

Abstract

Glioblastoma multiforme (GBM) tumors, which arise from glia in the central nervous system (CNS), are one of the most deadly forms of human cancer with a median survival time of ~1 year. Their high infiltrative capacity makes them extremely difficult to treat, and even with aggressive multimodal clinical therapies, outcomes are dismal. To improve understanding of cell migration in these tumors, three-dimensional (3D) multicomponent composite hydrogels consisting of collagen and hyaluronic acid, or hyaluronan (HA), were developed. Collagen is a component of blood vessels known to be associated with GBM migration; whereas, HA is one of the major components of the native brain extracellular matrix (ECM). We characterized hydrogel microstructural features and utilized these materials to investigate patient tumor-derived, single cell morphology, spreading, and migration in 3D culture. GBM morphology was influenced by collagen type with cells adopting a rounded morphology in collagen-IV versus a spindle-shaped morphology in collagen-I/III. GBM spreading and migration were inversely dependent on HA concentration; with higher concentrations promoting little or no migration. Further, noncancerous astrocytes primarily displayed rounded morphologies at lower concentrations of HA; in contrast to the spindle-shaped (spread) morphologies of GBMs. These results suggest that GBM behaviors are sensitive to ECM mimetic materials in 3D and that these composite hydrogels could be used to develop 3D brain mimetic models for studying migration processes.
Keywords: glioblastoma multiforme, collagen, hyaluronic acid, hydrogel
INTRODUCTION
Glioblastoma multiforme (GBM), a primary tumor of the glia and one of the most lethal forms of human cancer, affects ~22 500 individuals in the United States annually.1–4 GBMs are characterized by their extremely high invasion potential.5 For example, tumors can redevelop in the opposing brain hemisphere following surgical resection of the afficted hemisphere.6 Current treatment methods (e.g., surgery, radiation, and chemotherapy) have been largely unsuccessful, mainly because of the highly infiltrative nature of these tumors.7 Despite advances in these techniques, median overall survival time remains low (~12–15 months).1–3,8 This, in part, is a consequence of our poor understanding of the molecular and mechanical pathogenesis of GBMs. Thus, there is a need to develop new methods and models to understand the complex behavior of GBM tumors.
Many existing models to investigate tumor cell migration (e.g., scratch assay, microliter migration assay) utilize twodimensional (2D) substrates (e.g., plastic, glass) that do not recapitulate the complex in vivo tumor microenvironment.9 Several studies have demonstrated that cell behavior is drastically altered when exposed to three-dimensional (3D) microenvironments10,11 and that extracellular matrix (ECM) cues play a significant role in tumor progression.12 In recent years, there has been increasing interest in using hydrogels, 3D biomaterials commonly employed as tissue engineered scaffolds, to understand tumor cell biology. For example, seminal work by Bissell and co-workers using 3D Matrigel biomaterials to explore breast cancer has unraveled several tumor cell characteristics observed in vivo under in vitro conditions.13,14 For GBM studies, both naturally derived (e.g., Matrigel,15–17 collagen18–20) and synthetic (e.g., poly- (acrylamide)21) hydrogels have been utilized. Naturally derived materials present a rich in vitro GBM invasion platform but are limited in the tunability of their physicochemical properties. For example, ligand density, stiffness, and porosity cannot be varied beyond a certain range, which may prevent certain tumor cell characteristics from being captured. In addition, most of these assays do not employ hyaluronic acid/hyaluronan (HA), a significant component of the brain ECM.22–24 Synthetic hydrogels can overcome many of these limitations, providing highly tunable properties and user control over several parameters, but they often lack the complexity of naturally derived materials and therefore may not fully capture in vivo response. Additionally, regardless of the materials used, most of these studies have investigated behavior of well-established tumor cell lines isolated >25 years ago, which is undesirable because phenotypic and genotypic alterations have been reported after repeated culture of cell lines.25
To increase the complexity of the 3D tumor microenvironment beyond that provided by single component natural hydrogels,17–19,26 we investigated GBM behaviors in collagen–HA, multicomponent composite hydrogels. Similar constructs have been used in neural tissue engineering.27–30 Collagen was chosen because it is found in the cancer brain microenvironment. Specifically, collagen types I, III, and IV have been observed in the glial limitans externa and vascular basement membranes, with types I and III also found in the tumor ECM.31 Additionally, clinical observations suggest that GBMs migrate as single cells along these structures,6,32–35 and previous animal studies have shown the formation of a thicker collagen ECM around blood vessels in gliomas compared to normal tissue.36 HA, a high molecular weight, nonsulfated anionic, glycosaminoglycan (GAG),37 was chosen because in both normal and cancerous tissue it is the primary ECM component24 and is present at high levels in many gliomas when compared to normal tissues.38–40 HA in its unmodified form has previously been used as a transwell insert coating in a glioma cell motility assay41,42 and as an additive to Matrigel42,43 and fibrin44 in the traditional invasion assay.
Here, we combined these two ECM components (i.e., collagen types I/III or IV and HA) to yield protein–GAG composite hydrogels, characterized their architecture and examined 3D GBM response to altered HA composition, mimicking the increasing levels of HA typically observed in GBM tumors in vivo. Very few studies have examined GBM behavior in 3D,17–20,40,45,46 and even fewer have utilized collagen–HA composite hydrogels.45 Further, this work is the first to examine single cell morphology, spreading, and migration of GBM cells in 3D collagen–HA composites.
