and 10 associated with cell death. One gene (Fos-like antigen 2 (FosL2)) from the total list of 116 differentially regulated genes, and four (CTGF, NFκB1A, GADD45γ and IFI1) from the 6 hour minocycline cluster were selected for validation by quantitative real time-PCR (Q-RT-PCR).
Fig. 3. Supervised hierarchical clustering illustrates the effect of minocycline treatment on gene expression 6 and 12 hours after cortical ablation.
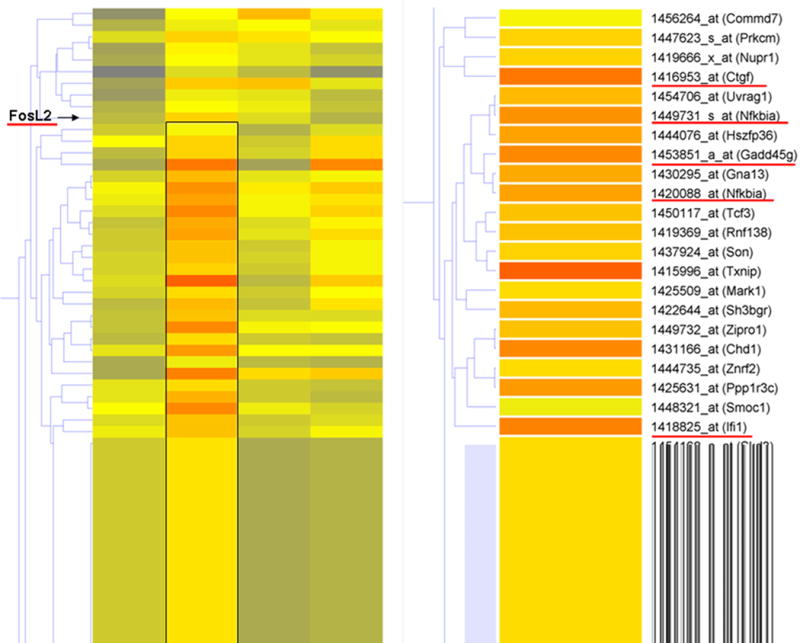
Supervised hierarchical clustering was conducted on 116 genes found to be differentially regulated by minocycline treatment after cortical ablation. Genes were clustered in GeneSpring via gene tree analysis. A cluster of genes up-regulated by minocycline at 6 hours post-injury was identified (box on left and enlarged on right) from the list of 116 genes. Expression of all probe sets was normalized to sham prior to analysis. Genes underlined in red were selected for validation by Q-RT-PCR based on biological and statistical relevance.
Q-RT-PCR was conducted on 8 independent LGN samples obtained at 6 hours after injury to compare the effects of these treatments on early mRNA expression (Fig. 4). Consistent with the expression changes identified by the microarray analyses (Table 1), Q-RT-PCR analyses confirmed the significant induction in CTGF, GADD45γ, IFI1, and FosL2 expression in the dLGN 6 hours after minocycline treatment. The induction of NFκB1A was not significantly different between minocycline and saline.
Fig. 4. Comparison of CTGF, GADD45γ, IFI1, NFκB1A and FosL2 expression by quantitative RT-PCR in saline and minocycline treated animals.
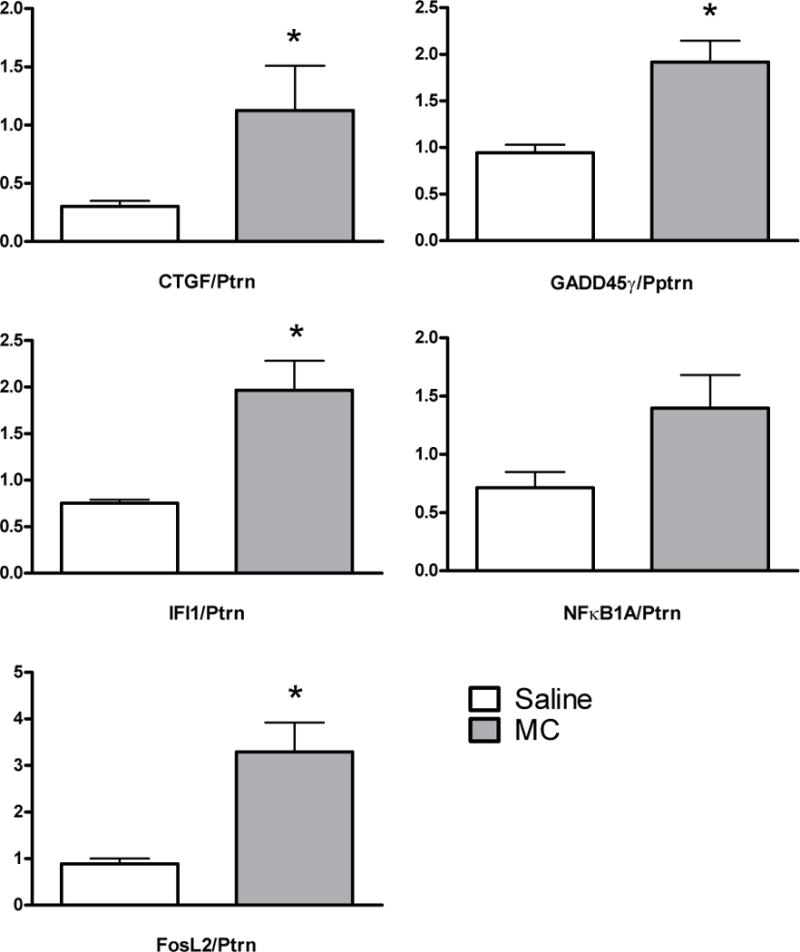
Genes were selected for validation based on biological and statistical relevance. RNA was extracted from independent samples (minocycline and saline treated mice, 6 hours post-injury, N=4/group). All values normalized to expression of a control gene, Ptprn. *P<0.05, Mann-Whitney U-test.
