

Abstract
The genetic code can be manipulated to reassign codons for the incorporation of non-standard amino acids (NSAA). Deletion of release factor 1 in Escherichia coli enhances translation of UAG (Stop) codons, yet may also extended protein synthesis at natural UAG terminated messenger RNAs. The fidelity of protein synthesis at reassigned UAG codons and the purity of the NSAA containing proteins produced require careful examination. Proteomics would be an ideal tool for these tasks, but conventional proteomic analyses cannot readily identify the extended proteins and accurately discover multiple amino acid (AA) insertions at a single UAG. To address these challenges, we created a new proteomic workflow that enabled the detection of UAG readthrough in native proteins in E. coli strains in which UAG was reassigned to encode phosphoserine. The method also enabled quantitation of NSAA and natural AA incorporation at UAG in a recombinant reporter protein. As a proof-of-principle, we measured the fidelity and purity of the phosphoserine orthogonal translation system (OTS) and used this information to improve its performance. Our results show a surprising diversity of natural AAs at reassigned stop codons. Our method can be used to improve OTSs and to quantify amino acid purity at reassigned codons in organisms with expanded genetic codes.
INTRODUCTION
The genetic code is nearly universal in living organisms, highly evolved and was assumed to be fixed (1–3). However, nature has revealed expanded genetic codes and researchers have even engineered new codes for non-standard amino acids (NSAA) (4–8). Expanding the genetic code beyond the standard amino acids (AAs) requires an aminoacyl-tRNA synthetase capable of efficiently ligating NSAAs to a tRNA that can decode an ‘open’ codon. These engineered orthogonal translation systems (OTSs) typically use the rare amber stop codon UAG as the ‘open’ codon to encode the NSAA of choice. Recently, new recoding strategies have emerged that have paved the way for converting amber codons into a dedicated NSAA sense codon (6,9–11). This advance creates new possibilities for NSAA insertion into proteins and has also opened up new pitfalls. Over 100 NSAAs have been encoded by diverse OTSs (5) and yet, methods that can quantify the purity and specificity of OTS-produced recombinant proteins and proteomes are limited.
We and others have observed natural AAs that can compete for insertion at amber codons (9,12–14) and also identified potential open reading frames (ORFs) that are at risk for natural extensions of their native protein products (6,10,11). These observations raise important concerns about the fidelity and purity of recombinant proteins containing NSAAs and require new methods to characterize NSAA containing proteins and proteomes. For example, NSAA containing proteins used for structural studies or therapeutics would benefit from methods that would help to diversify the OTS repertoire while maintaining the fidelity and purity of NSAA-containing protein synthesis.
Here we present a new set of proteomic methods that can quantify both standard and NSAAs in proteins and proteomes. We applied these tools to compare global amber codon readthrough in the proteomes of a variety of Escherichia coli strains. This required the creation of a new workflow that enabled the comprehensive and unbiased discovery of peptides reporting UAG readthrough from shotgun proteomic data. We validated this approach by examining UAG readthrough in the proteomes of three E. coli strains. The workflow also accommodates the characterization of phosphoserine (Sep) incorporation at UAG codons in a green fluorescent protein (GFP) reporter. Importantly, we developed a quantitative assay to measure the relative incorporation of both Sep and standard AAs at the UAG position. This application guided further optimization of the Sep-OTS system resulting in both increased phosphoprotein production and purity.