EXPERIMENTAL SECTION
Cell Culture
Patient Tumor Derived OSU-2 Cell Culture
Glioblastoma cells were directly procured from primary, patient brain tumors (OSU, Neurosurgery) in accordance with OSU approved IRB protocol 2005C0075 (dated 11/08/08). Written consent was obtained from participants involved in the study. These Ohio State University (OSU)-2 cells were subcultured for experimental use as described previously.47,48 Briefly, patient-derived tumors were prepared from discarded tissue by washing thoroughly with cell culture media (DMEM/F12 (Invitrogen)) containing 200 unit penicillin (Invitrogen), 200 μg streptomycin (Invitrogen), and 0.5 μg/mL amphotericin B (Invitrogen). Following this, samples were digested by treatment with 200 U/mL type 1A collagenase (Sigma) for ~4 h, triturated to eliminate cell aggregates, centrifuged at 250 g (~5 min), and resuspended in cell culture media (DMEM/F12 (Invitrogen)) containing 10% fetal bovine serum (Invitrogen), 100 units penicillin, 100 μg streptomycin, and 0.25 μg/mL amphotericin B. The well dispersed cell solution was then transferred into a Petri-dish and incubated at 37 °C in a 5% CO2 environment. Cells were fed 2–3 times per week and passaged on reaching confluency. Histopathology at the time of operation confirmed the type of tumor and grade (not shown). Further, to confirm astrocyte lineage indicative of GBM tumors, cells were stained for glial fibrillary acidic protein (GFAP), an astrocyte marker (Figure 1A and B).
Figure 1.

OSU-2 and noncancerous astrocytes in culture. (A) Hoechst stain labels the nucleus blue, whereas rhodamine-GFAP labels the cytoskeleton red. (B) Phase contrast image of OSU-2 cells in culture. (C) Noncancerous astrocytes in culture. Scale bar = 100 μm.
Normal (Noncancerous) Astrocyte Culture
Human astrocytes (Figure 1C) were obtained from Invitrogen (Gibco Human Astrocytes) and subcultured for one passage on Geltrex (Invitrogen) coated tissue culture plates (1:100 dilution in DMEM, ~200 μL/cm2). Cells were fed 2–3 times per week with complete astrocyte medium containing 88% DMEM, 1% N-2 supplement, 10% fetal bovine serum, and 1% penicillin–streptomycin (Invitrogen). For passaging, cells were washed with phosphate buffer saline (PBS), detached using Stem Pro Accutase (Invitrogen), centrifuged at 200 g for 4 min, and then transferred to new Geltrex coated plates or used for hydrogel experiments.
3D Cell Encapsulation in Collagen–HA Composite Hydrogels
Composite hydrogels were created using collagen (PureCol, pepsin solubilized bovine collagen composed of ~97% collagen type-I and ~3% type-III, Advanced BioMatrix Inc.) and thiolated hyaluronic acid (HA) (~ 250 kDa, Glycosan Biosystems Inc.). Collagen and thiolated HA both independently form hydrogels in situ at 37 °C providing permissible conditions for cell encapsulation. Sterile collagen (I/III) solution (1.5 mg/mL, pH ~ 7.4) was prepared with DMEM/F12 (Invitrogen) in a cold environment. Thiolated HA was sterilized using UV illumination (peak power 11.2 mW/cm2) for ~30 min and placed in a 96 well plate. OSU-2 cells prelabeled with Cell Tracker Green CMFDA (Invitrogen) at ~175 000 cells/mL in cell culture medium were then mixed with the diluted collagen solution and directly added to thiolated HA. Thus, cell-laden hydrogel constructs with a constant collagen-I/III concentration of 1 mg/mL and HA concentrations ranging from 0 to 20 mg/mL (0–2 wt/vol %) (N = 3; see Table 1 for all compositions) were created. Cell-laden composite hydrogels were incubated at 37 °C, 5% CO2 for ~1 h prior to the supplementation with additional OSU-2 cell culture media. In addition to OSU-2 cells, human derived, normal (noncancerous) astrocytes at a cell density of ~175 000 cells/mL in cell culture medium were also fluorescently labeled and encapsulated within these hydrogels for morphology observations.
Table 1.
Composition of Collagen–HA Composite Hydrogels
| collagen-I/III based |
collagen-IV based |
|||||||
|---|---|---|---|---|---|---|---|---|
| sample | Col | Col–0.1HA | Col–0.2HA | Col–0.5HA | Col–1HA | Col–2HA | Col-IV | Col-IV–HA |
| HA (mg/mL) | 0 | 1 | 2 | 5 | 10 | 20 | 0 | 9a/5b |
| HA (wt %) | 0 | 0.1 | 0.2 | 0.5 | 1 | 2 | 0 | 0.9a/0.5b |
| collagen (mg/mL) | 1 | 1 | 1 | 1 | 1 | 1 | 0.45a/0.3b | 0.45a/0.3b |
Cell studies.
Confocal reflectance microscopy, collagen-IV used at manufacturer supplied concentration.
OSU-2 cells prelabeled with Cell Tracker Green CMFDA (Invitrogen) were also encapsulated in human collagen-IV (Col-IV) (BD Biosciences) and Col-IV-HA composite hydrogels (Table 1). In both cases, a base gel layer was formed first to prevent cell settling through the loose hydrogel. Approximately 30 μL of 0.45 mg/mL (concentration as supplied by manufacturer) sterile Col–IV under neutral conditions was used independently or added to 0.27 mg presterilized thiolated HA and solidified at 37 °C, 5% CO2 in a 96 well plate for ~2 h to form Col-IV and Col-IV-HA base layers, respectively. Then, a cell laden solution was created using prelabeled OSU-2 cells at a density of ~350 000 cells/mL in Col-IV or Col-IV–HA solution. Approximately 30 μL of these solutions were added to the pregelled layer to yield cell-laden Col-IV (N = 2) and Col-IV–HA (N = 2) hydrogels with a final concentration of ~175 000 cells/mL, respectively. For Col-IV–HA hydrogels, the weight ratio of Col-IV to HA was held constant at ~1:20 (i.e., HA = 0.9 wt %), similar to Col–2HA hydrogels. Collagen IV is one of the primary components of blood vessels; however it forms very weak hydrogels. Therefore, multiple compositions of Col-IV–HA were not investigated. Cell-laden hydrogels were incubated for 1 h at 37 °C, 5% CO2 to permit gelation of the upper layer before supplementation with 60 μL additional cell culture media.
