

Summary
Gene-environment interactions are determining factors for the etiology of psychiatric disorders, diabetes and cancer, and are thought to contribute to disease inheritance across generations. Small non-coding RNAs (sncRNAs) are potential vectors at the interface between genes and environment. Here, we report that environmental conditions involving traumatic stress in early life in mice altered microRNAs (miRNAs) expression, and behavioral and metabolic responses in the progeny. Several miRNAs were affected in the serum and brain of both, the traumatized animals and their progeny when adult, but also in the sperm of traumatized males. Injection of sperm RNAs from these males into fertilized wild-type oocytes reproduced the behavioral and metabolic alterations in the resulting offspring. These results strongly suggest that sncRNAs are sensitive to environmental factors in early life, and contribute to the inheritance of trauma-induced phenotypes across generations. They may offer potential diagnostic markers for associated pathologies in humans.
While the genetic make-up of an individual contributes to disease risk and heritability 1, environmental factors, in particular, adverse and traumatic experiences in early life are also critical. How they mediate their influence is poorly understood but likely involves non-genetic mechanisms. Small non-coding RNAs (sncRNAs) are potential mediators of gene-environment interactions that can relay signals from the environment to the genome and exert regulatory functions on gene activity 2. They are implicated in gene dysregulation in many diseases including psychiatric and neurological conditions, cancer and metabolic disorders 2-4. Recent studies in C. elegans 5,6 and mice 7,8 have suggested that sncRNAs can mediate non-Mendelian inheritance of traits or phenotypes acquired across life. SncRNAs are abundant in mature sperm in mammals and may therefore convey transgenerational inheritance 9,10. Whether sncRNAs in germ cells are influenced by environmental factors like early traumatic stress and contribute to associated pathological traits is unknown.
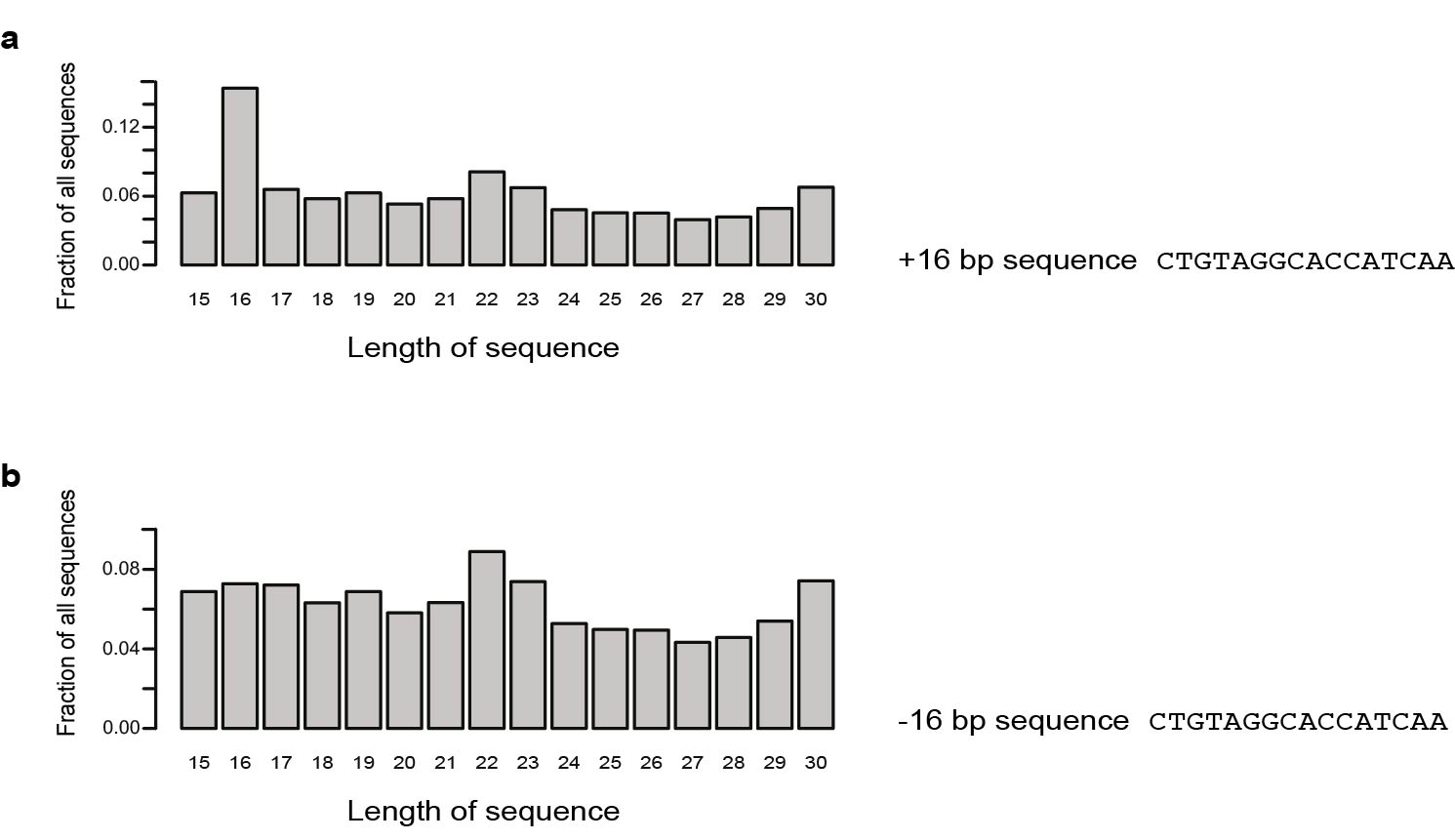
We investigated the involvement of sperm sncRNAs in the impact of traumatic stress in early life across generations and for this, first examined sncRNAs content of adult mouse sperm in normal conditions. Deep sequencing of purified sperm RNA (Supplementary Fig. 1) identified several populations of short RNAs mapping to the mouse genome to a different degree and fidelity (Fig. 1a, Supplementary Fig. 2a). Many had typical miRNA size (21–23bp), and others aligned to piRNA clusters and had typical piRNA size (26–31bp) (Supplementary Fig. 2b,c). Some 15–44bp reads mapped to ribosomal RNAs, to small cytoplasmic, nuclear or transfer RNAs, or to repeat regions such as retrotransposons (Fig. 1b,c). A 16bp read giving an unusually large peak mapped to a specific piRNA sequence (Supplementary Fig. 3). Reads of all sizes mapped uniquely to mitochondrial DNA sequences (Fig. 1d), consistent with the presence of mitochondria in adult sperm 11.