Table 1.
Fold Change in genes differentially expressed by minocycline.
| Gene Name | Affymetrix Probe ID | Microarray P-Value | Microarray Fold Change | Q-RT-PCR Fold Change |
|---|---|---|---|---|
| CTGF | 1416953_at | 0.0049 | 3.52 | 3.72 |
| IFI1 | 1418825_at | 0.0034 | 3.36 | 2.61 |
| GADD45γ | 1453851_a_at | 0.0002 | 2.4 | 2.03 |
| NFκB1A | 1449731_s_at | 0.0048 | 2.05 | 1.96 |
| FosL2 | 1437247_at | 0.0067 | 1.93 | 3.71 |
Comparison of average fold change in genes differentially expressed by minocycline treatment 6 hours after cortical ablation as determined by microarray analysis (PLIER algorithm) and, in independent samples by quantitative RT-PCR.
We next screened the microarray data to identify differentially regulated genes with functions directly related to the apoptotic process. At 6 and 12 hours post-injury, mRNA levels in saline treated mice were compared to those receiving minocycline (ANOVA, p<0.05). Using supervised hierarchical clustering of genes showing >1.5 fold (up or down) expression and with function directly related to apoptosis (GeneSpring supplied gene ontology), 11 pro-apoptotic and 3 anti-apoptotic genes were identified to be differentially regulated by minocycline treatment received 30 minutes after cortical injury (Fig. 5).
Fig. 5. Supervised hierarchical clustering illustrates the effect of minocycline on expression apoptosis-related genes 6 and 12 hours after cortical ablation.
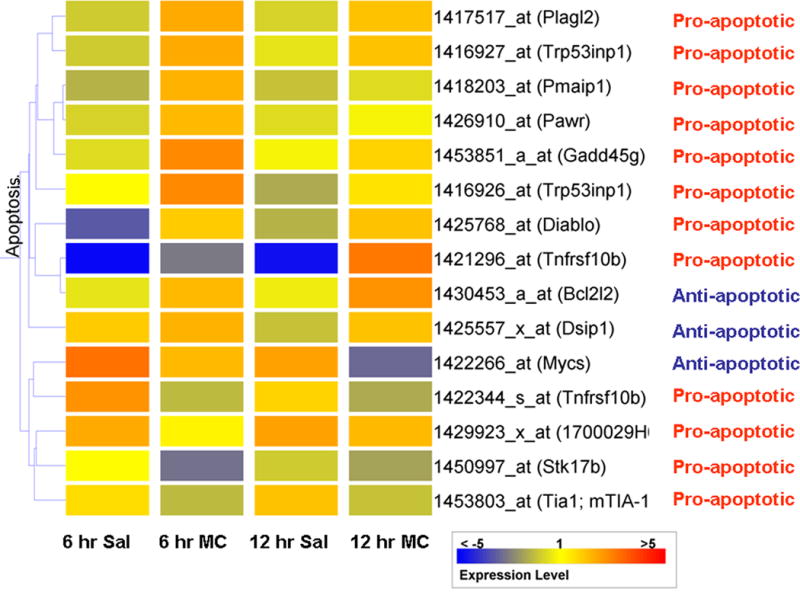
Expression of all probe sets were normalized to the average expression levels in the sham condition (N=4) prior to analysis. At 6 and 12 hours, saline treated animals (N=3) were compared to minocycline (N=3) animals using AVOVA. 15 genes showing differential expression > 1.5 fold (up or down) and with function relevant to apoptosis, underwent supervised hierarchical clustering.
GADD45γ is a stress-responsive protein identified here by both clustering schemes as up-regulated by minocycline treatment after injury (Figs. 3 & 5). By increasing expression of mitogen activated protein kinase kinase kinase (MAP3K4), GADD45 γ has been shown to increase phosphorylation of p38 (Takekawa and Saito 1998). Since phosphorylated p38 (P-p38) promotes a number of inflammatory and apoptotic processes, we compared levels of P-p38 in the LGN of saline and minocycline mice after injury. Although the abundance of P-p38 in the LGN was not significantly changed 24 hours after cortical injury with saline treatment (Fig. 6: compare 24 hour saline to sham), mice receiving minocycline showed levels of P-p38 that were almost three fold greater than following saline treatment (p=0.03).
Fig. 6. Minocycline induces MAPK p38 phosphorylation after cortical ablation.