Characterization of Composite Hydrogels
Rheological Characterization
Hydrogel mechanical properties were characterized using unconfined compression testing (RSAIII, TA Instruments). Acellular hydrogels (hydrogels without cells comprised of Col-I/III and Col-I/III–HA), N ≥ 3, were prepared as described above. After gelation, hydrogels were subjected to compression testing at a strain rate of 0.5 mm/min for ~20 s and then held for another 20 s in a multiple extension mode test. Stress–strain curves generated from the compression tests were used to obtain elastic moduli of hydrogels. All measurements were performed at room temperature (~25 °C).
Confocal Reflectance Microscopy
Acellular composite hydrogels were prepared as described above (i.e., Col, Col–0.5HA, and Col– 2HA, N = 3). Gels were formed in a cover well perfusion chamber gasket (8 chambers, 9 mm diameter, 2 mm depth, Invitrogen) glued to a glass slide. Gels were overlaid with cell culture media for imaging purposes. For Col-IV gels (N = 3), 80 μL at a concentration of ~0.3 mg/mL (concentration as supplied by manufacturer) was used and for Col-IV–HA gels (N = 2), ~0.4 mg thiolated HA was added to keep the weight ratio approximately equivalent to that used for cell experiments (i.e., 1:20, collagen:HA). Images were acquired at random gel positions using a laser scanning confocal microscope (Fluoview MPE) in reflected mode with a 25× objective, 3× zoom, and NA = 1.05. The excitation laser source was Alexa Fluor 488 nm, and the reflected light was detected using a photomultiplier tube detector (PMT).
Scanning Electron Microscopy
Acellular gels Col, Col–0.5HA, Col–2HA, and pure HA gels (N = 3) were prepared as described above. Gels were incubated with deionized water overnight and then flash frozen in liquid nitrogen to preserve the morphology and structure as described elsewhere.28,49 Samples were then lyophilized overnight and cut using a razor blade to observe the interior gel surface. Samples were mounted on aluminum stubs (Ted Pella Inc.), coated with gold for 30 s (Model 3 Sputter Coater 91000, Pelco, Reading, CA), and imaged using a scanning electron microscope (SEM, FEI XL-30 Sirion SEM, FEI Company, Hillsboro, OR) at an accelerating voltage of 2 kV.
Cell Studies
OSU-2 Morphology Analysis and Cell Spreading in Collagen–HA Composite Hydrogels
OSU-2 cell laden hydrogels were prepared as described above and imaged after ~24 h. Three still images per hydrogel (N = 3 hydrogels for every formulation) were randomly collected using a confocal microscope (LSM 510; Zeiss, Minneapolis, MN) and subjected to image analysis. OSU-2 morphology was quantitatively analyzed by examining the discrete area (μm2) and circularity (0–1, with 1 being completely spherical) of individual cells using NIH Image J software (available at http://rsbweb.nih.gov/ij/). Cell area and circularity were determined and reported as average ± SD (for three hydrogel replicates). In addition, the percent rounded cells in each hydrogel formulation was also examined (a cell was considered to be round if circularity ≥ 0.95).
Real Time OSU-2 Cell Tracking in 3D Composite Hydrogels
OSU-2 cell laden hydrogels were prepared as described above, and cell migration experiments were performed using confocal microscopy (LSM 510; Zeiss, Minneapolis, MN). After an initial 12 h incubation, a series of images were collected every 10 min for a total of 8 h using a confocal microscope equipped with a motorized stage and an incubation chamber. Some samples experienced considerable movement (i.e., swelling). This was corrected by applying the StackReg Plugin (available at http://bigwww.epfl.ch/thevenaz/stackreg/). Images were converted to movies using NIH Image J, and migration speeds were calculated using MTrack J by dividing the entire length traveled (μm) (i.e., distance traveled in 2D space within the 3D hydrogel) by the total time (h) of cell tracking. Also, only spread cells were motile, and hence, cell speeds in each gel formulation were calculated only for spread or “spindle” shaped cells. Migration speeds were computed from individual cells (n ≥ 40 cells per condition) for each gel formulation (N = 3 hydrogels) and are reported as box and whisker plots, showing mean, median, and outliers for each condition.
Statistical Analysis
Statistical analysis was performed using the JMP statistical software package. All samples were analyzed using ANOVA, and observations in collagen–HA composite hydrogel samples were compared to control collagen samples using Dunnett's Method (comparison to a control).
RESULTS AND DISCUSSION
Composite Hydrogel Modulus
The elastic moduli of composite hydrogels were obtained from stress–strain curves generated by unconfined compression testing. The modulus of a composite hydrogel has been shown to strongly influence cell migration50 and can alter with composition. Pure collagen (Col-I/III) hydrogels had an elastic modulus of ~300.48 ± 39.5 Pa, and the addition of HA increased this modulus to >1000 Pa with Col–1HA and Col–2HA samples having values statistically different from those of pure collagen (p < 0.001) (Figure 2A). Physiologically reported values for brain tissue are ~200– 1000 Pa51 for noncancerous brain (Figure 2B). Values for cancerous brain have not been conclusively determined; however, evidence suggests that tumor tissue is mechanically different from normal brain tissue.52 Thus, by changing the composition of HA in composites, mechanical stiffness could be controllably altered from 300 to 2065 Pa, with a maximum increase in modulus of 7× (e.g., Col–2HA) over pure collagen controls. This demonstrates that the collagen–HA composite hydrogel system is mechanically tunable, and its stiffiness can be modulated to span the physiological range for brain tissue while simultaneously permitting incorporation of GAGs (i.e., HA).