Figure 1. SncRNAs in adult sperm.

Mapping of 15–44bp sequencing reads to a) the mouse reference genome, b) ribosomal RNAs, c) other non-coding RNAs and repeat regions and d) mitochondrial DNA, with multiple (black) or unique (grey) hits (n=16 mice, pooled in 4 samples). % total reads represents the proportion of reads with a given size mapping to the mouse genome or selected sequences over the total number of same-size reads. (e) Heatmap showing miRNAs (>100 reads) in control libraries which are altered by MSUS in adult sperm (n=3 (each pooled from 5 mice) in each group). The blue-to-yellow scale is the number of normalized reads of a given sample over the mean normalized reads of all control samples for each miRNA. Bioinformatic analyses were performed twice using two independent methods. Data are mean ± s.e.m.
We next examined the impact of exposure to traumatic stress in early life on sperm sncRNAs using a mouse model of unpredictable maternal separation combined with unpredictable maternal stress (MSUS) (Supplementary Fig. 4) 12-14. In these mice, behavioral responses are affected by MSUS across generations. On the elevated plus maze, a test based on the natural avoidance of mice for open and unfamiliar space, MSUS males had shorter latency to first enter an open arm than controls (F1, Fig. 2a). This effect was not due to altered locomotor activity (Supplementary Fig. 5a), suggesting reduced avoidance and fear. In a light-dark box, a task based on the aversion of rodents for brightly lit areas, MSUS males spent more time in the illuminated compartment than controls (F1, Fig. 2b), suggesting altered response to aversive conditions. On a Porsolt forced swim test, a test of behavioral despair, MSUS males spent more time floating than controls (F1, Fig. 2c), suggesting depressive-like behaviors. Strikingly, these behavioral traits were transmitted to the F2 offspring. On the elevated plus maze, F2 MSUS mice had a shorter latency to first enter an open arm than F2 controls (Fig. 2a), but normal locomotor activity (Supplementary Fig. 5b). They spent more time in the bright compartment of the light-dark box, and had depressive-like behaviors on the forced swim test (Fig. 2b,c).
Figure 2. Behavioral responses in MSUS males across generations, and in mice derived from RNA-injected oocytes.

(a) Latency to first enter an open arm on an elevated plus maze in F1 (control, n=8; MSUS, n=18; t(24)=2.37) and F2 (control, n=30; MSUS, n=25; t(41.98)=3.74) mice. (b) Time spent in the bright compartment of the light dark box in F1 (control, n=16; MSUS, n=21; t(35)=−2.14) and F2 (control, n=33; MSUS, n=36; t(41.61)=−3) mice. (c) Time spent floating on the forced swim test in F1 (control n=14, MSUS n=16; t(28)=−2.34) and F2 (control n=19, MSUS n=20; t(37)=−2.36) mice. Results replicated in two independent experiments. (d) Insulin concentration in serum in F1 (control, n=5; MSUS, n=9; t(12)=0.28) and F2 (control, n=10; MSUS, n=10; t(18)=2.1) males. (e, f) Glucose level in blood in e) F2 MSUS (control, n=8; MSUS, n=7; F(1,13)=5.64) and f) Controls-RNAinj (n=8) and MSUS-RNAinj (n=8) (F(1,14)=9.72) males at baseline (time 0) and 15, 30 and 90 minutes after stress initiation. Data are mean ± s.e.m. *p<0,05, **p<0,01, ***p<0,001.
Since early stress can be a strong metabolic dysregulator 15, we next examined glucose metabolism. Insulin in serum was normal in F1 MSUS animals, but lower than controls in F2 MSUS progeny (Fig. 2d). Blood glucose was also normal in F1 animals (Supplementary Fig. 6a) but lower in F2 MSUS mice, both at baseline and following an acute restraint stress (Fig. 2e). Further, F1 MSUS males had normal baseline glucose level and clearance on a glucose tolerance test (GTT) but a larger decline in blood glucose on an insulin tolerance test (ITT) (Supplementary Fig. 6b,c). F2 MSUS animals had normal glucose at baseline, but lower glucose rise on GTT and normal glucose decrease on ITT (Supplementary Fig. 7a,b). These anomalies suggest insulin hypersensitivity. Further, F2 MSUS animals (not F1) also showed hypermetabolism as their body weight was lower than in controls despite higher caloric intake (Supplementary Fig. 6d,e and 7c,d). The alterations were overall more marked in F2 mice probably because the effects of stress are present starting at conception while they occur only after birth in F1.