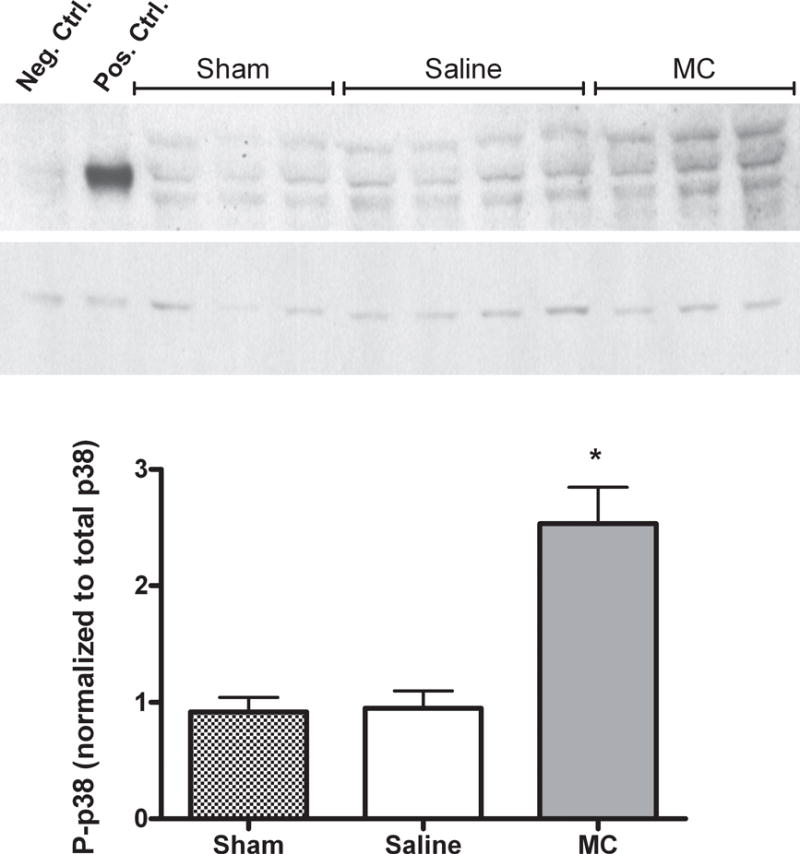
Phosopho-p38 (P-p38) levels and total p38 levels were determined in sham and ipsilateral LGN tissue derived from P10, MT−/− mice 24 hours after cortical ablation and either saline or minocycline treatment. A. Representative immunoblot showing P-p38 and total p38. Total extracts from C-6 glioma cells prepared with and without anisomycin treatment served respectively as positive and negative controls for P-p38. B. Quantification of P-p38 normalized to total p38. Minocycline induces a significant increase in the level of P-p38. *P= 0.03, Mann-Whitney U test. N≥ 3/group.
Minocycline transiently inhibits calpain activity after cortical injury, but promotes caspase-3 activity in MT−/− mice
The intracellular proteases calpain and caspase-3, have both been shown to play an important role in promoting neuronal loss following TBI (Liu et al. 2006; Raghupathi 2004; Ringger et al. 2004). While a number of studies have found minocycline’s neuroprotective effects to be in part mediated through inhibition of caspase-3 activity (Heo et al. 2006; Wei et al. 2005), its effect on calpain activity has been less well described. In order to investigate (1) the relative contribution made by each of these proteases after injury and (2) how that activity is affected by minocycline treatment, we analyzed the pattern of spectrin breakdown products (SBDPs).
When processed by caspase-3, the cytoskeletal protein α-II spectrin is cleaved to yield a specific 120 kDa breakdown product. Cleavage by calpain produces a 145 kDa breakdown product while both proteases contribute to the formation of a non-specific 150 kDa product. Figure 7 panels A and B show greater accumulation of the 145 compared to the 120 kDa product in saline treated mice 24 hours after injury, suggesting greater calpain than caspase-3 activity during this phase of injury. Examination of SBDPs at 12 hours after injury revealed significantly less calpain activity in geniculate tissue of mice who received minocycline (Fig. 7C), yet by 24 hours after injury, minocycline treatment tended to increase the 145 kDa product. Although caspase-3 related α-II spectrin degradation is relatively low early in this model, by 24 hours we also note a minocycline-mediated increase in accumulation of the 120 kDa product (Fig. 7D).
Fig. 7. Accumulation of spectrin break down products (SBDPs) in saline and minocycline treated mice after cortical ablation.
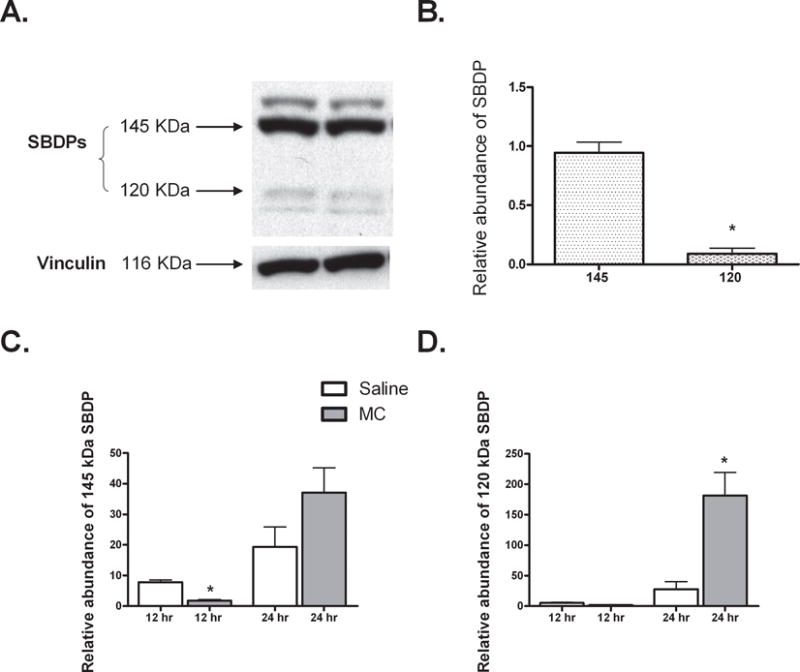
SBDPs were detected in sham and ipsilateral LGN protein derived from P10, MT−/− mice after cortical ablation and either saline (Sal) or minocycline (MC) treatment. A. Representative blot showing accumulation of the calpain specific 145 kDa product and the caspase-3 specific 120 kDa product 24 hours after injury in saline treated mice (N=2). B. The calpain-specific SBDP is significantly more abundant vs. the caspase-3 -specific product in saline treated mice 24 hours post-injury. C. 12 hours after injury, minocycline mediates a significant decrease in the accumulation of calpain-specific SBDPs. However, this trend is reversed by 24 hours with apparent increase in calpain activity in the minocycline treated animals. D. Greater caspase-3 activity is observed following minocycline treatment 24 hours after injury. Note: due to methodological differences (see methods section), the y-axes in figure C and D cannot be compared. *P < 0.05, Mann Whitney U. All data normalized to the abundance of a control protein, vinculin. N=4/group unless otherwise stated.
