

Abstract
Integrative analyses of epigenetic data promise a deeper understanding of the epigenome. Epidaurus is a bioinformatics tool used to effectively reveal inter-dataset relevance and differences through data aggregation, integration and visualization. In this study, we demonstrated the utility of Epidaurus in validating hypotheses and generating novel biological insights. In particular, we described the use of Epidaurus to (i) integrate epigenetic data from prostate cancer cell lines to validate the activation function of EZH2 in castration-resistant prostate cancer and to (ii) study the mechanism of androgen receptor (AR) binding deregulation induced by the knockdown of FOXA1. We found that EZH2's noncanonical activation function was reaffirmed by its association with active histone markers and the lack of association with repressive markers. More importantly, we revealed that the binding of AR was selectively reprogramed to promoter regions, leading to the up-regulation of hundreds of cancer-associated genes including EGFR. The prebuilt epigenetic dataset from commonly used cell lines (LNCaP, VCaP, LNCaP-Abl, MCF7, GM12878, K562, HeLa-S3, A549, HePG2) makes Epidaurus a useful online resource for epigenetic research. As standalone software, Epidaurus is specifically designed to process user customized datasets with both efficiency and convenience.
INTRODUCTION
Epigenetic mechanisms, including DNA methylation, histone modification and chromatin remodeling, play a critical role in various cell functions and processes. Epigenetic aberrations have been linked to the initiation and propagation of many diseases, and epigenetic dysregulation is currently recognized as one of the hallmarks of cancer (1). Different epigenetic mechanisms work cooperatively to regulate gene expression. For instance, it is well known that hyper-methylated DNA CpG islands (CGIs) function to maintain the repressed chromatin state and therefore silence transcriptional activity, whereas hypo-methylated CGIs are associated with active transcription (2–4). On the other hand, the acetylated histone is a marker of open chromatin and transcriptional activation whereas deacetylated histone is associated with condensed chromatin and gene silencing. Proteins binding to methylated DNA also form complexes with proteins involved in deacetylation of histones, suggesting that DNA methylation and histone acetylation act in concert to regulate gene expression (3). EZH2 (enhancer of homolog 2) represents another example of the collaboration between DNA methylation and histone modification. As the catalytic subunit of the PRC2 (Polycomb repression complex 2), EZH2 is a histone methyltransferase that methylates lysine-27 of histone 3 (H3K27me3) located in promoter regions, leading to the repression of target genes (5–8). In addition, EZH2 also serves as a recruitment platform for DNA methyltransferases (9). The above two examples highlight the connections between different epigenetic mechanisms especially the DNA methylation and histone modification, and suggests that the epigenome, as an integrated system, should be studied as a whole.
Driven by the Encylopedia of DNA Elements Consortium (ENCODE) and the NIH Roadmap Epigenomics Project, tremendous efforts have been spent to decipher the human epigenome. Large amounts of data have been generated to map transcription factors binding sites (TFBSs), characterize histone modifications and measure DNA methylation levels. For example, the Gene Expression Omnibus (GEO) database contains more than 10 000 ChIP-seq experiments, 50% of which were generated from human tissues. However, most of these datasets have been individually analyzed, although, in the context of epigenome study, analysis of the combined datasets can offer a much deeper understanding.
Combining different epigenetic data types requires two types of data combination methods, implemented in two consecutive steps. The first step, referred to as data aggregation, consists of accumulating the epigenomics information across many loci throughout the genome. Aggregation analysis is a holistic approach that summarizes epigenetic scores from many genomic regions and therefore provides a global view of the epigenomic landscape of these genomic regions. Such an analysis could be applied to genome regions such as TFBSs, histone modification sites, regions sharing a cognate DNA motif, transcription start sites (TSS). For example, genome-wide aggregation analysis on androgen receptor (AR) binding sites reveals the repositioning of nucleosomes from their original (central) positions to two flanking positions (10). Another study using the aggregation analysis approach finds that 20 nucleosomes are well-positioned around CCCTC-binding factor (CTCF) binding sites, highlighting the important role of CTCF in nucleosome positioning (11). These results demonstrate the power and usefulness of genome-wide aggregation analyses. The second step, referred as data integration, consists of integrating aggregated data of different types such as TF ChIP-seq, histone ChIP-seq, DNA methylation (MeDIP-seq) and DNase-seq. Data integration facilitates the side-by-side comparison of different data types.
Data visualization assists researchers in exploring relevance and differences among datasets, and in generating and validating hypotheses. UCSC genome browser, Ensembl and IGV provide user-friendly interfaces to visualize and compare genomic and epigenomic signals of many different types as vertically piled-up tracks for a single locus (12–14). However, they are not designed to visualize the results of genome-wide aggregation analyses. Therefore, complementary tools are needed to summarize and visualize epigenomic features and enable the identification of novel associations between these features. Spark is a tool designed to fulfill this goal (15). However, its visualization has limited capability to reveal the relevance and differences between datasets (see ‘Results and Discussion’ section).