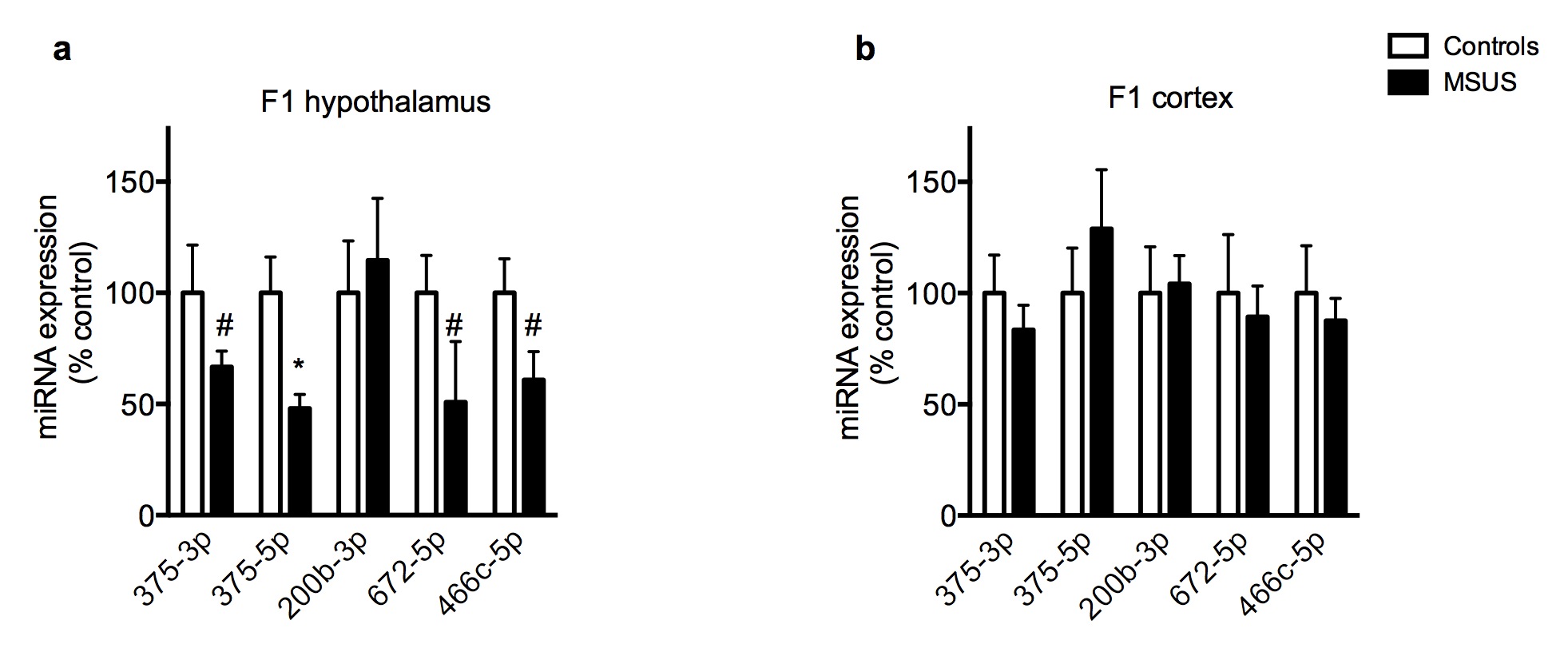
Because the traits induced by MSUS are transmitted to the progeny, we examined whether sperm sncRNAs are affected. Deep sequencing analyses revealed that several miRNAs were upregulated in F1 MSUS sperm (Fig. 1e), and 73 potential miRNAs targets, implicated in DNA/RNA regulation, epigenetic regulation or RNA binding and processing were identified (Supplementary Table 1). PiRNAs were also affected, in particular cluster 110 which was downregulated in MSUS sperm (Supplementary Fig. 8a,b). These results indicate an effect of MSUS on several sncRNA populations. Validation by quantitative RT-PCR confirmed that miR-375-3p and -5p, miR-200b-3p, miR-672-5p and miR-466-5p were up-regulated in F1 MSUS sperm (Fig. 3a). MiRNAs were also altered in serum, and in hippocampus and hypothalamus (not cortex), brain structures involved in stress response, in adult F1 MSUS animals (Fig. 3b,c and Supplementary Fig. 9a,b). Moreover, miRNAs were affected in the serum and hippocampus of adult F2 MSUS mice but not in F2 sperm (Fig. 3d-f). Consistently, miRNAs were normal in F3 hippocampus (Supplementary Fig. 10). Since F3 MSUS animals have behavioral symptoms similar to F1 and F2 animals 13 despite normal sperm miRNA level in F2 males, it is possible that the changes in miRNAs initially occurring in sperm cells are transferred to other non-genomic or epigenetic marks such as DNA methylation or histone posttranslational modifications, for maintenance and further transmission16,17.
Figure 3. Molecular effects of MSUS in adult F1 and F2 mice.