Minocycline promotes neuronal loss despite inhibiting microglia accumulation after cortical ablation in wild-type mice
Given the observed effects of minocycline on neuronal survival in MT−/− mice, we next asked whether it would have similar detrimental effects in wild-type mice. Using the same injury and treatment paradigm we found that minocycline did not significantly alter neuronal numbers 24 hours post-injury (Fig. 8A). However, by 48 hours after injury the group treated with minocycline exhibited a marked loss of neurons compared to the saline-treated group (p=0.03). Furthermore, while microglia progressively accumulated in the ipsilateral dLGN of saline treated mice (Fig. 8B), minocycline treated mice showed a much smaller (~1.3 fold) increase in microglial numbers from 24 to 48 hours post-injury. At 48 hours, minocycline treated mice therefore had approximately one half the number of microglia of their saline treated counterparts.
Fig. 8. Minocycline promotes neuronal loss despite inhibition of microglial accumulation in the ipsilateral dLGN 48 hours after cortical ablation in WT P10 mice.
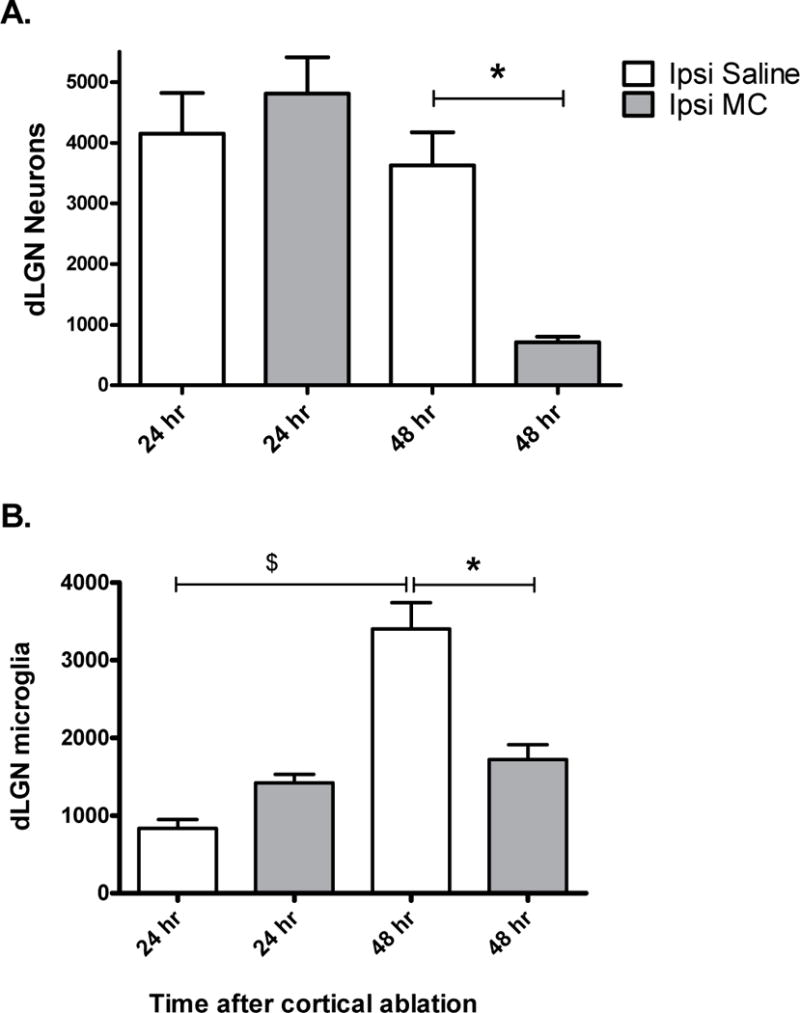
Using unbiased stereological methods, the number of cresyl violet stained neurons (A) and the number of Iba1 positive microglia (B) in the ipsilateral dLGN was estimated by an investigator blinded to the treatment condition. No significant difference was found between numbers of neurons in the saline (SAL) vs. minocycline (MC) treated mice 24 hours post-injury. 48 hours post-injury MC treated mice had fewer neurons remaining in the dLGN compared to their SAL treated counterparts. *P=0.03, Saline vs. Minocycline, Mann-Whitney U.
Minocycline inhibited microglial accumulation in the ipsilateral dLGN 48 hours post-injury. $ P=0.03, 24 hr vs. 48 hr Saline, Mann-Whitney U. *P= 0.02 Saline vs. Minocycline, Mann-Whitney U. N= 5 for each group.
Discussion
The main finding of this study is that minocycline promotes neuronal loss after in vivo target deprivation in the immature, metallothionein-deficient brain. Minocycline has been shown in multiple, but not all, studies to exert a strong inhibitory effect on inflammatory and apoptotic processes. The overall effect of the drug may be influenced by many factors including dosing, brain maturity, and the nature of the injury itself. This study highlights a previously unrecognized influence on minocycline’s activity in neurological injury: expression of MT I&II. It is therefore essential to further our understanding of the mechanisms that underlie both minocycline’s protective and deleterious effects.