Figure 2.
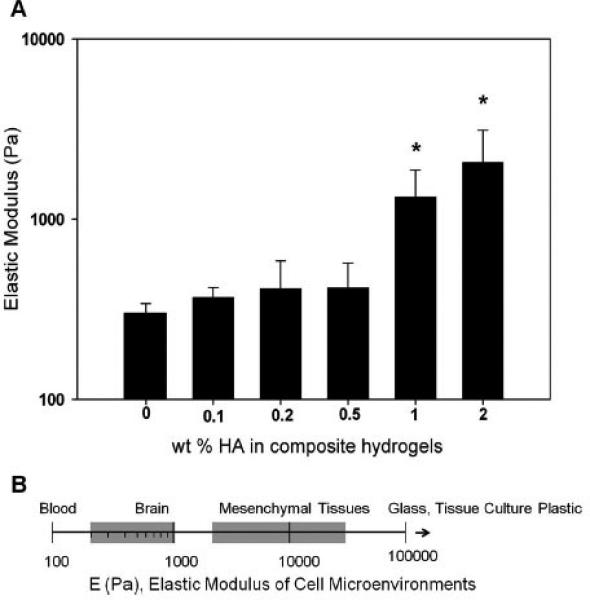
Mechanical characterization of collagen (I/III) and collagen (I/III)–HA composite hydrogels. (A) Elastic modulus of collagen (I/ III)–HA composite hydrogels. * indicates statistically significant from collagen controls (p < 0.0001, as reported from ANOVA). (B) Elastic modulus values reported for various tissues. Adapted from the work of Buxboim et al.51 Copyright 2010 IOPScience.
Composite Hydrogel Microarchitecture
Composite hydrogel microarchitectures were assessed using confocal reflectance microscopy (CRM) and scanning electron micros-copy (SEM). CRM images showed that pure collagen (I/III) hydrogels demonstrated a strong fibrillar character, which was also present in composite hydrogels formed with HA, but declined with increasing HA composition (Figure 3).
Figure 3.

Confocal reflectance microscopy (CRM) images of collagen (I/III) and collagen (I/III)–HA composite hydrogels.
In contrast, Col-IV gels, as well as composites of Col-IV with HA, had little to no fibrillar character (Supporting Information Figure 1). The structure of Col I/III hydrogels and composites was further confirmed using SEM (Figure 4). The fibrillar structure of pure collagen hydrogels is evidenced.
Figure 4.

Scanning electron microscopy (SEM) imaging of collagen (I/III), collagen (I/III)–HA composite, and HA hydrogels. Scale bar = 50 μm.
In comparison, pure HA hydrogels had a more flat, smooth, sheet-like, dense architecture as observed previously,40,53 and this structure was more evident as HA composition increased in collagen–HA composite hydrogels. Thus, composite hydrogels demonstrated characteristics of both material components, similar to collagen–HA interpenetrating networks (IPNs)28,53 and presented a combination of unique architectures representative of blood vessels (i.e., fibrillar collagen) and brain ECM (i.e., flat sheetlike HA). Thus, collagen–HA hydrogels exhibit many brain mimetic features, including tunable control of collagen and HA composition, distinct microarchitectures, and physiologically relevant mechanical properties, which make them suitable 3D biomaterial scaffolds for the investigation of neural cells.
OSU-2 and Normal Astrocyte Behaviors in 3D Composite Hydrogels
The morphology of patient derived OSU-2 cells encapsulated in composite hydrogels was characterized, including cell spreading, circularity, and percentage of rounded cells (Figure 5). OSU-2 cells adopted a spread or “spindle” morphology in collagen hydrogels (control) (Figure 5A). This morphology was maintained at lower HA concentrations (≤0.5 wt % HA) and is representative of the morphology observed in vivo.54–57 As the concentration of HA increased, OSU-2 cells transitioned to a rounded morphology. For example, cells in 2 wt % HA were mostly rounded (~92.77 ± 6.73%) compared to those in 1 wt % HA (~54 ± 9.6%; Figure 5A and D). This was further confirmed by quantification of cell area (Figure 5B) and circularity (Figure 5C), with higher HA concentration reducing cell spreading and increasing cell circularity, indicating minimal interaction of OSU-2 cells with higher HA weight percent composite hydrogels.
Figure 5.

(A) OSU-2 morphologies in collagen-(I/III)–HA composite hydrogels. Scale bar = 100 μm. (B) OSU-2 cell area in composite hydrogels. (C) OSU-2 cell circularity in composite hydrogels. A value of 1 indicates a completely spherical cell. (D) Percentage of rounded cells in composite hydrogels. * indicates pairs that are statistically significant compared to Col (control) (p < 0.0001, as reported from ANOVA).
Cell morphology in Col IV and Col IV–HA hydrogels was also examined; however, gel integrity was not sufficient to prevent cell settling over the time period investigated (e.g., 24 h). Thus, cells contacted the bottom of the dish and displayed typical 2D culture behaviors. However, at shorter time points (i.e., 6 h), OSU-2 cells in Col-IV and Col-IV–HA maintained a rounded shape (Supporting Information Figure 2), suggesting that cells preferred Col-I/III environments to Col-IV environments. [For comparison, cells in the Col-I/III environment exhibited spread or spindle shaped morphologies at shorter time points as well (i.e., 6 h) (data not shown)].