(a-f) RT-qPCR in (a) sperm of F1 control and MSUS adult males (miR-375-3p: control, n=10; MSUS, n=9; t(7.679)=−2.79. miR-375-5p: control, n=10; MSUS, n=10; t(18)=−3.19. miR-200b-3p: control, n=10; MSUS, n=10; t(11.17)=−2.46. miR-672-5p: control, n=10; MSUS, n=10; t(9.38)=−2.92. miR-466c-5p: control, n=10; MSUS, n=10 t(13.05)=−2.4), (b) serum of F1 control and MSUS adult males (miR-375-3p: control, n=8; MSUS, n=8; t(7.06)=−5.17. miR-375-5p: control, n=8; MSUS, n=8; t(7.01)=4.33. miR-200b-3p: control, n=8; MSUS, n=7; t(9.3)=0.90. miR-672-5p: control, n=8; MSUS, n=8; t(8.8)=2.24. miR-466c-5p: control, n=8; MSUS, n=7; t(7.90)=2.26), (c) hippocampus of F1 control and MSUS adult males (miR-375-3p: control, n=8; MSUS, n=6; t(12)=−2.34. miR-375-5p: control, n=8, MSUS, n=6; t(6.045)=0.59. miR-200b-3p: control, n=8; MSUS, n=6; t(5.8)=−1.1. miR-672-5p: control, n=8; MSUS, n=6; t(12)=−0.54. miR-466c-5p: control, n=8; MSUS, n=6; t(11)=−2.79), (d) serum of F2 control and MSUS adult males (miR-375-3p: control, n=6; MSUS, n=6; t(9)=0.93. miR-375-5p: control, n=5; MSUS, n=6; t(9)=0.93. miR-200b-3p: control, n=6; MSUS, n=6; t(10)=1.38. miR-672-5p: control, n=6; MSUS, n=6; t(5.29)=2.08. miR-466c-5p: control, n=6; MSUS, n=6; t(10)=2.21), (e) hippocampus of F2 control and MSUS adult males (miR-375-3p: control, n=7; MSUS, n=8; t(8.62)=−2.74. miR-375-5p: control, n=14; MSUS, n=15; t(17,89)=−2,14. miR-200b-3p: control, n=8; MSUS, n=8; t(14)=−1.47. miR-672-5p: control, n=7; MSUS, n=8; t(13)=−2.01). miR-466c-5p: control, n=7; MSUS, n=8; t(13)=−2.15), (f) sperm of F2 control and MSUS adult males (miR-375-3p: control, n=8; MSUS, n=8; t(14)=0.26; miR-375-5p: control, n=8; MSUS, n=8; t(14)=0.94; miR-200b-3p: control, n=4; MSUS, n=3; t(5)=0.44; miR-672-5p: control, n=4; MSUS, n=4; t(6)=−0.24; miR-466c-5p: control, n=4; MSUS, n=4; t(6)=−1.16). (g, h) Level of Ctnnb1 (g) mRNA (control n=7; MSUS n=7; t(12)=0.4) and (h) protein (control n=7; MSUS n=6; t(11)=3.26) in hippocampus of F2 control and MSUS males. Results replicated in an independent experiment using samples from a different batch of animals. Data are mean ± s.e.m. #p<0.1, *p≤0.05, **p<0.01, ***p≤0.001.
MiR-375 is implicated in stress response and metabolic regulation 18,19. We found that mimicking the effect of stress by injecting corticosterone in vivo increases miR-375 expression in the hippocampus (Saline n=14, corticosterone n=14; t(26)=2.27). One of miR-375 predicted targets is catenin β1 (Ctnnb1), a protein implicated in stress pathways 20. Cultured cells transfected with a miR-375 expression vector showed downregulation of Ctnnb1 (Control n=3; transfected n=3, mRNA t(4)=2.78 protein: t(4)=5,14), confirming that miR-375 targets Ctnnb1. Consistently, Ctnnb1 was decreased in F2 MSUS hippocampus (Fig. 3g,h), suggesting that miR-375 alteration has functional consequences on Ctnnb1 expression in vivo.
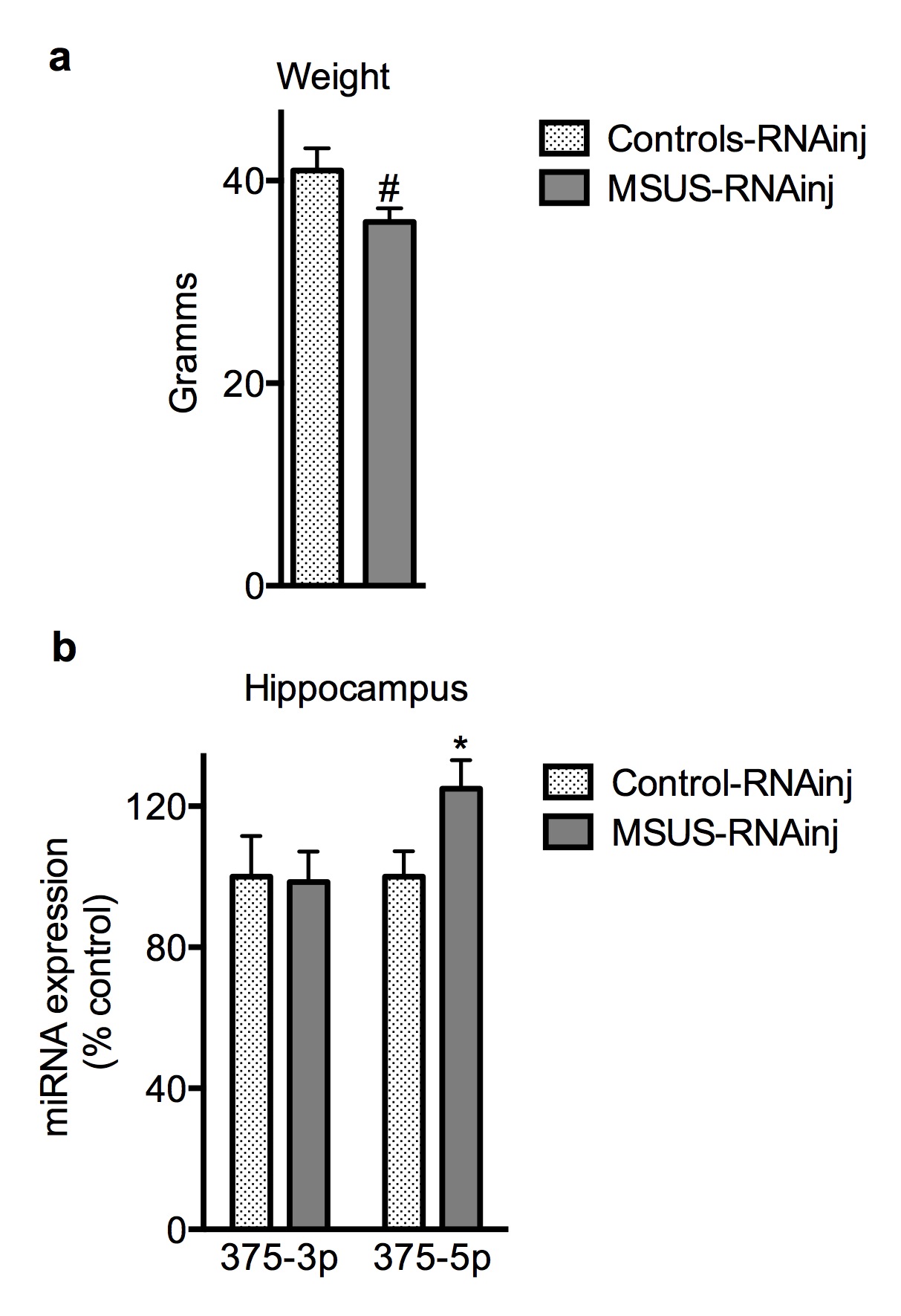
Finally, we tested the causal link between sperm RNAs and the effects of MSUS across generations by microinjecting RNAs purified from sperm from MSUS males (Supplementary Fig. 1) into wild-type fertilized mouse oocytes. On the elevated plus maze, MSUS-RNAinj animals (n=20) had a lower latency to first enter an open arm than controls-RNAinj (n=19) animals (t(37)=2.67) but normal locomotion (Supplementary Fig. 5c). In the light dark box, they spent more time in the bright compartment (Controls-RNAinj (n=15), MSUS-RNAinj (n=17) males, t(30)=−3.77), and on the forced swim test (Controls-RNAinj (n=18), MSUS-RNAinj, (n=20) males (t(37)=−2.19), they floated longer than controls-RNAinj. Further in MSUS-RNAinj animals, insulin (Controls-RNA-inj (n=4) and MSUS-RNAinj (n=7) (t(9)=2.31)) and glucose at baseline and after acute stress (Fig. 2f) were decreased, body weight was lower, and miR-375-5p was up-regulated in the hippocampus (Supplementary Fig. 11a,b). These results indicate comparable behavioral, metabolic and molecular effects by either, direct exposure to MSUS during early postnatal life or injection of sperm RNAs from MSUS males. Notably, the offspring of MSUS-RNAinj animals also showed depressive-like behaviors (Supplementary Fig. 12), indicating transmission of the effects of injected sperm RNAs.