Expression of MT I&II has also been shown to influence the inflammatory response in the setting of a number of CNS pathologies (Giralt et al. 2002; Penkowa et al. 2001; Penkowa et al. 2005). In particular, deficient expression of these factors was associated with an enhanced microglial response (Penkowa et al. 2001; Potter et al. 2007). It was on this basis that we hypothesized inhibition of microglial accumulation by minocycline would compensate for lack of MT I&II and lead to less secondary neurodegeneration.
Our results show that accumulation of microglia within the ipsilateral dLGN of MT−/− mice was unaffected by minocycline treatment in this model of injury but that the proportion of amoeboid microglia 48 hours post- injury in minocycline treated mice was increased. Although all microglia have the ability to phagocytose, amoeboid microglia are generally considered to do so most effectively and their presence is associated with more severe tissue damage (Streit et al. 1999). Analysis of gene expression patterns after injury were consistent with this observation and revealed a minocycline-induced up-regulation of IFI1. IFI1 is a member of the interferon-responsive p47 family of GTPases whose expression is required for maturation and acidification of phagosome vesicles within macrophages. Its increased expression 6 hours post-injury further supports the idea that microglia are more highly activated with minocycline treatment.
In order to explain the mechanisms responsible for increased neuronal loss after injury, we focused our studies on changes in gene expression patterns induced by minocycline and identified four plausible mechanisms. First, using a stringent selection strategy, we found GADD45γ to be significantly up-regulated by minocycline 6 hours post-injury in the LGN (Takekawa and Saito 1998), as well as a minocycline-induced increase in phospho-p38. Previous in vitro studies have found minocycline to inhibit GADD45γ-induced phosphorylation of MAPK p38, and thus provide neuroprotection (Du et al. 2001; Suk 2004; Wei et al. 2005). In microglia, phosphorylation of MAPK p38 increases production of inflammatory mediators (Saccani et al. 2002; Suk 2004), while in neurons, it has been shown to cause direct activation of cell death pathways (Du et al. 2001; Lin et al. 2001). It therefore seems likely that, under the conditions of the present study, a minocycline-mediated increase in MAPK p38 phosphorylation contributed to greater neuronal loss after injury.
Second, we found minocycline to up-regulate the expression of a number of pro-apoptotic and pro-inflammatory factors, however we also noted an increase in FosL2 and Bcl2l2, both of which have been associated with increased cell survival. FosL2 is a transcription factor implicated in the adaptation of surviving neurons to injury. A number of studies have found injury to induce a delayed, but prolonged increased in the expression of this factor (Butler and Pennypacker 2004; Lee et al. 2004; Pennypacker et al. 2000). A recent study found FosL2 to induce transcription of a number of neuroprotective genes following hypoxic insult (Butler and Pennypacker 2005). Bcl2l2 is a member of the anti-apoptotic Bcl-2 family that maintains cell viability through caspase inhibition (O’Reilly et al. 2001). Although it seems that expression of these factors should promote neuronal survival, the final outcome is of course a balance in the activity of both pro- and anti-apoptotic factors.
Third, our microarray data suggest that minocycline may affect transcriptional events in astrocytes following brain injury. We observed a significant increase in the expression of CTGF, a cytokine predominately generated by astrocytes in response to neuronal insults such as trauma and oxidative stress (Park et al. 2001; Schwab et al. 2001). CTGF promotes scar formation through deposition of extracellular matrix, thereby impairing regenerative events such as neuronal sprouting and oligodendrocyte migration. Such effects may further contribute to a negative outcome.
Fourth, while we found minocycline inhibited calpain activity 12 hours post-injury, by 24 hours post-injury, we observed a trend for increased calpain as well as caspase-3 activity, with minocycline treatment. Caspase-3 is a well-documented molecular target of minocycline. While patterns of SBDPs did indicate caspase-3 activity after in vivo target deprivation, this appeared to be significantly less than calpain activity. The ability of minocycline to exert a neuroprotective influence is dependent on the contribution made by its molecular targets to the cell death process. For example, in a 3-nitropropionic model of Huntington’s disease where calpain is known to be the major cell death effector (Bizat et al. 2003), minocycline was found to be an ineffective neuroprotective agent as it failed to inhibit calpain activity (Bantubungi et al. 2005). The authors of this study surmised that minocycline would not be protective against calpain-dependent degeneration. Our results do not permit us to determine whether there was a trend for increased calpain activity at the 24 hour time-point due to time-dependent cumulative effects of the drug, or the natural course of the injury. The former suggests that a lower dosing regimen may prove protective, while if the latter is true, minocycline may be inherently toxic in this injury model.
In light of the observed deleterious effects of minocycline on neuronal survival in MT−/− mice after injury, we decided to further explore the effects of minocycline in P10 wild-type mice. Surprisingly, we found that despite an effective inhibition of the microglial response, minocycline promoted significant neuronal loss. These results raise a number of important questions that warrant further study. First, the contrasting effects of minocycline on microglia in MT−/− and wild-type mice leads us to speculate that MT I&II may be involved in regulating the microglial response to injury. Given that expression of MT I&II has been shown to alter the inflammatory response in multiple studies (Giralt et al. 2002; Potter et al. 2007; Potts et al. 2006) this effect invites further mechanistic exploration. Second, a minocycline-mediated inhibition of microglia was associated with decreased neuronal survival in wild-type animals. Concurrently, we observed an approximately two-fold decrease in the microglial response. While curtailment of an overactive microglial response may reduce neuronal exposure to pro-inflammatory cytokines, it is possible that decreasing the microglial response may have lessened protective effects such as secretion of neurotrophic factors and clearance of cellular debris and thus contributed to neuronal insult. These results should be further explored in the future.