The altered response may have been a consequence of the unique architecture of these hydrogels. Collagen-I monomers self-assemble at physiological temperature and pH to form hydrogels, first forming aggregates and then filaments that eventually form fibrils by lateral cross-linking. Three dimensional hydrogels are created when these fibrils entangle in a noncovalent fashion.46,58 In contrast, collagen-IV is less fibrillar (as confirmed using confocal reflectance microscopy, Supporting Information Figure 1) and is weaker than collagen-I fibrillar networks.59 The inherent, weak nature of collagen-IV hydrogels might hinder cell attachment and spreading, which in turn could result in extremely weak traction forces. This is further supported by results of 2D culture on Col-I/III and Col-IV coated surfaces. Whereas, initial cell adhesion does not differ, 2D spreading is significantly higher for Col-I/III versus Col-IV (p < 0.0001) demonstrating that the differences in cell spreading do not result from differences in adhesion (Supporting Information Figure 3).
The behavior of normal human astrocytes was also examined in these materials. In almost all compositions, normal astrocytes displayed rounded morphologies occasionally with short processes (Figure 6). This behavior is in contrast with that of tumor cells, which showed a spindle-shaped morphology at lower HA concentration. This could result from altered integrin expression in normal versus cancer cells. For example, β1 integrins, which mediate attachment to collagen I, are rare in normal astrocytes but commonly found in glioblastomas.60 These contrasting behaviors, including normal astrocyte migration and their adhesion-dependent 3D biology, will be more fully investigated in future studies.
Figure 6.

Noncancerous (normal) astrocyte morphology in collagen-(I/III)–HA composite hydrogels. Arrows indicate small processes extending from the astrocyte cell body.
OSU-2 Migration in 3D Composite Hydrogels
The migration capacity of OSU-2 cells encapsulated in 3D composite hydrogels was characterized using time-lapse confocal imaging. Since cells did not spread in Col-IV or Col-IV–HA gels, migration in these hydrogels was not investigated. Cells in pure collagen (I/III) exhibited the fastest migration speeds at 9.4 ± 3.4 μm/h (Supporting Information Video 1). OSU-2 cells in composite hydrogels with lower concentrations of HA migrated in a similar fashion to pure collagen (I/III) controls.
Individual tumor cells are known to migrate via mesenchymal or amoeboid migration modes in 3D matrices.61 In mesenchymal mode, cells attach to the ECM via formation of focal contacts that are eventually dissolved upon migration to an adjacent site;61 whereas in amoeboid mode, cells squeeze through the matrix pores with minimal attachment to the matrix.61 At low HA concentration (i.e., ≤0.2 wt % HA), migration was mesenchymal in nature for both composites and collagen I/III controls. Further, OSU-2 cells in Col–0.1HA hydrogels migrated at 7.7 ± 3.9 μm/h, speeds that were statistically indistinguishable from that of pure collagen. (Supporting Information Video 2).
However, as HA concentration increased (i.e., ≥0.2 wt % HA), cell migration speeds showed a decreasing trend and eventually cells failed to migrate. (Figure 7, Supporting Information Videos 1–4, 5.1, 5.2, and 6, and Figure 8). This behavior is clearly evidenced in Supporting Information Video 5.2, which shows some rounded cells unable to migrate through the matrix.
Figure 7.

OSU-2 cell migration in an example collagen-(I/III)–HA composite hydrogel (Col–0.2HA) as shown through stills from time lapse microscopy movies. Time stamp reported in hours (h). Scale bar = 100 μm.
Figure 8.

Quantification of single cell migration speeds (average) in collagen-(I/III)–HA composite hydrogels. The blue * indicates statistical significance when compared to collagen (control) (p < 0.0001, as reported from ANOVA). Representative cell morphologies are presented as insets. Red lines within the box indicate mean and black lines indicate median values.
Both collagen31 and HA24 are important components of the tumor microenvironment, with increased HA expression evidenced over levels in normal tissues.38–40 OSU-2 cells cultured in these 3D materials exhibited greater spreading and migration at lower HA wt% (i.e., ≤0.2 wt % HA), demonstrating patient derived glioma sensitivity to HA concentration. As the concentration of HA was increased, OSU-2 cells transitioned to a rounded morphology and lost the ability to migrate through the gel structure. These findings further corroborate the view that tumor cell behaviors are regulated by the structural and mechanical properties of 3D ECMs.40,45 Furthermore, in contrast to prior 3D hydrogel studies,17–20,40,45,46 this study is the first to explore the behaviors of patient-derived GBM cells rather than well-established cell culture lines.
Previous reports using collagen-only hydrogels have shown that collagen is a supportive matrix for 3D glioma invasion.62 Thus, the primary focus of this study was examining the addition of HA to collagen matrices. Consistent with our observations, David et al. showed collagen type I and III to be strong stimulators of glioma invasion even after HA incorporation.63 However, these studies investigated the invasion of surface-seeded tumor cells into collagen–HA gels using static methods, rather than the dynamic, single cell tracking methods reported here. In addition, cell morphology in these constructs was primarily rounded as only one composition of HA hydrogel coated with collagen was employed, whereas our constructs display both spindle-shaped and rounded morphologies as a function of HA content in 3D, consistent with the morphologies observed in migratory versus non migratory gliomas in vivo.54–57 These results are also consistent with spheroid migration observations in other HA- based systems (i.e., HA gels incorporating the RGD peptide)40 but contradict reports using HA alone42 or in a composite with Matrigel41,42 or collagen.45 For example, HA incorporation (10 mg/mL) into a collagen-I hydrogel (1 mg/mL) had no significant effect on glioma spheroid invasion over a 3 day period. This may result from the difference in HA used; here, cross-linked thiolated HA was used versus the free polymer. These differences may also result from the tumor cell type or source used. A detailed investigation of cell migration as a function of unmodified HA molecular weight or as a function the molecular weight of its degradation products would further elucidate the interactions of GBM tumor cells with different forms of HA. Nonetheless, these results demonstrate that material presentation is crucial in dictating tumor cell spreading and migration, further enhancing our understanding of the role of specific physiological ECM components (i.e., HA) on tumor progression in 3D.