These findings provide novel evidence in mammals that RNA-dependent processes contribute to the transmission of acquired traits. They underscore the importance of sncRNAs in germ cells, and highlight their sensitivity to early traumatic stress. They newly demonstrate the consequences of their exposure to such traumatic experience in early life across generations. The identification of several miRNAs and putative targets as mediators of these effects provide novel molecular markers of traumatic stress for potential diagnostic of stress predisposition and stress-induced disorders in humans.
Sequencing data were deposited to NCBI’s Gene Expression Omnibus with the accession number: GSE50132.
Methods
Animals
C57Bl/6J mice were maintained under a reverse light-dark cycle in a temperature and humidity-controlled facility with food and water ad libitum. All experimental manipulations were performed during the animals’ active cycle in accordance with guidelines and regulations of the cantonal veterinary office, Zurich.
Mice treatment
For unpredictable maternal separation combined with maternal stress (MSUS), C57Bl/6J dams (2-3 months-old) and litters were selected at random and subjected to daily 3hr proximal separation from postnatal day 1 to 14 as described previously 13. Control animals were left undisturbed apart from a cage change once a week until weaning (postnatal day 21). Once weaned, pups were reared in social groups (4-5 mice/cage) composed of animals subjected to the same treatment but from different dams to avoid litter effects. To obtain second and third generations, adult F1 males (>5 months old) were bred with primiparous C57Bl/6J females.
Preparation of sperm samples
Mature sperm cells extracted from cauda epididymis from males were separated from somatic cells by counterflow centrifugal elutriation using a Beckman JE-5.0 elutriation rotor in a Sanderson chamber and a Beckman Avanti J-26 XPI Elutriation Centrifuge. Briefly, cauda epidydimis and epidydimis were collected in culture dishes in PBS pH 7.4 (0.2 M phosphate, 1.5 M NaCl) containing 5% BSA, 5% non-fat dry milk powder, 1M CaCl2, 1M MgCl2 filtered through a cellulose acetate membrane (Sartorius) and cut into small pieces to release sperm cells. The suspension was loaded into the elutriation chambers, which form part of the centrifuge rotor, using a rotor speed of 3500 revolutions per minute (rpm) and a pump rate of 7ml/min. Mature sperm was eluted by increasing the pump rate to 31ml/min. Purity of the elutriate was confirmed by inspecting the eluted sperm cells under a light microscope.
RNA sequencing
Total RNA was prepared from adult mouse sperm using a standard Trizol protocol. The quantity and quality of RNAs were determined by Agilent 2100 Bioanalyser (Agilent Technologies), Qubit® fluorometer (Life Technologies) and mass spectrometry (Supplementary Fig. 1 and see below). Pure RNA preparations with no DNA or protein contamination were used for sequencing. Sequencing was done using an Illumina Genome Analyzer (Illumina, San Diego, USA) at Fasteris AG, Geneva, Switzerland. Small RNA libraries were prepared according to a modified Illumina v1.5 protocol. Briefly, small RNAs of <50 nt were purified on an acrylamide gel. Universal miRNA cloning linker (New England Biolabs) instead of 3′ adapters and then 5′ Illumina adapters were single-stranded ligated with T4 truncated RNA and T4 ligase respectively. The constructs were purified on an acrylamide gel to remove empty adapters then reverse-transcribed and PCR-amplified. The primers used for cDNA synthesis and PCR were designed to insert an index in the 3- adapter. This index enables assignation of a specific read to the corresponding library, among the multiplexed libraries of one sequencing lane. High-throughput sequencing was performed on a Genome Analyzer HiSeq 2000 for 50 cycles plus 7 cycles to read the indexes. After demultiplexing and adapter removal, an average of 16067416 pass filter reads was obtained in the libraries.. Sequencing results were validated by RT-qPCR on the same samples as those used for deep sequencing.