MATERIALS AND METHODS
Methanol, formic acid (FA) 88%, trifluoroacetic acid (TFA) and hydrochloric acid (HCl) were from J.T. Baker (Philipsburg, NJ). 2-amino-2-(hydroxymethyl)-1,3-propanediol (Tris), NaCl, CaCl2, bovine serum albumin and iodoacetamide (IAA) were from Sigma-Aldrich (St. Louis, MO, USA). Dithiothretitol (DTT) and ethylenediaminetetraacetic acid (EDTA) were from American Bioanalytical (Natick, MA, USA). The anionic acid labile surfactant II (ALS-110) was obtained from Protea Biosciences, Inc. (Morgantown, WV, USA) as a dry powder. The detergent stock solution was prepared by dissolving 5 mg of the lyophilized ALS-110 in 50 μl methanol and 50 μl 100 mM Tris–HCl buffer pH = 8.0 (23°C) prior to use. Premade (Optima quality) liquid chromatography-mass spectrometry (LC-MS) grade water and acetonitrile (ACN) containing 0.1% FA was obtained from Fisher Chemicals (Pittsburgh, PA, USA). The modified porcine trypsin (sequencing grade) was obtained from Promega (Madison, WI) as a frozen solution with 0.5 μg/μl trypsin. UltraMicroSpin columns (C18) were from The Nest Group Inc. (Southborough, MA, USA). All buffers were prepared at 23°C unless noted otherwise. The peptide library of stable isotope labeled peptides (Maxi SpikeTides L) was obtained from JPT Technologies GmbH (Berlin, Germany) and used without further purification. Library peptides (∼0.5 mg) were individually dissolved in a mixture of 1:9 by volume of 70% FA and 0.1% FA to generate stock solutions with an estimated 500 pmol/μl peptide. Stable isotope labeled AAs in peptides sequences are underlined.
Protein expression and sample preparation
Detailed methods for plasmid construction, bacterial cell culture, protein purification and sample preparation for proteomics were followed as described previously (13). Strains and genotypes of all strains used in this study are listed in the supplement (See Supplementary Table S1).
Databases
We re-annotated all E. coli ORFs that contain a TAG codon. This was accomplished using a series of automated Practical Extraction and Report Language (PERL) scripts. After removing all non-coding RNA genes from the database, all annotated E. coli genes (IMG DATABASE (15)) with in-frame or terminal TAG codons were identified. The complete E. coli genome sequence was used to lengthen these genes to the next in-frame TGA or TAA stop codon. Some genes containing frame shifts were also found to contain in-frame UAG codons. These sequences were identified and included in the database in the event that the presence of a TAG decoding system (i.e. the Sep-OTS) could alter the expected frame-shifting behavior. The output of the scripts is a list of re-annotate E. coli genes in which TAG is treated as a sense codon. This nucleotide sequence database is then converted to a database designated TAG = X containing 328 protein sequences representing 386 TAG codons in FASTA file format (see Supplementary Table S9). In this database, X is substituted for any of the possible 20 natural AAs at TAG codons and protein sequences are extended to the next in-frame non-TAG stop codon. To generate 20 individual databases representing incorporation of any of the 20 standard AAs, we simply substituted X with any of the 20 standard AAs. All databases are provided in the supplement (Aerni et al. protein databases). A concatenated database containing all possible sequences was also generated. In this database, each protein was assigned a unique accession number enabling the simultaneous detection of any natural AA at any of the TAG sites in a single search. Although many of these hypothetical protein sequences will not be found in E. coli cells, this database is comprehensive and contains all possible TAG read-through events that might occur. These custom databases were always used in conjunction with an E. coli protein database to ensure a sufficiently large search space for matching of peptides and calculation of false discovery rates.
Peptides reporting incorporation of NSAAs or suppression by canonical AAs were identified by matching all peptide spectra matches (PSM) with a filter list containing all possible tryptic peptides from the TAG = X database. To generate the filter list, we first performed in silico digestion of the TAG = X protein database using Protein Digestion Simulator software v.2.2.5053 (retrieved from http://omics.pnl.gov/software/protein-digestion-simulator). Up to five missed cleavages were considered and all fully tryptic peptides in the mass range between 400 and 8000 Da were written in a text file. All peptides containing X in their sequence were then extracted in Microsoft Excel. The final filter list contained 4484 peptides representing all potential tryptic peptides reporting UAG suppression in any of the 328 UAG containing ORF's. Differences of ORFs reported by Lajoie (6) and our database are listed in Supplementary Table S10. PSM deposited in Yale Protein Expression Database (YPED) were then matched with a custom Java routine using the regular expression functions to generate a customizable text file with all matching identified peptides. The Java routine consisted of the following steps. Step 1: given a list of peptides which contain the letter ‘X’ representing a potential position for a novel AA substitution, create a list of regular expressions, ‘Rexp list’, from these peptides where each ‘X’ is matched with a single character, i.e. the peptide THYPXVG -> THYP\wVG$. Step 2: find all the distinct peptides, ‘result peptides’, with score greater than or equal to the homology score from a single result or a series of results. Step 3: each ‘result peptide’ is matched against each regular expression in the ‘Rexp list’ and peptides where the matching is successful are exported into a .txt file.