In this study, we presented Epidaurus, a bioinformatics tool that can simultaneously perform aggregation analysis of thousands of genome regions and integrative analysis for many epigenetic datasets. To demonstrate its usefulness, we used Epidaurus to analyze the epigenome of castration resistant prostate cancer (CRPC) in Abl cells (16). Use of Epidaurus enabled us to confirm that transcription repressor EZH2 works in solo to activate gene expression in CRPC (16). When applying Epidaurus to another prostate cancer epigenome dataset in LNCaP cells (17), we revealed a novel regulating mechanism of AR. Specifically, knockdown of the pioneer factor FOXA1 selectively induced AR to bind promoters, thus reprograming AR to regulate a set of genes including EGFR that are not normally androgen stimulated (17). We therefore exemplified in this study that Epidaurus cannot only validate hypotheses, but can also generate novel biological insights, leading to a deeper understanding of the epigenetic landscape.
MATERIALS AND METHODS
Data collection
Epigenetic data for LNCaP, VCaP, LNCaP-Abl (Abl), MCF7, GM12878, K562, HeLa-S3, A549 and HePG2 cells were assembled from published data deposited into GEO and Sequence Read Archive. Histone ChIP-seq datasets include H3K4me1, H3K4me2, H3K4me3, H3K9me2, H3K9me3, H3K27me3, H3K36me3, H3K79me2, H4K20me1 H4K5ac, H3K27ac, H2A.Z, H2AZac and H3K122ac. Transcription factor ChIP-seq datasets include AR, CTCF, FOXA1, MED12, P300, EZH2, SUZ12, NKX3.1, Pol2, CEBPB, ELF1, FOSL2, FOXM1, GABP, GATA3, E2F1, HDAC2, JUND, MAX, NR2F2, ERG1, CMYC, etc. Chromatin accessibility datasets include DNase-seq and FAIRE-seq. DNA methylation datasets include MeDIP-seq and RRBS. Gene expression datasets include RNA-seq, small RNA-seq and GRO-seq. MNase-seq data from hematopoietic stem cells (CD34+ cells) and their differentiated erythroid lineage cells (CD36+ cells), a leukemia cell line (K562) and a lymphoblastoid cell line (GM12878) were collected. We also prepared genome feature datasets including sequence conservation (PhastCon and PhyloP score), GC content and CpG density. The current Epidaurus database contains 233 datasets (Supplementary Tables S1–S9). This number will increase as more data become available.
Software implementation
Epidaurus was implemented in Python and C: the source code and documentation are freely available from our website (http://epidaurus.sourceforge.net/). Epidaurus can be invoked from the command line as well as from our online web server (http://bioinformaticstools.mayo.edu:8080/Epidaurus/). When running from the command line, Epidaurus took two files as input: a configuration file specifying the parameters and paths of all BigWig files (18), and a BED file containing genome regions of interest such as TFBS. Epidaurus was configured by four parameters: HALF_WINDOW_SIZE, specifying the window size added to both sides of the middle point of regions defined in the BED file (default = 1000 bp), HEAD_ROWS, specifying number of rows Epidaurus would take into calculation (default = 2000), HM_FORMAT, specifying the output graphic format (pdf, png or tiff, default = pdf) and DIST_METRIC, specifying metric to measure distance between two datasets (Pearson, Kendall, Spearman, Euclidean, default = Kendall). The conceptual design of Epidaurus is illustrated in Supplementary Figure S1. Briefly, Epidaurus analysis procedure is detailed in the following steps:
For each row in input BED files, Epidaurus built the genomics window by extending HALF_WINDOW_SIZE (bp) up- and downstream from the middle point. If HALF_WINDOW_SIZE was set to 0, Epidaurus used the original regions provided in BED file without extension: in this case all genomic regions in input BED file must be the same size.
Using parameters defined in the configuration file, Epidaurus extracted signals from BigWig files. For example, if there were K BigWig files representing K datasets, n (specified by HEAD_ROWS) rows in the BED file and the HALF_WINDOW_SIZE was set to w (note the total window size will be 2w + 1). After signal extraction, Epidaurus generated K data matrixes with each matrix having n × (2w + 1) values.
For each data matrix, Epidaurus calculated the mean of each column resulting K lists with each list having 2w + 1 values. The K lists represented signal profiles of K datasets
- Epidaurus then built the matrix K × (2w + 1). Values in each row were scaled into range (0,1) using:
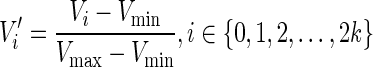
Finally, the heatmap and line graph were generated. The order of datasets displayed in the heatmap was determined by the distances to ‘seed’ dataset specified in configuration file. Distance was measured by one of the four metrics: Pearson correlation coefficient, Kendall rank correlation coefficient, Spearman rank correlation coefficient or Euclidean distance. Details are provided in the section below.
Measuring similarity between two epigenetic datasets
The majority of high-throughput sequencing data (such as RNA-seq, ChIP-seq, MNase-seq) could be represented as a set of genomic positions and the associated scores. Regardless of the data type, Epidaurus computed an epigenetic profile for each selected dataset with single nucleotide resolution. When visualizing these profiles using heatmap, we grouped similar profiles together to facilitate comparison and interpretation.