Mass spectrometry
In-solution trypsin digestion of protein
Tryptic digestions were performed as previously described with slight modifications21. In brief, 1 ul of sperm RNA samples was made up to 100μl with 25 mM ammonium bicarbonate, pH 8.0. Samples were reduced with 10 mM dithiothreitol (DTT) for 45 min at 56°C and alkylated with 40 mM iodoacetamide for 30 min. Samples were digested overnight with trypsin (Promega) at 37 °C and desalted using C18 Ziptip prior to MS analysis using ESI-LTQ-Orbitrap Velos. MS and MS/MS data were searched using Mascot and searches of MS/MS spectra used a Swiss-Prot protein database.
miRNA targets prediction
The DIANA-microT CDS miRNA target prediction algorithm 22 which is based on potential binding site in the 3′ untranslated region of the mRNA and predicted stable thermodynamic binding, was used to predict target genes of miRNAs. Binding score threshold was set to 0.9 (1=highest potential binding predicted, 0=no binding predicted) and only the top 100 targets were considered for each miRNA, to only consider predictions with highest probability.
RNAs injection in fertilized oocytes
Fertilized oocytes were collected from B6D2F1 (Janvier) females superovulated by intraperitoneal (ip) injection of 5 IU pregnant mare serum gonadotropin followed by 5 IU human chorionic gonadotrophin 48 hours later, then mated with B6D2F1 males. One to two picoliters of 0,5 ng/μl solution of total RNA isolated and pooled from sperm from 5 adult MSUS or control males (same samples used for sequencing) dissolved in 0,5 mM Tris-HCl, pH 8.0, 5 uM EDTA were microinjected into the male pronucleus of fertilized eggs using a standard microscope and DNA microinjection method 23.
Behavioral testing
The experimenter was blind to treatment, and behaviors were monitored by direct observation and videotracking (Viewpoint, France). All behavioral tests were conducted in adult male animals.
Elevated plus maze
The elevated plus maze consisted of a platform with two open (without walls) and two closed (with walls) arms (dark gray PVC, 30cm × 5cm) elevated 60 cm above the floor. All experiments were performed in red light (15W). Each mouse was placed on the central platform, facing a closed arm, and observed for a 5-min period. The latency to enter an open arm, the time spent in each arm and the total distance moved were automatically recorded by a videotracking system. The number of rearing, protected (body in closed arm) and unprotected (body in opened arm) stretch-attend postures in the center of the maze were manually recorded.
Light/dark box
Each mouse was placed in the lit compartment (white walls, 130 lux) of a plastic box (40 × 42 × 26cm) split into two unequal compartments (2/3 lit, 1/3 dark compartment with black walls and covered by a black lid) by a divider with an opening (5×5cm). The animal can move freely from the lit to the dark compartment during a 10 mins session. The time spent in each compartment and the latency to enter the dark compartment were measured manually.
Forced swim test
Mice were placed in a small tank of water (18 cm high, 13 cm diameter, 18 ± 1°C, filled up to 12 cm) for 5 min. Floating duration was scored manually.
Serum insulin and blood glucose analyses
For non-fasted baseline measurements of insulin, blood was collected, stored overnight at 4°C, centrifuged for 10 minutes at 2,000 g at 4°C, then serum was collected and stored at −80°C until analyzed. Insulin was measured in serum using a mouse insulin ELISA (Alpco). The sensitivity of the assay was 0.06 ng/mL, and the intra-assay coefficient of variation was 3.7%. Glucose in non-fasted animals was measured in blood samples at baseline and after acute stress. Mice were restrained for 30 minutes (between 14h00 and 16h00) in a plastic tube and blood samples were collected from a tail nick 0, 15, 30 and 90 minutes after initiation of restraint. For the glucose tolerance test, mice were fasted for 6 hours. Glucose was measured in blood samples at baseline, and 0, 15, 30 and 90 minutes after i.p. injection of 2mg/g body weight glucose in 0.45% saline (injection started at 2 pm). For the insulin tolerance test, mice were fasted for 6 hours. Glucose was measured in blood samples at baseline, and 0, 15, 30, 90 and 120 minutes after i.p. injection of 1mU/g body weight insulin (NovoRapid Novo Nordisk A/S) in sterile 0.9% saline. If blood glucose fell below 1.7 mM/ml, animals were rescued with i.p. injection of 2mg/g glucose and were removed from the experiment. Glucose level was determined in fresh tail blood using an Accu-Chek Aviva device (Roche).
Caloric intake measurement
The amount of consumed food was measured for each mouse (4 months old) every 24h. Caloric intake was calculated as the mean amount of food intake over 48 hours in relation to mean body weight (caloric intake = mean food intake/mean body weight).
Cell culture
Mouse neuroblastoma (N2a) cells were obtained from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified eagle medium (DMEM) with 10% fetal bovine serum. Approximately 300,000 cells from three different passage number stocks were simultaneously platted in 6-well culture plates. Cells were treated with miScript miRNA mimic (Qiagen) and a negative control siRNA with no known targets in mammalian genome (All Stars Negative siRNA, Qiagen) at 60 nM for 48 hours. Transfections were carried out using lipid-based HiPerfect transfection reagent (Qiagen). Cells were harvested 48 hours after transfection and total RNA was isolated using standardized Trizol protocol.