Inconsistencies in the effects of minocycline in different species and various models of injury warrant explanation. While other investigators have reported negative outcomes with minocycline (Diguet et al. 2004; Tsuji et al. 2004), the possible mechanisms underlying these effects have not been addressed. MAPK p38 is a known target for minocycline and inhibiting its phosphorylation after injury confers neuroprotection (Lin et al. 2001; Suk 2004; Tikka et al. 2001; Xie et al. 2004). In the immature, metallothionein deficient brain, we find that minocycline increases phosphorylation MAPK p38. Moreover, we speculate that minocycline-mediated changes in the expression level and activity of GADD45γ may drive phosphorylation of MAPK p38 and should be examined in other models of injury where minocycline is harmful.
Study limitations
Results reported here are subject to several limitations. First, we selected only one, relatively high dose of minocycline for these experiments. However, other studies, including one in which minocycline was reported to limit tissue injury and neurological deficits following TBI (Sanchez Mejia et al. 2001), have demonstrated neuroprotective effects from similarly high-dose regimens (Dommergues et al. 2003; Lee et al. 2003; Yrjanheikki et al. 1998). Second, using non-biased stereology in a blinded, well powered study added strength to the present investigation, but we did not examine whether microglial accumulation occurred as a result of infiltration or proliferation of the resident population. Since, as mentioned, Fendrick et al. (Fendrick et al. 2005) found minocycline to exert no effect on microglial proliferation future studies should examine the conditions under which inhibition occurs and whether these affects preferentially effect microglial infiltration or proliferation. If we are to improve our understanding of how neuronal survival is influenced by the microglial population after injury, then it is vital to use reliable and sensitive techniques to quantify microglial numbers and activity levels.
Conclusions
Deficient expression of MT I&II in the immature brain is an important factor contributing to enhanced microglial reactivity and increased neuronal loss after injury. Minocycline has been shown to be neuroprotective in the setting of a number of CNS insults. Its ability to inhibit detrimental processes such as caspase activity and production of pro-inflammatory factors by microglia, afford it significant therapeutic potential. However, it is essential to consider the molecular mechanisms relevant to injury and determine how minocycline may influence the balance of neuroprotective and neurotoxic factors. As this study reveals, in the immature brain MT I&II play a role in minocycline’s neuroprotective balance.
Acknowledgments
This work was supported by the March of Dimes (5-FY01-535), NIH (K08NS41273), Children’s National Medical Center (CNMC) Research Advisory Board (#8929), and CNMC Board of Lady Visitors. Emily G. Potter was a predoctoral student in the Neuroscience Program of the Institute for Biomedical Sciences at the George Washington University. This work is from a dissertation presented to the above program in partial fulfillment of the requirements for the Ph.D. degree.
References
- 1.Expression profiling–best practices for data generation and interpretation in clinical trials. Nat Rev Genet. 2004;5(3):229–237. doi: 10.1038/nrg1297. [DOI] [PubMed] [Google Scholar]
- 2.Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 2002;52(1):54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- 3.Bantubungi K, Jacquard C, Greco A, Pintor A, Chtarto A, Tai K, Galas MC, Tenenbaum L, Deglon N, Popoli P, Minghetti L, Brouillet E, Brotchi J, Levivier M, Schiffmann SN, Blum D. Minocycline in phenotypic models of Huntington’s disease. Neurobiol Dis. 2005;18(1):206–217. doi: 10.1016/j.nbd.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 4.Bizat N, Hermel JM, Boyer F, Jacquard C, Creminon C, Ouary S, Escartin C, Hantraye P, Kajewski S, Brouillet E. Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: implications for Huntington’s disease. J Neurosci. 2003;23(12):5020–5030. doi: 10.1523/JNEUROSCI.23-12-05020.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Butler TL, Pennypacker KR. Temporal and regional expression of Fos-related proteins in response to ischemic injury. Brain Res Bull. 2004;63(1):65–73. doi: 10.1016/j.brainresbull.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Butler TL, Pennypacker KR. The transcriptional response to hypoxic insult controlled by FRA-2. Gene Expr. 2005;12(2):61–67. doi: 10.3727/000000005783992160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol. 2006 doi: 10.1016/j.expneurol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Dailey ME, Waite M. Confocal imaging of microglial cell dynamics in hippocampal slice cultures. Methods. 1999;18(2):222–230. 177. doi: 10.1006/meth.1999.0775. [DOI] [PubMed] [Google Scholar]
- 10.Diguet E, Fernagut PO, Wei X, Du Y, Rouland R, Gross C, Bezard E, Tison F. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur J Neurosci. 2004;19(12):3266–3276. doi: 10.1111/j.0953-816X.2004.03372.x. [DOI] [PubMed] [Google Scholar]
- 11.Domercq M, Matute C. Neuroprotection by tetracyclines. Trends Pharmacol Sci. 2004;25(12):609–612. doi: 10.1016/j.tips.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Dommergues MA, Plaisant F, Verney C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121(3):619–628. doi: 10.1016/s0306-4522(03)00558-x. [DOI] [PubMed] [Google Scholar]