The similarity between two profiles (X and Y) was measured by the distance between the corresponding arrays of values (x ∈ {x0, x1, x2, … x2k} and y ∈ {y0, y1, y2, … y2k}). We used four different metrics to measure such distance. Euclidean distance (d), Pearson's correlation coefficient (r), Spearman's rank correlation coefficient (ρ) and Kendall's rank correlation coefficient (τ) are defined as:
 |
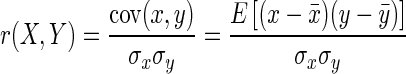 |
 |
 |
Where cov is the covariance, σx is the standard deviation of X,  is the mean of X, x′ is the rank of X, and E is the expectation operator. A pair of observations, (xi, yi) and (xj, yj), were considered concordant if the ranks for both elements agreed (i.e. if both xi > xj and yi > yj or if both xi < xj and yi < yj), and they were considered as discordant, if xi > xj and yi < yj or if xi < xj and yi > yj. If xi = xj or yi = yj, the pair was neither concordant nor disconcordant.
is the mean of X, x′ is the rank of X, and E is the expectation operator. A pair of observations, (xi, yi) and (xj, yj), were considered concordant if the ranks for both elements agreed (i.e. if both xi > xj and yi > yj or if both xi < xj and yi < yj), and they were considered as discordant, if xi > xj and yi < yj or if xi < xj and yi > yj. If xi = xj or yi = yj, the pair was neither concordant nor disconcordant.
RESULTS AND DISCUSSION
Validate noncanonical transcription activation function of EZH2 in castration-resistant prostate cancer cells
EZH2 is a well-known transcription repressor that cooperates with other PRC2 components including SUZ12, EED and RBBP4 (19). However, Xu et al. demonstrated that EZH2 switches its transcriptional repressive function in androgen-dependent prostate cancer to a gene activating function in CRPC, using LNCaP cells as a model of androgen-dependent prostate cancer and Abl cells as a model of CRPC (16). In Abl cells, Xu et al. identified two groups of EZH2 binding sites based on H3K27me3 enrichment. Ensemble peaks (i.e. EZH2 binding peaks with H3K27me3 enrichment) repress gene expression and solo peaks (i.e. EZH2 binding peaks lacking H3K27me3 enrichment) activate gene expression. Using Epidaurus, we systematically reanalyzed the epigenome landscapes at both EZH2 ensemble and solo peaks.
Upon reanalyses, we found that solo peaks were narrow and sharp and ensemble peaks were wide and flat (Figures 1 and 2a). The sequence conservation profiles of these two types of peaks further confirmed this observation (Figure 2b). This was presumably due to the fact that physical dimension of EZH2 protein alone was much smaller than the PRC2 complex, which consists of EZH2 and other cofactors. Here we showed that genome-wide aggregation analysis was able to provide new evidence to validate existing findings.
Figure 1.

Distinct epigenome landscapes between EZH2 ensemble (a) and solo peaks (b). Ensembl and solo peaks were defined by Xu et al. from prostate cancer cell line Abl (16). For each EZH2 peak, we took the peak center and then extended 1-kb to up- and downstream. In the heatmap, each row is a dataset and each column is genomic position around EZH2 peak center. Signals of each dataset were normalized into range (0,1). Magenta and cyan colors indicate high and low signals, respectively. All ChIP-seq data were generated from Abl cell line. CpG density was computed from the human reference genome (hg19/GRCh37) and PhastCon score was downloaded from UCSC annotation database. Both heatmaps were generated by Epidaurus.
Figure 2.

Comparison of signal intensity between EZH2 ensemble peaks (red curves) and solo peaks (blue curves). (a) EZH2 ChIP-seq signal intensity profile, (b) PhastCon conservation score profile, (c) - (f) ChIP-seq signal intensity profiles for Pol2, AR, H3K4me1, H3K4me2, H3K4me3, H3K9me3 and H3K27me3 , respectively. All ChIP-seq data were generated from Abl cell line. EZH2 ensemble (red) and solo (blue) andpeaks were defined by Xu et al. (16). In each panel, the x-axis is the distance to peak center (bp) and the y-axis is tag intensity.
From the perspective of the epigenome, we reaffirmed that EZH2 ensemble peaks were primarily associated with transcription repression and that solo peaks were associated with transcription activation with multiple evidence. First, Pol II and AR binding signals were much higher in solo peaks than those in ensemble peaks (Figure 2c and d). Second, we found that in agreement with its repressive role, EZH2 ensemble peaks were mainly located in closed chromatin (Figure 1a), and consistent with its activating role, EZH2 solo peaks were located in nucleosome-free regions delineated by the decreased signal in the middle of H3K4me1, H3K4me2, H3K4me3 and H3K9me3 peaks (Figures 1b and 2e–h). Third, H3K4me2 and H3K4me3, promoter-specific histone modifications associated with active transcription (20–22), had much higher enrichment in solo peaks than that in ensemble peaks (Figure 2f and g). Similarly, enhancer-specific histone marker H3K4me1 signals were much higher in solo peaks than in ensemble peaks (Figure 2e). Finally, H3K9me3 is well known for its repressive role in transcriptional regulation (23,24). Our data showed that H3K9me3 signals were almost undetectable in solo peaks but were enriched in ensemble peaks (Figure 2h). The distinct epigenetic landscapes between EZH2's ensemble and solo binding sites strongly supported the dual role of EZH2 in transcription regulation in prostate cancer. These results also highlighted the usefulness of Epidaurus and the strength of integrative analysis.