The observed migration response is a result of a number of factors: (1) cell response to increasing mechanical stiffness, (2) reduced porosity and steric barriers resulting from increased HA density (as observed using SEM) [it is recognized that mechanics and matrix pore size are inextricably linked], (3) inability of tumor cells to secrete HA degrading enzymes, or (4) repellent chemical interactions between HA and cell surface adhesion proteins. Using a coaxial, 2D electrospun fibers model, in which the core and shell of a fiber could be independently controlled, we previously investigated the independent effects of chemistry (i.e., HA) and mechanics on cell migration, identifying both as contributing factors and perhaps utilizing different mechanisms of activation.48 The current data, collected in a more physiologically relevant 3D model, confirm and corroborate our previous results. This study specifically examined the role of mechanics and porosity in GBM migration behaviors; additional studies examining chemical contributions to migration, such as the secretion of HA degrading enzymes in 3D and expression of HA receptors (i.e., CD44 and RHAMM)38 and collagen receptors, along with their intracellular signaling cascades, should provide additional insight. Indeed, a systematic study of the interdependence of these factors will be crucial in outlining the role of physiologically relevant 3D microenvironment on tumor cell behaviors, enabling better design of 3D biomimetic tumor cell culture systems.
Our notion that physical factors, independent of traditional ligand–receptor signal transduction pathways, regulate cell migration in the nervous system concords with data from forebrain development. As the forebrain develops, neuronal progenitors from the ventricular/subventricular zone attach and migrate along fibrillar, radial scaffolds to reach the cerebral cortex through a process known as radial migration. These scaffolds express a spectrum of ECM molecules, including many glycosaminoglycans such as HA. Yet, in mice lacking glycoproteins or proteoglycans comprised of brevican, neurocan, tenascin-R, and tenascin-C, radial migration proceeds unperturbed.64 These data suggest that redundancy exists between ECM proteins and also suggests that, provided a physical scaffold is preserved, neural progenitors will migrate to their target location. Indeed, cell shape and the physical characteristics of cellular microenvironments are well-known modulators of biological processes. For example, low stiffness gels promote pluripotency of mouse embryonic stem cells.65 In summary, a tissue's physical microenvironment plays key roles in a spectrum of cellular processes. Our data demonstrate that modulating HA concentration changes the physical characteristics of hydrogels, that HA concentration in hydrogels induces morphological changes in cells, and that high HA concentration decreases glioma cell migration. We propose that the physical properties of glioblastoma microenvironments in situ therefore facilitate elongated cell morphology and glioblastoma cell migration.
CONCLUSIONS
The potential of collagen–HA hydrogels as 3D biomimetic systems with tunable mechanical and chemical properties to explore the role of microenvironment on the migration of patient derived brain tumor cells was examined. To our knowledge, this is one of the first studies to examine the morphology and migration behaviors of human, patient-derived tumor cells in a 3D, protein–GAG composite hydrogel in real time. Tumor cells adopted in vivo-like spindle-shaped morphologies at lower HA concentrations, in contrast to normal human cells that maintained rounded morphologies at all concentrations investigated. GBM migration was an inverse function of HA concentration, with HA impeding and eventually stopping cell movement. Three dimensional materials that combine relevant ECM molecules, such as those described here, could greatly enhance our understanding of GBM migration, which is crucial to the development of improved therapeutic options. Also, these composite hydrogels offer great potential to investigate migration capacity of other cancers, as HA and collagen are widely found in the extracellular environment of many tissues, demonstrating the broad applicability of these materials. Further, in contrast to other collagen–HA interpenetrating networks,28,29,53 the HA employed was chemically cross-linked, adding to the existing library of multicomponent 3D hydrogels that can be employed for soft tissue engineering and regenerative medicine
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge financial support from the National Science Foundation (CBET BME 0854015, to J.O.W. and A. S.), Women in Philanthropy, OSU (to J.O.W), the H.C. “Slip” Slider Professorship (to J.O.W.), and a Pelotonia Graduate Fellowship (to S.S.R). Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the Pelotonia Fellowship Program. The authors would like to thank Prof. Bharat Bhushan (Mechanical Engineering, OSU) for use of his microbalance, Prof. Kurt Koelling (Chemical Engineering, OSU) for use of his RSAIII Dynamic Mechanical Analysis Instrument, Dr. Sara Cole (Campus Microscopy and Imaging Facility, OSU) for assistance with confocal reflectance microscopy, and C. Jenny Dorcena (Chemical Engineering, OSU) for assistance with scanning electron microscopy.
Footnotes
The authors declare no competing financial interest.