Western blotting
Western blotting was performed as previously described 24. Briefly, 30-60 μg proteins were resolved on 10-12% SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad). Membranes were blocked (milk 5%) then incubated in primary and secondary antibodies. Band intensity was determined and quantified using an Odyssey IR scanner (Li-Cor Biosciences). βactin (1:15000; mouse monoclonal; Sigma #A5316) was used as internal control. The following antibodies were used: Ctnnb1 (1:2000, mouse monoclonal; BD Transduction Laboratories #610153); and goat anti-mouse (IRDye 680 nm, 1:10,000; Li-Cor Biosciences). Samples from different groups were processed on the same blots. Data are expressed as percent relative to controls.
Reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR)
For miRNAs, DNaseI-treated RNA isolated from pure sperm cells or hippocampal samples (Trizol) was reverse-transcribed (RT) using the miScript reverse transcription kit (Qiagen). RT-qPCR were performed in a LightCycler 480 qPCR (Roche) using miScript probes (Qiagen) according to the manufacturer’s recommendations. For normalization of Ct values for miRNAs, we used miR-101b for sperm, ribosomal Rrnu6 for hippocampus and miR-195 for serum. For mRNAs, RT-qPCR were performed using SYBR green (Roche) on a Light-Cycler II 480 (Roche) according to the manufacturer’s recommendations. Data for tissue samples were normalized to two endogenous controls (Tubd1 and Hprt), and data for cell samples were normalized to GAPDH. Cycling conditions: 5 min at 95°C, 45 cycles with denaturation (10 sec at 95°C), annealing (10 sec at 60°C) and elongation (10 sec at 72°C). Primers were as follows: Tubd1 forward: TCTCTTGCTAACTTGGTGGTCCTC reverse: GCTGGGTCTTTAAATCCCTCTACG, Hprt forward: GTTGGGCTTACCTCACTGCTTTC reverse: CCTGGTTCATCATCGCTAATCACG, Ctnnb1 forward: ATGGAGCCGGACAGAAAAGC reverse: CTTGCCACTCAGGGAAGGA, GAPDH forward: CCACTGGTGCTGCCAAGGCT reverse: GGCAGGTTTCTCCAGGCGGC.
Deep sequencing data analyses
Overall analysis of small RNAs libraries
After adapter trimming, sequence reads were sorted based on length (number of nucleotides) and only 15-44bp reads were used for analysis. The number of reads of each size was counted and normalized to the total number of reads. The obtained counts were averaged across control libraries. Reads were aligned to the mouse genome (UCSC mm9, http://hgdownload.cse.ucsc.edu/goldenPath/mm9/bigZips/) 25, ncRNAs and repeat elements from rmsk (UCSC mm9, http://hgdownload.cse.ucsc.edu/goldenPath/mm9/bigZips/) and mitochondrial DNA (UCSC mm9, http://hgdownload.cse.ucsc.edu/goldenPath/mm9/bigZips/) using a BWA software 26 with mismatch tolerance of up to 2bp for 15–17bp inserts, and 3 and 4bp for 18–38bp and 37–44bp inserts respectively. Sequencing reads were aligned to different selected features (ncRNAs, mtDNA) separately to “force” mapping (sequences of the mouse genome other than the respective feature were masked to prevent alignment to featured regions), and the different features matching each read were determined. The percentage of reads of a given size mapping once or multiple times (unique or multiple hits) to the mouse genome or to a given feature (100% represents all reads of a given size within a library) was determined and averaged across all control libraries. In figure 1a, un-mapped reads may result from PCR pre-sequencing amplification artifacts, incomplete trimming of adapters or sequencing errors, or may reflect the presence of RNA splicing products. In Figure 1b, some reads map to ribosomal RNAs (cleaved) with multiple hits, reflecting ribosomal RNAs cleavage and no functional ribosomes in sperm cells (transcriptionally quiescent) 27. Reads mapping to mitochondrial DNA showing only unique hits allow unambiguous attribution to mitochondria DNA. For quantitative comparison, one control library showing a substantially lower number of total reads, possibly due to a bias in library preparation prior to sequencing, was excluded from the analyses.
Analysis of miRNA and piRNA sequences
Perfect matches to mature miRNA sequences downloaded from miRBase 28 were identified using custom Perl scripts. One sample was removed from the analysis due to much lower total read count (see Supplementary Figure 3a). Read counts were identified for each miRNA and normalized using DESeq 29. A Wilcox unpaired test on the normalized data was used to identify miRNAs showing a statistically significant difference after MSUS treatment. piRNAs were identified by determining sequences that aligned to annotated piRNA clusters 30 using Bowtie 31. Alignments to piRNA cluster sequences were conducted as a custom-built “genome” with parameters −k 1 −v 0 —best (to select only the best aligning read with 0 mismatch). After inspection to confirm enrichment of piRNA-like sequences (Supplementary Figure 3 c), all sequences with a length of 26–32 nucleotides and a T as the first nucleotide were selected from the libraries and used for alignment to piRNA clusters. DEseq was then used to normalize read counts for each cluster and the differential expression and statistical significance of the differential expression was calculated using a negative binomial test within the DESeq package.