Altered epigenetic landscape of AR binding induced by FOXA1 knockdown
FOXA1 is a transcription factor involved in embryonic development and establishment of tissue-specific gene expression and acts as a pioneer factor in chromatin remodeling. As a master regulator of AR, FOXA1 has been extensively studied in prostate cancer (10,17,25–29). It was reported that FOXA1 opened the local chromatin to facilitate AR binding (30). Wang et al. defined three groups of AR binding sites after FOXA1 knockdown: lost binding (1881 loci, referred as lost AR program), conserved binding (1234 loci, referred as conserved AR program) and gained binding (10 869 loci, referred as gained AR program) (17).
Using Epidaurus, we reanalyzed the genome and epigenome datasets generated from LNCaP cells for these three groups of AR binding sites. We demonstrated that all three groups of AR binding regions were highly conserved across 100 vertebrate genomes, hypersensitive to DNase I and enriched for AR and P300 ChIP-seq signals, suggesting the reliability of these AR binding sites (Figure 3, Supplementary Figure S2k and l, o and p). From the Epidaurus results, we observed dramatically different epigenome landscapes between the gained and lost AR programs. In the lost AR program, all histone ChIP-seq data consistently delineated a nucleosome free region flanking the center of AR binding sites (Figure 3a). In particular, active enhancer markers H3K4me1, H3K4me2, H3K27ac and histone variant H2A.Z clearly exhibited a symmetrical, bimodal pattern with reduced nucleosome occupancy at the central nucleosome and concomitant increased occupancy at two flanking nucleosomes (Supplementary Figure S2g–j). In contrast, for the gained AR program, most AR binding sites were located to the central, well-positioned nucleosome, as shown by the unimodal signal of H3K4me1, H3K4me2, H3K27ac, H3K36me3, H4K5ac, H2A.Z and H3K9me2 (Figure 3b, Supplementary Figure S2d–j). This distinct nucleosome architecture between gained and lost AR programs highlighted the dynamics of nucleosome and the role of FOXA1 as pioneer factor in chromatin remolding (27,31,32). The epigeneitc profile of the conserved AR program was very similar to that of the lost AR program (Figure 3c). However, signals of enhancer-specific histone markers such as H3K4me1, H3K4me2 and H3K27ac were much higher in the conserved AR program than those in the lost AR program, suggesting the conserved AR binding sites had much higher intrinsic enhancer activity and thus were independent of the pioneer effect of FOXA1 on gene activation (Supplementary Figure S2g, i and j). This assumption was substantiated by the observation that the eRNA abundances as measured by GRO-seq as well as the Pol II ChIP signals were also much higher in the conserved AR program than those in the lost AR program (17,33–35) (Supplementary Figure S2m and n).
Figure 3.

Distinct epigenome landscapes between ‘lost’, ‘conserved’ and ‘gained’ AR programs. Comparison of epigenome landscapes between lost (a), gained (b) and conserved (c) AR programs. The loss, gained and conserved AR binding sites induced by siFOXA1 were defined by Wang et al. in LNCaP cells (17). For each AR binding site, we extended 1-kb to up- and downstream of the peak center. In the heatmap, each row is a dataset and each column is genomic position. Signals of each dataset were normalized into range (0,1). Magenta and cyan colors indicate high and low signals, respectively. MeDIP-seq, DNaseI-seq, GRO-seq and all ChIP-seq data were generated from LNCaP cells treated with dihydrotestosterone. CpG density was computed from the human reference genome (hg19/GRCh37) and the PhastCon score was downloaded from the UCSC annotation database. All heatmaps were generated by Epidaurus.