GFP reporter for quantification of NSAA incorporation
Affinity purified GFP was separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and gel bands were visualized by Coomassie staining. Bands representing full length GFP (28 kDa) were quantitated by densitometry using a bovine serum albumin (BSA) dilution series as a reference. In-gel digestion of excised GFP bands was performed as described by Shevchenko (16). The resulting peptides were quantified by UV280 on a nanodrop and 100 ng of the digest was injected for liquid chromatography-tandem-mass spectrometry (LC-MS/MS) analysis using a 90-min gradient method (see below). Peptides reporting the suppression site E17 were extracted using the TAG = X workflow.
In-solution digestion of affinity purified GFP for multiple reaction monitoring (MRM)
An aliquot corresponding to 25 μg GFP was transferred into a 1.5-ml PCR tube and the composition of the sample was adjusted using stock solutions consisting of 100-mM Tris–HCl buffer pH = 8.0, 100-mM DTT, 100 mM EDTA, 5% ALS-110 and water. The final sample composition was 25 μg GFP dissolved in 40 μl 10 mM Tris–HCl pH = 8.0 (23°C), 0.5% ALS-110, 10 mM DTT and 1 mM EDTA. Cysteines were reduced for 35 min at 55°C in a water bath. The reaction was briefly quenched on ice and 16 μl of a 60 mM IAA was added for alkylation of cysteines. The reaction proceeded for 30 min at room temperature and in the dark. Excess IAA was quenched with 14 μl of 25 mM DTT. The digest was then diluted with 40 μl of 1M Tris–HCl buffer pH = 8.0 and 310 μl of 70 mM Tris–HCl pH = 8.0 containing 2 mM CaCl2. Sequencing grade trypsin prepared at 0.5 μg/μl was added to obtain a trypsin/protein ratio of 1:15 by weight and protein was digested for 16 h at 37°C in an incubator. The digest was quenched with 64 μl of a 20% TFA solution, which lowered the sample pH below 2. Cleavage of the acid cleavable detergent was performed for 15 min at room temperature and peptides were desalted on a C18 UltraMicroSpin Column (The Nest Group Inc., Southborough, MA, USA). Peptides were dried in a rotary vacuum centrifuge operated without additional heat. Dried peptides were dissolved by vortex with 2.6 μl 1-propanol, 2 μl 70% FA and 15.4 μl 0.5% acetic acid. The peptide concentration of these stock solutions was estimated by UV280 on a nanodrop. The typical peptide concentration was in the range of 0.2–1.3 μg/μl depending on the sample analyzed. Samples were frozen at −80°C until further analysis.