Supporting Information
CRM images of Col-IV and Col-IV–HA hydrogels, OSU-2 cell morphologies in Col-IV and Col-IV–HA hydrogels, and OSU-2 cell adhesion and spreading on Col (I/III) and Col-IV coated surfaces (supplementary Figures 1, 2, and 3). Additionally, videos of OSU-2 cell migration within Col (I/III)–HA composite hydrogels [supplementary Videos 1 (am402097j_-si_005.avi), 2 (am402097j_si_006.avi), 3 (am402097j_-si_007.avi), 4 (am402097j_si_008.avi), 5.1 (am402097j_-si_009.avi), 5.2 (am402097j_si_010.avi), and 6 (am402097j_-si_011.avi)] are available. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Wen PY, Kesari SN. Engl. J. Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Thoman WJ, Ammirati M, Caragine LP, Jr., McGregor JM, Sarkar A, Chiocca EA. Top. Magn. Reson. Imaging. 2006;17:121–6. doi: 10.1097/RMR.0b013e31802f5de8. [DOI] [PubMed] [Google Scholar]
- 3.Sarkar A, Chiocca EA. Glioblastoma and malignant astrocytoma. In: Kaye AH, Laws ER, editors. Brain tumors: An encyclopedic approach. 3rd ed. Churchill Livingstone; Edinburgh, New York: 2011. pp. 384–407. [Google Scholar]
- 4.Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME. Cell. Mol. Life Sci. 2007;64:458–78. doi: 10.1007/s00018-007-6342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. CA. Cancer J. Clin. 2010;60:166–93. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. Int. J. Biochem. Cell Biol. 2004;36:1046–1069. doi: 10.1016/j.biocel.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Goldbrunner RH, Bernstein JJ, Tonn JC. Acta Neurochir. (Wien) 1999;141:295–305. doi: 10.1007/s007010050301. discussion 304–5. [DOI] [PubMed] [Google Scholar]
- 8.Sarkar A, Chiocca EA. J. Neurosurg. 2011;114:574–5. doi: 10.3171/2010.3.JNS10194. discussion 575. [DOI] [PubMed] [Google Scholar]
- 9.Valster A, Tran NL, Nakada M, Berens ME, Chan AY, Symons M. Methods. 2005;37:208–15. doi: 10.1016/j.ymeth.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Cukierman E, Pankov R, Stevens DR, Yamada KM. Science. 2001;294:1708–12. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 11.Yamada KM, Cukierman E. Cell. 2007;130:601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Bissell MJ, Radisky D. Nat. Rev. Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee GY, Kenny PA, Lee EH, Bissell MJ. Nat. Methods. 2007;4:359–65. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein JJ, Laws ER, Jr., Levine KV, Wood LR, Tadvalkar G, Goldberg WJ. Neurosurgery. 1991;28:652–8. doi: 10.1097/00006123-199105000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein JJ, Goldberg WJ, Laws ER., Jr. J. Neurooncol. 1994;18:151–61. doi: 10.1007/BF01050421. [DOI] [PubMed] [Google Scholar]
- 17.Gordon VD, Valentine MT, Gardel ML, Andor-Ardo D, Dennison S, Bogdanov AA, Weitz DA, Deisboeck TS. Exp. Cell Res. 2003;289:58–66. doi: 10.1016/s0014-4827(03)00256-8. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman LJ, Brangwynne CP, Kasza KE, Filippidi E, Gordon VD, Deisboeck TS, Weitz DA. Biophys. J. 2005;89:635–50. doi: 10.1529/biophysj.105.061994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HD, Guo TW, Wu AP, Wells A, Gertler FB, Lauffenburger DA. Mol. Biol. Cell. 2008;19:4249–59. doi: 10.1091/mbc.E08-05-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang YL, Motte S, Kaufman LJ. Biomaterials. 2010;31:5678–5688. doi: 10.1016/j.biomaterials.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 21.Ulrich TA, de Juan Pardo EM, Kumar S. Cancer Res. 2009;69:4167–74. doi: 10.1158/0008-5472.CAN-08-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laurent TC, Fraser JR. FASEB J. 1992;6:2397–404. [PubMed] [Google Scholar]
- 23.Toole BP. Nat. Rev. Cancer. 2004;4:528–39. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 24.Newton HB. Expert Rev. Anticancer Ther. 2004;4:803–21. doi: 10.1586/14737140.4.5.803. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Maestro RD, Shivers R, McDonald W, Maestro AD. J. Neurooncol. 2001;53:87–98. doi: 10.1023/a:1012236830230. [DOI] [PubMed] [Google Scholar]
- 27.Wang TW, Spector M. Acta Biomater. 2009;5:2371–2384. doi: 10.1016/j.actbio.2009.03.033. [DOI] [PubMed] [Google Scholar]
- 28.Suri S, Schmidt CE. Acta Biomater. 2009;5:2385–97. doi: 10.1016/j.actbio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Suri S, Schmidt CE. Tissue Eng. Part A. 2010;16:1703–1716. doi: 10.1089/ten.tea.2009.0381. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt CE, Leach JB. Annu. Rev. Biomed. Eng. 2003;5:293–347. doi: 10.1146/annurev.bioeng.5.011303.120731. [DOI] [PubMed] [Google Scholar]
- 31.Giese A, Westphal M. Neurosurgery. 1996;39:235–250. doi: 10.1097/00006123-199608000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Jung S, Ackerley C, Ivanchuk S, Mondal S, Becker LE, Rutka JT. J. Neurosurg. 2001;94:80–9. doi: 10.3171/jns.2001.94.1.0080. [DOI] [PubMed] [Google Scholar]