Cytosines in CpG dinucleotides can be methylated to 5-methylcytosine, which spontaneously deaminate to form thymidine residues over time. Therefore, the CpG dinucleotide is greatly under-represented in the human genome at only about one-fifth than would be expected (36) (Supplementary Figure S3). Genome regions with a high concentration of CpG sites are known as CGIs (37). About 70% of CGIs are located within 2-kb regions flanking TSS (Supplementary Figure S4). On the other hand, the majority (>85%) of AR binding sites are distal from the TSSs of AR regulated genes (26,27,38). Because of this, the chance of observing overlaps between AR binding sites and CGIs is conceivably very slim. To estimate how many AR binding sites overlapped with CGIs by chance, we shuffled AR binding sites and then overlapped them with CGIs (28 691 regions, total 21 842 742 bp or 0.7% of the human reference genome) downloaded from the UCSC annotation database. We estimated that 0.97 ± 0.09% of the AR binding sites would overlap with CGIs by chance (Supplementary Figure S5). Interestingly, we found that the average CpG density was much higher in gained AR binding sites than that in the lost and conserved AR binding sites (Figure 3, Supplementary Figure S6). This suggested the co-localization of CGIs with AR binding sites in the gained AR program. As shown in Figure 4a, 3.06% (333/10 867) of gained AR binding sites overlapped with CGIs, which is 3.15-fold enrichment (P < 2.2E-16, 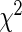 test) compared to background (0.97%), suggesting gained AR binding sites tended to colocalize with CGIs. As a comparison, only 0.05% (1/1860) of lost AR binding sites overlapped with CGIs, a 23.4-fold depletion (P = 1.02E-4,
test) compared to background (0.97%), suggesting gained AR binding sites tended to colocalize with CGIs. As a comparison, only 0.05% (1/1860) of lost AR binding sites overlapped with CGIs, a 23.4-fold depletion (P = 1.02E-4,  test) compared to the background control, suggesting that lost AR binding sites tended to localize outside of CGIs. The overlap between conserved AR binding sties and CGIs was about 1.0% (13/1297), approximating background control.
test) compared to the background control, suggesting that lost AR binding sites tended to localize outside of CGIs. The overlap between conserved AR binding sties and CGIs was about 1.0% (13/1297), approximating background control.
Figure 4.

Characteristics of gained AR binding induced by siFOXA1. (a) Percentage of AR binding sites that overlapped with CpG islands. Conserved, lost and gained AR bindings are indicated by black, red and purple colors respectively. (b) Genomic distribution of AR binding sites that overlapped with CpG islands. TSS = transcription start site. (c) Disease ontology analysis of AR binding sites that overlapped with CpG islands. X-axis indicates the FDR-adjusted binomial P-values were calculated using GREAT (http://bejerano.stanford.edu/great/public/html/). (d) Transcription factor targets oncology analysis of AR binding sites overlapped with CpG islands. X-axis indicates the FDR-adjusted binomial P-values calculated using GREAT. P-values are calculated using  test with continuity correction. (e) Expression analysis for KLK3, EGFR, SUN2 and GAS2L1 using Illumina Human-6 v2.0 expression beadchip in LNCaP cells treated with dihydrotestosterone. Three biological replicates of siFOXA1 (siFOXA1_r1, siFOXA1_r2, siFOXA1_r3) were compared with three biological replicates of siControl (siCtrl_r1, siCtrl_r2, siCtrl_r3). (f) Percentage of AR binding sites containing FOXA1 motif. Conserved, lost and gained AR bindings are indicated by black, red and purple bars, respectively. AR bindings that overlapped with CGI are indicated by blue bar and random control is indicated by gray bar.
test with continuity correction. (e) Expression analysis for KLK3, EGFR, SUN2 and GAS2L1 using Illumina Human-6 v2.0 expression beadchip in LNCaP cells treated with dihydrotestosterone. Three biological replicates of siFOXA1 (siFOXA1_r1, siFOXA1_r2, siFOXA1_r3) were compared with three biological replicates of siControl (siCtrl_r1, siCtrl_r2, siCtrl_r3). (f) Percentage of AR binding sites containing FOXA1 motif. Conserved, lost and gained AR bindings are indicated by black, red and purple bars, respectively. AR bindings that overlapped with CGI are indicated by blue bar and random control is indicated by gray bar.
We further investigated the genome position of gained AR binding sites overlapped with CGIs. Since the majority of CGIs are located in promoters, as expected, we found, as expected, that these AR binding sites were primarily located in promoter regions (Figure 4b). De novo motif search using MEME-ChIP (39) showed significant enrichment of palindromic AR motifs (E-value = 2.4E−158), suggesting the reliability of these AR binding sites (Supplementary Figure S7). We then investigated the genes targeted by gained AR binding sites that overlapped with CGIs. Disease ontology analysis suggested that the target genes were significantly associated with various types of cancers as well as other transcription factors such as SOX2, ETS1, HNF4α and CREB (Figure 4c and d). Genes such as KLK3, EGFR and GAS2L1 play critical roles in prostate cancer pathogenesis and progression. Overexpression of KLK3 was widely used as a marker for early prostate cancer detection (PSA test) for decades until its recent suspension. Activation of EGFR is one of the mechanisms accounting for the maintenance of AR signaling in hormone poor environments (such as CRPC) (40,41), and intense efforts have been focused on the development of therapeutic strategies to block EGFR signaling in prostate cancer (42–47). GAS2L1 is an ERG-dependent AR activated gene and frequently silenced in prostate cancers (48–50). We analyzed the expression of these four genes using Illumina human-6 v2.0 beadchip (GSE27682) (17), and found that their expressions were significantly up regulated in siFOXA1 LNCaP cells treated with androgen. The t-test P-values for KLK3, EGFR, SUN2 and GAS2L1 were 0.0003, 0.049, 0.01 and 0.044, respectively (Figure 4e). The upregulation of EGFR protein in CRPC was also reported in a previous study (51). As illustrated in Figure 5, AR bound to the H3K4me3-positive promoters of KLK3, EGFR, SUN2 and GAS2L1 in FOXA1 knockdown cells. The promoter regions of three of the four genes (EGFR, SUN2 and GAS2L1) contained CGIs. Increased AR binding at the promoters of all four genes was also confirmed by ChIP-qPCR in LNCaP cells (Figure 6). Genes targeted by gained AR binding sites overlapping with CGI are listed in Supplementary Table S10, and more examples are shown in Supplementary Figure S8.