Mass spectrometry
LC-MS/MS was performed on an Orbitrap Velos as described previously (6) with the following changes. Nano liquid chromatography was performed with a vented split setup consisting of a homemade fused silica trap column (30 mm x 150 μm ID) fitted with a Kasil frit following a protocol by Link (17). The trap column was connected to a metal nano T that was connected to an external switching valve. The valve was switched into the open position during sample trapping and was closed at the start of the gradient program. This setup reduced sample loading times and greatly reduced ion source contamination. The electrospray voltage, typically 1500–1800 V was applied to the metal T using an alligator cable. The trap column was packed in-house with MAGIC C18AQ resin with 200Å pore size and 3-μm particle size (Bruker Daltonics). Trapping was performed for 3.75 min at a flow rate of 4 μl/min with 5% eluent B (defined below). The capillary column was a 75 μm ID PicoFrit column (New Objectives, Woburn, MA, USA) packed in-house with 20 cm of 1.9 μm diameter Reprosil-Pur 120 C18-AQ C18 particles (Dr Maisch GmBH, Ammerbuch Germany) using methanol as the packing solvent. Peptides were separated at a flow rate of 300 nl/min using a 90 min gradient with 0.1% FA (Eluent A) and 0.1% FA in acetonitrile (Eluent B). The linear gradient was as follows (min/%B): 0.0/5.0, 0.1/5.0, 45.0/25.0, 65.0/50.0, 66.0/95.0, 71.0/95.0 72.0/5.0, 90.0/5.0. Mass spectrometry was performed on an Orbitrap Velos instrument (Thermo Scientific) configured with a top10 HCD method. An estimated 100 ng of the peptide digest dissolved in 2% Eluent B was injected for analysis. Database searching was performed with MASCOT as described below but using a custom database of GFP E17 containing all potential AA at position E17.
Synthetic peptide library and generation of a spectral library in Skyline
A custom synthetic peptide library for the development of a multiple reaction monitoring (MRM) assay enabling the quantitation of AA incorporation at position E17 = TAG in GFP was designed and then synthetized by JPT Technologies GmbH (Berlin Germany). All peptides in the library were C-terminally labeled with stable isotope labeled lysine (13C615N2) or arginine (13C615N4) depending on the sequence of the peptide. Reporter peptides corresponding to the sequence SKGEELFTGVVPILVXLDGDVNGHK (one missed cleavage site) and GEELFTGVVPILVXLDGDVNGHK with X designating any of the 20 natural AAs and the NSAA (Sep) were included in the synthetic peptide library. Peptides SKGEELFTGVVPILVX and GEELFTGVVPILVX were also included in the library to accommodate the special case of K or R incorporation. K or R incorporation at X followed by digestion with trypsin is expected to generate these truncated forms of the reporter peptide. Finally, the heavy reference peptides FEGDTLVNR and FSVSGEGEGDATYGK were synthetized to assist with assay development and the relative quantitation of GFP. We recognize that SpikeTides_L while being a cost-effective strategy for MRM assay development and relative quantitation of peptides are not suited for absolute quantitation of peptides because the exact absolute quantity of each peptide supplied in the library is not available.
Quality control of library peptides was performed with matrix-assisted laser desorption/ionization (MALDI) MS on a 4800 MALDI TOF/TOF instrument. For this experiment, 2 pmol of each peptide was manually spotted on a stainless steel target using the dried droplet method and using α-cyano-4-hydroxycinnamic acid (CHCA), prepared at 3 mg/ml in 50% acetonitrile 0.1% TFA, as the matrix. Accurate mass measurement obtained from the analysis of an estimated 2 pmol/spot peptide showed that the library was synthesized correctly (data not shown). Further validation of synthetic peptides was performed by shotgun proteomics on the Orbitrap Velos as described above. For this purpose four peptide mixtures were prepared ensuring that each containing only peptides with unique precursor ion masses. An estimated 400 fmol of each peptide was injected for this analysis. Database searching was performed with MASCOT using the EcoCyc (v.16) E. coli protein database (18) (Strain K12 sub strain MG1655) containing 4567 unique sequence entries and a custom database with protein sequences considering the possibility of any of the 20 natural AAs at position E17 of GFP. The following variable modifications were considered in these searches: deamidation (N/Q), phosphorylation (S,T,Y), oxidation (M) and C-terminal labeling with 13C615N2 and 13C615N4 for the AAs K and R respectively. The precursor mass tolerance was set to 20 ppm and the fragment ion mass tolerance was 0.02 Da for all searches. All PSM were imported into Skyline (19) software v. 1.3 and a spectral library was generated. The 10 most intense fragment ions in the spectral library were selected to generated 10 MRM transitions for each peptide. Only b and y and neutral loss ions for phosphorylated peptides in the mass range between 300–1400 Da were considered. The collision energy (CE) for precursor fragmentation was calculated in silico with Skyline software. The calculation was based on the mass-to-charge (m/z) ratio of the precursor ion using equations optimized for Agilent QQQ instruments (19): CE = 0.051*m/z − 15.563 and CE = 0.037*m/z − 9.784 for doubly and triply charged precursor ions, respectively.