- 13.Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98(25):14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan LW, Pang Y, Lin S, Rhodes PG, Cai Z. Minocycline attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neuroscience. 2005a;133(1):159–168. doi: 10.1016/j.neuroscience.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 15.Fan LW, Pang Y, Lin S, Tien LT, Ma T, Rhodes PG, Cai Z. Minocycline reduces lipopolysaccharide-induced neurological dysfunction and brain injury in the neonatal rat. J Neurosci Res. 2005b;82(1):71–82. doi: 10.1002/jnr.20623. [DOI] [PubMed] [Google Scholar]
- 16.Fendrick SE, Miller KR, Streit WJ. Minocycline does not inhibit microglia proliferation or neuronal regeneration in the facial nucleus following crush injury. Neurosci Lett. 2005;385(3):220–223. doi: 10.1016/j.neulet.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 17.Fox C, Dingman A, Derugin N, Wendland MF, Manabat C, Ji S, Ferriero DM, Vexler ZS. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2005;25(9):1138–1149. doi: 10.1038/sj.jcbfm.9600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giralt M, Penkowa M, Lago N, Molinero A, Hidalgo J. Metallothionein-1+2 protect the CNS after a focal brain injury. Exp Neurol. 2002;173(1):114–128. doi: 10.1006/exnr.2001.7772. [DOI] [PubMed] [Google Scholar]
- 19.Heo K, Cho YJ, Cho KJ, Kim HW, Kim HJ, Shin HY, Lee BI, Kim GW. Minocycline inhibits caspase-dependent and -independent cell death pathways and is neuroprotective against hippocampal damage after treatment with kainic acid in mice. Neurosci Lett. 2006;398(3):195–200. doi: 10.1016/j.neulet.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 20.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32(5):1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 21.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 22.Lee HK, Choi SS, Han KJ, Han EJ, Suh HW. Roles of adenosine receptors in the regulation of kainic acid-induced neurotoxic responses in mice. Brain Res Mol Brain Res. 2004;125(1–2):76–85. doi: 10.1016/j.molbrainres.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Lee SM, Yune TY, Kim SJ, Park DW, Lee YK, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma. 2003;20(10):1017–1027. doi: 10.1089/089771503770195867. [DOI] [PubMed] [Google Scholar]
- 24.Lin S, Zhang Y, Dodel R, Farlow MR, Paul SM, Du Y. Minocycline blocks nitric oxide-induced neurotoxicity by inhibition p38 MAP kinase in rat cerebellar granule neurons. Neurosci Lett. 2001;315(1–2):61–64. doi: 10.1016/s0304-3940(01)02324-2. [DOI] [PubMed] [Google Scholar]
- 25.Liu MC, Akle V, Zheng W, Dave JR, Tortella FC, Hayes RL, Wang KK. Comparing calpain- and caspase-3-mediated degradation patterns in traumatic brain injury by differential proteome analysis. Biochem J. 2006;394(Pt 3):715–725. doi: 10.1042/BJ20050905. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Milligan CE, Levitt P, Cunningham TJ. Brain macrophages and microglia respond differently to lesions of the developing and adult visual system. J Comp Neurol. 1991;314(1):136–146. doi: 10.1002/cne.903140113. [DOI] [PubMed] [Google Scholar]
- 27.Morimoto N, Shimazawa M, Yamashima T, Nagai H, Hara H. Minocycline inhibits oxidative stress and decreases in vitro and in vivo ischemic neuronal damage. Brain Res. 2005;1044(1):8–15. doi: 10.1016/j.brainres.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 28.Muessel MJ, Klein RM, Wilson AM, Berman NE. Ablation of the chemokine monocyte chemoattractant protein-1 delays retrograde neuronal degeneration, attenuates microglial activation, and alters expression of cell death molecules. Brain Res Mol Brain Res. 2002;103(1–2):12–27. doi: 10.1016/s0169-328x(02)00158-4. [DOI] [PubMed] [Google Scholar]
- 29.Natale JE, Cheng Y, Martin LJ. Thalamic neuron apoptosis emerges rapidly after cortical damage in immature mice. Neuroscience. 2002;112(3):665–676. doi: 10.1016/s0306-4522(02)00098-2. [DOI] [PubMed] [Google Scholar]
- 30.Natale JE, Knight JB, Cheng Y, Rome JE, Gallo V. Metallothionein I and II mitigate age-dependent secondary brain injury. J Neurosci Res. 2004;78(3):303–314. doi: 10.1002/jnr.20265. [DOI] [PubMed] [Google Scholar]
- 31.O’Reilly LA, Print C, Hausmann G, Moriishi K, Cory S, Huang DC, Strasser A. Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ. 2001;8(5):486–494. doi: 10.1038/sj.cdd.4400835. [DOI] [PubMed] [Google Scholar]
- 32.Park SK, Kim J, Seomun Y, Choi J, Kim DH, Han IO, Lee EH, Chung SK, Joo CK. Hydrogen peroxide is a novel inducer of connective tissue growth factor. Biochem Biophys Res Commun. 2001;284(4):966–971. doi: 10.1006/bbrc.2001.5058. [DOI] [PubMed] [Google Scholar]
- 33.Penkowa M, Espejo C, Martinez-Caceres EM, Poulsen CB, Montalban X, Hidalgo J. Altered inflammatory response and increased neurodegeneration in metallothionein I+II deficient mice during experimental autoimmune encephalomyelitis. J Neuroimmunol. 2001;119(2):248–260. doi: 10.1016/s0165-5728(01)00357-5. [DOI] [PubMed] [Google Scholar]
- 34.Penkowa M, Florit S, Giralt M, Quintana A, Molinero A, Carrasco J, Hidalgo J. Metallothionein reduces central nervous system inflammation, neurodegeneration, and cell death following kainic acid-induced epileptic seizures. J Neurosci Res. 2005;79(4):522–534. doi: 10.1002/jnr.20387. [DOI] [PubMed] [Google Scholar]
- 35.Pennypacker KR, Yang X, Gordon MN, Benkovic S, Miller D, O’Callaghan JP. Long-term induction of Fos-related antigen-2 after methamphetamine-, methylenedioxymethamphetamine-, 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine- and trimethyltin-induced brain injury. Neuroscience. 2000;101(4):913–919. doi: 10.1016/s0306-4522(00)00381-x. [DOI] [PubMed] [Google Scholar]