Figure 5.

Examples of gained AR binding (highlighted in light blue) at promoters after FOXA1 knockdown. Screenshots taken from UCSC genome browser showing 4 genes that had AR binding at their promoters. From top to bottom: KLK3 (a), EGFR (b), SUN2 (c) and GAS2L1 (d). Six tracks are displayed for each panel: AR binding in normal condition (red), AR binding with siFOXA1 (brown), H3K4me3 promoter marker (blue), CpG density in 50-bp window (green), CpG islands defined by UCSC (dark green) and gene model (dark blue). AR, AR (siFOXA1), FOXA1 and H3K4me3 ChIP-seq data were all generated from LNCaP cells treated with dihydrotestosterone.
Figure 6.

ChIP qPCR validations. ChIP (Chromatin immunoprecipitation) qPCR analyses to confirm gained AR binding on promoters of KLK3 (a), EGFR (b), SUN2 (c) and GAS2L1 (d). NT, non-target shRNA; shFOXA, shRNA knockdown FOXA1.
Despite the higher CpG density in gained AR binding sites, these CpGs were mostly hypo-methylated compared to flanking regions as measured by MeDIP-seq, and the DNA methylation levels in gained AR binding sites were comparable to those in lost and conserved AR binding sites (Supplementary Figure S9). On the other hand, the FOXA1 motif was under represented (P = 1.09E-10, 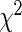 test) in gained AR binding sites that overlapped with CGIs, confirming that these binding events were independent of FOXA1 (17) (Figure 4f).
test) in gained AR binding sites that overlapped with CGIs, confirming that these binding events were independent of FOXA1 (17) (Figure 4f).
Although FOXA1 has been extensively studied, its functions in prostate cancer are controversial and not fully understood. FOXA1 expression levels have been associated with both good and bad clinical outcomes depending on the patient cohort (17,26,28). It was reported that FOXA1 expression is slightly up-regulated in localized prostate cancer because cell proliferation is the main feature in this stage, but is remarkably down-regulated in CRPC because cell motility and epithelial-to-mesenchymal transition are essential at this stage (52). Systematic analysis also suggested that FOXA1 is a key factor in the initiation of lung cancer metastasis (53). Therefore, FOXA1 plays different roles in cancer development and progression. Through integrative analysis of the prostate cancer epigenome using Epidaurus, we revealed a novel mechanism for FOXA1 regulation of AR binding to promoter regions. Specifically, we found that knockdown of FOXA1 increased binding of AR to promoter regions. Consistent with our finding, Sharma et al. also found that a larger proportion of AR binding sites were associated with promoter regions in CRPC than in castration-responsive prostate tumor or cell lines (54). We found that the knockdown of FOXA1 induced AR to bind the promoter of EGFR and up-regulate its expression. Interestingly, EGFR activation is one of the mechanisms to activate AR via phosphorylation in androgen-poor conditions such as CRPC to maintain AR signaling (40,41). However, the exact regulatory mechanisms of this feedback loop remained unclear.
Comparing Epidaurus to the existing platform Spark
When compared to the existing data exploration platform, Spark, Epidaurus produced a more accurate representation for the same datasets using the same list of lost AR bindings (Figure 7). This was most likely due to the capability of Epidaurus to normalize each dataset independently, compared to Spark, which normalizes the whole datasets. In practice, it is difficult to render data from different datasets and different types of data comparable for several reasons. First, sequencing depth and DNA fragment size can be considerably different between datasets, often with an order of magnitude difference between ChIP-seq data published years ago and that published recently. Second, even though sequencing depths and DNA fragment size can be normalized onto the same scale, the signals of a particular locus (or a list of loci) are still not comparable between diffuse, broad-peak (e.g. H3K36me3) ChIP-seq experiments and localized, narrow-peak (eg H3K4me3) ChIP-seq experiments. Third, the total binding sites can be considerably different between different transcription factors. Finally, high throughput sequencing-derived epigenetic datasets and genome features (such as PhastCon conservation score, CpG dinucleotide density) cannot be normalized onto the same scale. However, independent normalization has its own drawbacks: for example, color depth is not comparable between different datasets or between different heatmaps. To overcome these limitations, Epidaurus generated a raw data table to facilitate the direct comparison of the absolute values between datasets (such as in Figure 2).
Figure 7.

Comparison of the visualization effect of Spark to Epidaurus. Comparison of Epidaurus with Spark (15) using the same epigenetic datasets and genome coordinates. (a) Heatmap generated by Spark. (b) Heatmap generated by Epidaurus.