Statistical analyses
No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in our previous publications on th the MSUS model12-14. Two-tailed Student t tests were used to assess statistical significance for behavioral, quantitative RT- qPCR, insulin, bodyweight and caloric intake measurements. The remaining metabolic experiments were analyzed using repeated measurements ANOVAs. All analyzed data matched the requirements for parametric statistical tests (normal distribution). If variance was not homogenous between groups (determined by Levene’s test), adjusted p-value, t-value and degree of freedom were determined. miRNAs were analysed using Mann Whitney U test and piRNAs were analyzed using negative binomial test with and without Bonferoni multiple test correction. Values over two standard deviations away from the mean of each group were considered outliers and excluded from analysis. All statistics were computed with SPSS. All reported replicates were biological replicates, or pooled samples from biological replicates in the case of sequencing samples. Significance was set at p < 0.05 for all tests. Error bars represent SEM in all figures.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the Austrian Academy of Sciences, the University Zürich, the Swiss Federal Institute of Technology, Roche, the Swiss National Science Foundation, and The National Center of Competence in Research “Neural Plasticity and Repair”. P.S. was supported by a Gonville and Caius College fellowship. We thank Minoo Rassoulzadegan and Valérie Grandjean for help with the sperm purification, Francesca Manuella and Heiko Hörster for assistance with the MSUS paradigm, Hans Welzl for help with behavior, Grégoire Vernaz for help with Western blotting, Ry Tweedie-Cullen and Paolo Nanni for help with mass spectrometry, Andrea Patrignani for advice on DNA/RNA quality assessment, and Alon Chen and Andrea Brunner for constructive discussions.
References
- 1.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nature reviews. Neuroscience. 2012;13:528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abe M, Bonini NM. MicroRNAs and neurodegeneration: role and impact. Trends in cell biology. 2013;23:30–36. doi: 10.1016/j.tcb.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nature reviews. Molecular cell biology. 2012;13:239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burton NO, Burkhart KB, Kennedy S. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2011;108:19683–19688. doi: 10.1073/pnas.1113310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu SG, et al. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat Genet. 2012;44:157–164. doi: 10.1038/ng.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu WM, et al. Sperm-borne microRNA-34c is required for the first cleavage division in mouse. Proc Natl Acad Sci U S A. 2012;109:490–494. doi: 10.1073/pnas.1110368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rassoulzadegan M, et al. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441:469–474. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- 9.Kawano M, Kawaji H, Grandjean V, Kiani J, Rassoulzadegan M. Novel small noncoding RNAs in mouse spermatozoa, zygotes and early embryos. PLoS One. 2012;7:e44542. doi: 10.1371/journal.pone.0044542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krawetz SA, et al. A survey of small RNAs in human sperm. Hum Reprod. 2011;26:3401–3412. doi: 10.1093/humrep/der329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pena FJ, et al. Mitochondria in mammalian sperm physiology and pathology: a review. Reproduction in domestic animals = Zuchthygiene. 2009;44:345–349. doi: 10.1111/j.1439-0531.2008.01211.x. [DOI] [PubMed] [Google Scholar]
- 12.Franklin TB, Linder N, Russig H, Thony B, Mansuy IM. Influence of early stress on social abilities and serotonergic functions across generations in mice. PLoS One. 2011;6:e21842. doi: 10.1371/journal.pone.0021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franklin TB, et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry. 2010;68:408–415. doi: 10.1016/j.biopsych.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 14.Weiss IC, Franklin TB, Vizi S, Mansuy IM. Inheritable effect of unpredictable maternal separation on behavioral responses in mice. Front Behav Neurosci. 2011;5:3. doi: 10.3389/fnbeh.2011.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rose AJ, Herzig S. Metabolic control through glucocorticoid hormones: an update. Molecular and cellular endocrinology. 2013;380:65–78. doi: 10.1016/j.mce.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Drake AJ, Seckl JR. Transmission of programming effects across generations. Pediatric endocrinology reviews: PER. 2011;9:566–578. [PubMed] [Google Scholar]
- 17.Sharma A. Transgenerational epigenetic inheritance: Focus on soma to germline information transfer. Progress in biophysics and molecular biology. 2013;113:439–446. doi: 10.1016/j.pbiomolbio.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 18.El Ouaamari A, et al. miR-375 targets 3′-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes. 2008;57:2708–2717. doi: 10.2337/db07-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang N, et al. MicroRNA 375 mediates the signaling pathway of corticotropin-releasing factor (CRF) regulating pro-opiomelanocortin (POMC) expression by targeting mitogen-activated protein kinase 8. J Biol Chem. 2013;288:10361–10373. doi: 10.1074/jbc.M112.425504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maguschak KA, Ressler KJ. A role for WNT/beta-catenin signaling in the neural mechanisms of behavior. Journal of neuroimmune pharmacology: the official journal of the Society on NeuroImmune Pharmacology. 2012;7:763–773. doi: 10.1007/s11481-012-9350-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplementary References
- 21.Tweedie-Cullen RY, Reck JM, Mansuy IM. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. Journal of proteome research. 2009;8:4966–4982. doi: 10.1021/pr9003739. [DOI] [PubMed] [Google Scholar]
- 22.Maragkakis M, et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics. 2009;10:295. doi: 10.1186/1471-2105-10-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogan BC, F., Lacy L. Manipulating the Mouse Embryo - A Laboratory manual. Second Edition Cold Spring Harbor Laboratory; NY: 1994. [Google Scholar]
- 24.Koshibu K, et al. Protein phosphatase 1 regulates the histone code for long-term memory. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:13079–13089. doi: 10.1523/JNEUROSCI.3610-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic acids research. 2008;36:D173–177. doi: 10.1093/nar/gkm696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson GD, et al. Cleavage of rRNA ensures translational cessation in sperm at fertilization. Mol Hum Reprod. 2011;17:721–726. doi: 10.1093/molehr/gar054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Betel D, Sheridan R, Marks DS, Sander C. Computational analysis of mouse piRNA sequence and biogenesis. PLoS computational biology. 2007;3:e222. doi: 10.1371/journal.pcbi.0030222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.