- 33.Holland EC. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6242–4. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gladson CL. J. Neuropathol. Exp. Neurol. 1999;58:1029–40. doi: 10.1097/00005072-199910000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Scherer HJ. Am. J. Cancer. 1938;34:333–351. [Google Scholar]
- 36.di Tomaso E, London N, Fuja D, Logie J, Tyrrell JA, Kamoun W, Munn LL, Jain RK. PLoS One. 2009;4:e5123. doi: 10.1371/journal.pone.0005123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shu XZ, Ahmad S, Liu Y, Prestwich GD. J. Biomed. Mater. Res. A. 2006;79:902–12. doi: 10.1002/jbm.a.30831. [DOI] [PubMed] [Google Scholar]
- 38.Viapiano MS, Lawler SE. Glioma Invasion: Mechanisms and Therapeutic Challenges. In: Van Meir EG, editor. CNS Cancer: Models, Markers, Prognostic Factors, Targets, and Therapeutic Approaches. 1st ed. Humana Press (Springer); New York: 2009. pp. 1219–1252. [Google Scholar]
- 39.Delpech B, Maingonnat C, Girard N, Chauzy C, Maunoury R, Olivier A, Tayot J, Creissard P. Eur. J. Cancer. 1993;29A:1012–7. doi: 10.1016/s0959-8049(05)80214-x. [DOI] [PubMed] [Google Scholar]
- 40.Ananthanarayanan B, Kim Y, Kumar S. Biomaterials. 2011;32:7913–23. doi: 10.1016/j.biomaterials.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa T, Kubota T, Kabuto M, Kodera T. Anticancer Res. 1996;16:2917–22. [PubMed] [Google Scholar]
- 42.Koochekpour S, Pilkington GJ, Merzak A. Int. J. Cancer. 1995;63:450–4. doi: 10.1002/ijc.2910630325. [DOI] [PubMed] [Google Scholar]
- 43.Radotra B, McCormick D. J. Pathol. 1997;181:434–8. doi: 10.1002/(SICI)1096-9896(199704)181:4<434::AID-PATH797>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 44.Hayen W, Goebeler M, Kumar S, Riessen R, Nehls V. J. Cell Sci. 1999;112(Pt 13):2241–51. doi: 10.1242/jcs.112.13.2241. [DOI] [PubMed] [Google Scholar]
- 45.Yang YL, Sun C, Wilhelm ME, Fox LJ, Zhu JL, Kaufman LJ. Biomaterials. 2011;32:7932–7940. doi: 10.1016/j.biomaterials.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulrich TA, Jain A, Tanner K, MacKay JL, Kumar S. Biomaterials. 2010;31:1875–84. doi: 10.1016/j.biomaterials.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 47.Rao SS, Bentil S, Dejesus J, Larison J, Hissong A, Dupaix R, Sarkar A, Winter JO. PLoS One. 2012;7:e35852. doi: 10.1371/journal.pone.0035852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao SS, Nelson MT, Xue R, Dejesus JK, Viapiano MS, Lannutti JJ, Sarkar A, Winter JO. Biomaterials. 2013;34:5181–90. doi: 10.1016/j.biomaterials.2013.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Xu C, Wang C, Kopecek J. Biomacromolecules. 2006;7:1187–95. doi: 10.1021/bm051002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10889–94. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buxboim A, Rajagopal K, Brown AE, Discher DE. J. Phys.: Condens. Matter. 2010;22:194116. doi: 10.1088/0953-8984/22/19/194116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Unsgaard G, Rygh OM, Selbekk T, Muller TB, Kolstad F, Lindseth F, Hernes TA. Acta Neurochir. (Wien) 2006;148:235–53. doi: 10.1007/s00701-005-0688-y. discussion 253. [DOI] [PubMed] [Google Scholar]
- 53.Brigham MD, Bick A, Lo E, Bendali A, Burdick JA, Khademhosseini A. Tissue Eng. Part A. 2009;15:1645–53. doi: 10.1089/ten.tea.2008.0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson J, Nowicki MO, Lee CH, Chiocca EA, Viapiano MS, Lawler SE, Lannutti JJ. Tissue Eng. Part C. 2009;15:531–540. doi: 10.1089/ten.tec.2008.0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caspani EM, Echevarria D, Rottner K, Small JV. Neuron Glia Biol. 2006;2:105–14. doi: 10.1017/S1740925X06000111. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki SO, Iwaki T. Neuropathology. 2005;25:254–62. doi: 10.1111/j.1440-1789.2005.00626.x. [DOI] [PubMed] [Google Scholar]
- 57.Guillamo JS, Lisovoski F, Christov C, Le Gúerinel C, Defer GL, Peschanski M, Lefrançois T. J. Neurooncol. 2001;52:205–215. doi: 10.1023/a:1010620420241. [DOI] [PubMed] [Google Scholar]
- 58.Pedersen JA, Swartz MA. Ann. Biomed. Eng. 2005;33:1469–90. doi: 10.1007/s10439-005-8159-4. [DOI] [PubMed] [Google Scholar]
- 59.van der Rest M, Garrone R. FASEB J. 1991;5:2814–23. [PubMed] [Google Scholar]
- 60.Paulus W, Baur I, Schuppan D, Roggendorf W. Am. J. Pathol. 1993;143:154–163. [PMC free article] [PubMed] [Google Scholar]
- 61.Friedl P, Wolf K. Nat. Rev. Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 62.Hegedüs B, Marga F, Jakab K, Sharpe-Timms KL, Forgacs G. Biophys. J. 2006;91:2708–2716. doi: 10.1529/biophysj.105.077834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.David L, Dulong V, Coquerel B, Le Cerf D, Cazin L, Lamacz M, Vannier JP. Cell Prolif. 2008;41:348–64. doi: 10.1111/j.1365-2184.2008.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rauch U, Zhou XH, Roos G. Biochem. Biophys. Res. Commun. 2005;328:608–17. doi: 10.1016/j.bbrc.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 65.Chowdhury F, Li Y, Poh YC, Yokohama-Tamaki T, Wang N, Tanaka TS. PLoS One. 2010;5:e15655. doi: 10.1371/journal.pone.0015655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.