CONCLUSION
We exemplified in this study that large-scale integrative analyses of prostate epigenome could validate previous findings as well as generate novel biological insights and lead to a deeper understanding of prostate cancer. Obviously, the application of Epidaurus is not limited to prostate cancer epigenome studies. Tremendous epigenetic data for other cancer types have been generated, and the data volume is growing even faster thanks to the dramatic decrease of sequencing cost. The interactions between different type of epigenetic data have not been fully explored partially due to the lack of convenient bioinformatic tools. Epidaurus is such a tool that facilitates the holistic analysis and provides informative visualization of the epigenome. By assembling epigenetic data from public resources, the Epidaurus web server is a useful centralized data hub for epigenetic research projects that use cancer cell lines including LNCaP, VCaP, LNCaP-Abl (Abl), MCF7, GM12878, K562, HeLa-S3, A549 and HePG2. For most of these cell lines, ChIP-seq data of extensively used histone markers (such as H3K4me1, H3K4me2, H3K4me3 and H3K27ac) and chromatin accessibility data (such as DNase-seq and FAIRE-seq) were prebuilt into our online database. However, since epigenome is highly dynamic and tends to be cell-type-specific, standalone Epidaurus is specifically designed to process user customized, arbitrary datasets with both efficiency and convenience.
AVAILABILITY
Source code and comprehensive documentation of Epidaurus are available at: http://epidaurus.sourceforge.net/. Online web server is available at: http://bioinformaticstools.mayo.edu:8080/Epidaurus/
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank Drs Han Xu and Shirley X. Liu from Harvard Medical School for providing the EZH2 solo peaks identified from Abl cell. The authors also thank Michael T. Kalmback for the help in building Epidaurus web server.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors’.
FUNDING
Center for Individualized Medicine at Mayo Clinic [CA15083-40C19 to J.-P.A.K.]; National Institute of Health [CA130908, CA134514 to H.H.]. Funding for open access charge: Center for Individualized Medicine at Mayo Clinic [CA15083-40C19].
Conflict of interest statement. None declared.
REFERENCES
- 1.Baylin S.B., Ohm J.E. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction. Nat. Rev. Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki M.M., Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 3.Nan X., Ng H.H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 4.Bird A.P., Wolffe A.P. Methylation-induced repression–belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 5.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 6.Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 7.Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller J., Hart C.M., Francis N.J., Vargas M.L., Sengupta A., Wild B., Miller E.L., O'Connor M.B., Kingston R.E., Simon J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 9.Viré E., Brenner C., Deplus R., Blanchon L., Fraga M., Didelot C., Morey L., Van Eynde A., Bernard D., Vanderwinden J.-M., et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 10.He H.H., Meyer C.A., Shin H., Bailey S.T., Wei G., Wang Q., Zhang Y., Xu K., Ni M., Lupien M., et al. Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Y., Sinha M., Peterson C.L., Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn R.M., Haussler D., Kent W.J. The UCSC genome browser and associated tools. Brief. Bioinformatics. 2013;14:144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spudich G.M., Fernández-Suárez X.M. Touring Ensembl: a practical guide to genome browsing. BMC Genomics. 2010;11:295. doi: 10.1186/1471-2164-11-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson J.T., Thorvaldsdóttir H., Winckler W. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen C.B., Younesy H., O'Geen H., Xu X., Jackson A.R., Milosavljevic A., Wang T., Costello J.F., Hirst M., Farnham P.J., et al. Spark: a navigational paradigm for genomic data exploration. Genome Res. 2012;22:2262–2269. doi: 10.1101/gr.140665.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu K., Wu Z.J., Groner A.C., He H.H., Cai C., Lis R.T., Wu X., Stack E.C., Loda M., Liu T., et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D., Garcia-Bassets I., Benner C., Li W., Su X., Zhou Y., Qiu J., Liu W., Kaikkonen M.U., Ohgi K.A., et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010;26:2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chase A., Cross N.C.P. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011;17:2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 20.Bernstein B.E., Kamal M., Lindblad-Toh K., Bekiranov S., Bailey D.K., Huebert D.J., McMahon S., Karlsson E.K., Kulbokas E.J., Gingeras T.R., et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 22.Koch C.M., Andrews R.M., Flicek P., Dillon S.C., Karaöz U., Clelland G.K., Wilcox S., Beare D.M., Fowler J.C., Couttet P., et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyer L.A., Plath K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 24.Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K.-I., et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C., Wang L., Wu D., Chen H., Chen Z., Thomas-Ahner J.M., Zynger D.L., Eeckhoute J., Yu J., Luo J., et al. Definition of a FoxA1 Cistrome that is crucial for G1 to S-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res. 2011;71:6738–6748. doi: 10.1158/0008-5472.CAN-11-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sahu B., Laakso M., Ovaska K., Mirtti T., Lundin J., Rannikko A., Sankila A., Turunen J.