- 36.Pi R, Li W, Lee NT, Chan HH, Pu Y, Chan LN, Sucher NJ, Chang DC, Li M, Han Y. Minocycline prevents glutamate-induced apoptosis of cerebellar granule neurons by differential regulation of p38 and Akt pathways. J Neurochem. 2004;91(5):1219–1230. doi: 10.1111/j.1471-4159.2004.02796.x. [DOI] [PubMed] [Google Scholar]
- 37.Potter EG, Cheng Y, Knight JB, Gordish-Dressman H, Natale JE. Metallothionein I and II Attenuate the Thalamic Microglial Response following Traumatic Axotomy in the Immature Brain. J Neurotrauma. 2007;24(1):28–42. doi: 10.1089/neu.2006.0056.R1. [DOI] [PubMed] [Google Scholar]
- 38.Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, Manvelyan HM, Noble-Haeusslein LJ. Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. NeuroRx. 2006;3(2):143–153. doi: 10.1016/j.nurx.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14(2):215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray SK, Dixon CE, Banik NL. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol. 2002;17(4):1137–1152. doi: 10.14670/HH-17.1137. [DOI] [PubMed] [Google Scholar]
- 41.Repici M, Atzori C, Migheli A, Vercelli A. Molecular mechanisms of neuronal death in the dorsal lateral geniculate nucleus following visual cortical lesions. Neuroscience. 2003;117(4):859–867. doi: 10.1016/s0306-4522(02)00968-5. [DOI] [PubMed] [Google Scholar]
- 42.Ringger NC, O’steen BE, Brabham JG, Silver X, Pineda J, Wang KK, Hayes RL, Papa L. A novel marker for traumatic brain injury: CSF alphaII-spectrin breakdown product levels. J Neurotrauma. 2004;21(10):1443–1456. doi: 10.1089/neu.2004.21.1443. [DOI] [PubMed] [Google Scholar]
- 43.Robertson CL. Mitochondrial dysfunction contributes to cell death following traumatic brain injury in adult and immature animals. J Bioenerg Biomembr. 2004;36(4):363–368. doi: 10.1023/B:JOBB.0000041769.06954.e4. [DOI] [PubMed] [Google Scholar]
- 44.Robertson CL, Soane L, Siegel ZT, Fiskum G. The potential role of mitochondria in pediatric traumatic brain injury. Dev Neurosci. 2006;28(4–5):432–446. doi: 10.1159/000094169. [DOI] [PubMed] [Google Scholar]
- 45.Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3(1):69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48(6):1393–1399. doi: 10.1097/00006123-200106000-00051. discussion 1399–1401. [DOI] [PubMed] [Google Scholar]
- 47.Schwab JM, Beschorner R, Nguyen TD, Meyermann R, Schluesener HJ. Differential cellular accumulation of connective tissue growth factor defines a subset of reactive astrocytes, invading fibroblasts, and endothelial cells following central nervous system injury in rats and humans. J Neurotrauma. 2001;18(4):377–388. doi: 10.1089/089771501750170930. [DOI] [PubMed] [Google Scholar]
- 48.Seo J, Gordish-Dressman H, Hoffman EP. An interactive power analysis tool for microarray hypothesis testing and generation. Bioinformatics. 2006;22(7):808–814. doi: 10.1093/bioinformatics/btk052. [DOI] [PubMed] [Google Scholar]
- 49.Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24(9):2182–2190. doi: 10.1523/JNEUROSCI.5275-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57(6):563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 51.Suk K. Minocycline suppresses hypoxic activation of rodent microglia in culture. Neurosci Lett. 2004;366(2):167–171. doi: 10.1016/j.neulet.2004.05.038. [DOI] [PubMed] [Google Scholar]
- 52.Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95(4):521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 53.Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A. 2004;101(9):3071–3076. doi: 10.1073/pnas.0306239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21(8):2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166(12):7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- 56.Tsuji M, Wilson MA, Lange MS, Johnston MV. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp Neurol. 2004;189(1):58–65. doi: 10.1016/j.expneurol.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 57.Wang CX, Yang T, Shuaib A. Effects of minocycline alone and in combination with mild hypothermia in embolic stroke. Brain Res. 2003a;963(1–2):327–329. doi: 10.1016/s0006-8993(02)04045-3. [DOI] [PubMed] [Google Scholar]
- 58.Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279(19):19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci U S A. 2003b;100(18):10483–10487. doi: 10.1073/pnas.1832501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei X, Zhao L, Liu J, Dodel RC, Farlow MR, Du Y. Minocycline prevents gentamicin-induced ototoxicity by inhibiting p38 MAP kinase phosphorylation and caspase 3 activation. Neuroscience. 2005;131(2):513–521. doi: 10.1016/j.neuroscience.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 61.West MJ, Ostergaard K, Andreassen OA, Finsen B. Estimation of the number of somatostatin neurons in the striatum: an in situ hybridization study using the optical fractionator method. J Comp Neurol. 1996;370(1):11–22. doi: 10.1002/(SICI)1096-9861(19960617)370:1<11::AID-CNE2>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 62.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231(4):482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 63.Xie Z, Smith CJ, Van Eldik LJ. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia. 2004;45(2):170–179. doi: 10.1002/glia.10314. [DOI] [PubMed] [Google Scholar]
- 64.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95(26):15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96(23):13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zemke D, Majid A. The potential of minocycline for neuroprotection in human neurologic disease. Clin Neuropharmacol. 2004;27(6):293–298. doi: 10.1097/01.wnf.0000150867.98887.3e. [DOI] [PubMed] [Google Scholar]
- 67.Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417(6884):74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]