Validation of the predicted collision energies was performed with selected peptides using a Skyline supported workflow for optimization of collision energies. This generated individual MRM transitions for each peptide that varied the CE systematically around the predicted CE using a step size of 3 V and considering five distinct collision energies on either side of the predicted CE for each precursor/product ion pair. The instrument was configured with an external syringe pump and the JetStream electrospray ion source. Peptides were prepared at 5–20 pmol/μl in 30% acetonitrile 0.1% FA were infused at a flow rate of 3 μl/min. The spray voltage was set to 4.5 kV and the ion source temperature was 250°C. Data were processed with Skyline software to extract collision energies for the 2–5 most sensitive transitions for detection of the NSAA Sep and the AAs Q, K,Y,E,S, G and T were selected for the final MRM method (See Supplementary Table S7).
LC-MRM-MS
Relative quantitation of tryptic peptides from E17GFP was performed by MRM on an Agilent 6490 triple quadrupole instrument that was equipped with a ChipCube ion source. Nano liquid chromatography (nanoLC) of peptides was performed with an Agilent 1260 HPLC system configured with a temperature controlled (4°C) model 1260 HiP micro ALS autosampler, a model 1260 cap pump and a model 1260 nano pump. A complete description of the instrument configuration and the method used for quantitation of GFP peptides is provided in a supplementary Excel file (LC_MRM_MS method). Briefly, peptides were separated on a Polaris-HR-Chip 3C18 (Agilent #G4240–62030) incorporating a 360 nl trap column and a 75 μm ID x 150 mm capillary column packed with 3 μm C18 particles. Both pumps were operated with 0.1% FA and 90% acetonitrile with 0.1% FA as eluent A and B respectively. The trap column was operated in a vented split design with a flow rate of 1.5 μl/min for trapping. The eluent composition during trapping was 2% B and the injection volume was 2 μl unless mentioned otherwise. The sample flush volume for the ChipCube was 8 μl. The carryover reduction function of the ChipCube was activated to perform two wash cycles prior to the next sample injection. Each wash cycle switched the injection valve and flushed the sample loop and needle assembly at 300 bar with 20 and 80% eluent B, respectively.
The capillary column was operated at a flow rate of 0.4 μl/min using the following optimized linear gradient (min/%B): 0/2, 0.2/2, 1.5/9, 13/17, 14.5/29, 27/32, 37/40, 38.5/100, 40/100, 42/2. At 43 min, the trap column was switched back to sample trapping which ensured adequate equilibration of the trap column prior to the next sample injection. The mass spectrometer was operated with a scheduled MRM method with Q1 and Q3 operated at unit resolution. The source temperature was set to 200°C, the fragmentor voltage was 380 V, the cell accelerator voltage was 5 V and the electron multiplier offset was set to 350 V for all MRM experiments. The cycle time for the longest MRM time segment was 2.86 s which ensured collection of at least 10 data points across each peak in the chromatogram. The position of the chip and the spray voltage (1700–2000 V) were optimized routinely to ensure stable and reproducible spray performance. The sensitivity and reproducibility of the platform was evaluated daily by injecting mixtures of stable isotope labeled GFP reporter peptides. For quality control, we injected BSA digests and solvent blanks between samples. This provided continuous monitoring of instrument sensitivity, chromatographic reproducibility and absence of carryover.