-P., Lundin M., Konsti J., et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962–3976. doi: 10.1038/emboj.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q., Li W., Zhang Y., Yuan X., Xu K., Yu J., Chen Z., Beroukhim R., Wang H., Lupien M., et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerhardt J., Montani M., Wild P., Beer M., Huber F., Hermanns T., Müntener M., Kristiansen G. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am. J. Pathol. 2012;180:848–861. doi: 10.1016/j.ajpath.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 29.Sahu B., Laakso M., Pihlajamaa P., Ovaska K., Sinielnikov I., Hautaniemi S., Jänne O.A. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013;73:1570–1580. doi: 10.1158/0008-5472.CAN-12-2350. [DOI] [PubMed] [Google Scholar]
- 30.Cirillo L.A., Lin F.R., Cuesta I., Friedman D., Jarnik M., Zaret K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 31.Lupien M., Eeckhoute J., Meyer C.A., Wang Q., Zhang Y., Li W., Carroll J.S., Liu X.S., Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z., Wang L., Wang Q., Li W. Histone modifications and chromatin organization in prostate cancer. Epigenomics. 2010;2:551–560. doi: 10.2217/epi.10.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Natoli G., Andrau J.-C. Noncoding transcription at enhancers: general principles and functional models. Annu. Rev. Genet. 2012;46:1–19. doi: 10.1146/annurev-genet-110711-155459. [DOI] [PubMed] [Google Scholar]
- 34.Kim T.-K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J., Harmin D.A., Laptewicz M., Barbara-Haley K., Kuersten S., et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y., Sun L., Chen Z., Whitaker J.W., Wang T., Wang W. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res. 2013;41:10032–10043. doi: 10.1093/nar/gkt826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 37.Gardiner-Garden M., Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 38.Yu J., Yu J., Mani R.-S., Cao Q., Brenner C.J., Cao X., Wang X., Wu L., Li J., Hu M., et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Machanick P., Bailey T.L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinlein C.A., Chang C. Androgen receptor in prostate cancer. Endocr. Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 41.Scher H.I., Sawyers C.L. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 42.Di Lorenzo G., Tortora G., D'Armiento F.P., De Rosa G., Staibano S., Autorino R., D'Armiento M., De Laurentiis M., De Placido S., Catalano G., et al. (2002) Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin. Cancer Res. 8:3438–3444. [PubMed] [Google Scholar]
- 43.Salomon D.S., Brandt R., Ciardiello F., Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 44.Normanno N., Bianco C., De Luca A., Maiello M.R., Salomon D.S. Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr. Relat. Cancer. 2003;10:1–21. doi: 10.1677/erc.0.0100001. [DOI] [PubMed] [Google Scholar]
- 45.Scaltriti M., Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin. Cancer Res. 2006;12:5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 46.Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science. 2006;312:1175–1178. doi: 10.1126/science.1125951. [DOI] [PubMed] [Google Scholar]
- 47.Bianco R., Troiani T., Tortora G., Ciardiello F. Intrinsic and acquired resistance to EGFR inhibitors in human cancer therapy. Endocr. Relat. Cancer. 2005;12(Suppl. 1):S159–S171. doi: 10.1677/erc.1.00999. [DOI] [PubMed] [Google Scholar]
- 48.Taniya T., Tanaka S., Yamaguchi-Kabata Y., Hanaoka H., Yamasaki C., Maekawa H., Barrero R.A., Lenhard B., Datta M.W., Shimoyama M., et al. A prioritization analysis of disease association by data-mining of functional annotation of human genes. Genomics. 2012;99:1–9. doi: 10.1016/j.ygeno.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Cai C., Wang H., He H.H., Chen S., He L., Ma F., Mucci L., Wang Q., Fiore C., Sowalsky A.G., et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J. Clin. Invest. 2013;123:1109–1122. doi: 10.1172/JCI66666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lodygin D., Epanchintsev A., Menssen A., Diebold J., Hermeking H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 2005;65:4218–4227. doi: 10.1158/0008-5472.CAN-04-4407. [DOI] [PubMed] [Google Scholar]
- 51.Pignon J.-C., Koopmansch B., Nolens G., Delacroix L., Waltregny D., Winkler R. Androgen receptor controls EGFR and ERBB2 gene expression at different levels in prostate cancer cell lines. Cancer Res. 2009;69:2941–2949. doi: 10.1158/0008-5472.CAN-08-3760. [DOI] [PubMed] [Google Scholar]
- 52.Jin H.-J., Zhao J.C., Ogden I., Bergan R.C., Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 2013;73:3725–3736. doi: 10.1158/0008-5472.CAN-12-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H., Meyer C.A., Fei T., Wang G., Zhang F., Liu X.S. A systematic approach identifies FOXA1 as a key factor in the loss of epithelial traits during the epithelial-to-mesenchymal transition in lung cancer. BMC Genomics. 2013;14:680. doi: 10.1186/1471-2164-14-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma N.L., Massie C.E., Ramos-Montoya A., Zecchini V., Scott H.E., Lamb A.D., MacArthur S., Stark R., Warren A.Y., Mills I.G., et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35–47. doi: 10.1016/j.ccr.2012.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.