Optimization of gradient conditions for MRM
Gradient conditions for the separation of the reporter peptides were optimized using mixtures of stable isotope labeled reporter peptides (labeled residue underlined) with the general sequence SKGEELFTGVVPILVXLDGDVNGHK and GEELFTGVVPILVXLDGDVNGHK with position 17 X = G, S, Sep, Q, T, E, Y and peptides SKGEELFTGVVPILVX and GEELFTGVVPILVX with X = K. Peptides were dissolved in the LC-MS solvent and 66 fmol were injected for LC-MRM-MS. Systematic optimization of the gradient was performed providing separation of peptides with similar precursor masses. For example, reporter peptides (position 17 in bold) GEELFTGVVPILVQLDGDVNGHK and GEELFTGVVPILVELDGDVNGHK, which are only separated by one mass unit, were base line separated with retention times of 35.01 and 36.67 min, respectively. Retention times for all peptides are listed in the Supplementary Table S8).
MRM assay for relative quantitation of GFP
The peptide stock solution was diluted to a concentration of 100 ng/μl using 2% acetonitrile 0.1% FA and the solution was mixed 1:1 by volume with a peptide solution containing 200 fmol/μl of the stable isotope labeled peptides FEGDTLVNR and FSVSGEGEGDATYGK prepared in 2% acetonitrile 0.1% FA. Two microliters of this mixture corresponding to 100 ng digest and an estimated 200 fmol of the internal standard peptides were injected and analyzed by LC-MRM-MS to quantify the concentration of GFP in the samples. The stable isotope labeled peptide FEGDTLVNR was used to determine the relative concentration of GFP in the digests providing excellent sensitivity and reproducibility across all runs as judged by the intensity of the stable isotope labeled version of this peptide (see Supplementary Figure S3). The relative concentration of GFP in the samples depended on the overall GFP expression level in the bacterial culture. We standardized the total amount of GFP injected in subsequent LC-MRM-MS experiments to the sample with the lowest concentration of GFP, which was the GFP-OTS sample in biological replicate 1. This ensured equal loading of GFP peptides across all experiments and enabled a direct comparison of relative peak intensities of the reporter peptide between samples.
RESULTS
Enhanced natural codon readthrough in the absence of release factor 1
We hypothesized that release factor 1 (RF1) deletion would promote increased UAG codon readthrough and increased NSAA incorporation while competition by misincorporation of natural AAs may also increase. Indeed, we and others have observed natural AA incorporation at UAG and we were motivated to explore this in greater depth (6,9,10,20). E. coli contains 321 natural amber stop codons (6,9,11) and thus RF1 deficient strains could potentially extend natural protein synthesis beyond the UAG- and onto the next in-frame stop codon (Figure 1A). Mass spectrometry based proteomics is an ideal tool to characterize these new protein forms. Standard proteomic workflows cannot readily identify extended proteins because extended protein sequences are not present in sequence databases used for proteomics. Furthermore, tools that rapidly identify peptides reporting UAG readthrough from the thousands of identified peptides observed in a proteomic experiment were previously nonexistent.
Figure 1.
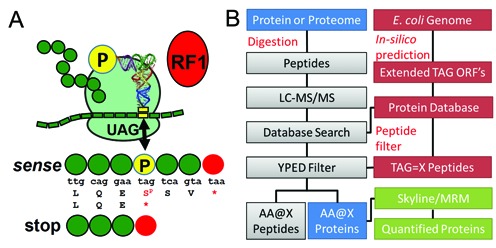
A proteomics workflow to identify natural AAs and Sep at native UAG codons in Escherichia coli. (A) Incorporation of Sep (P) at the stop codon (UAG) competes with RF1. Sep incorporation extends protein synthesis to the next in-frame stop codon (UAA or UGA) thus extending the natural ORF. (B) Proteomics workflow for the detection and quantitation of UAG stop codon readthrough. Custom open reading frame (ORF) databases are generated from the E. coli genome. ORFs are extended in silico past the natural UAG stop codon and these extended ORFs are appended to the standard E. coli proteome. These databases interface with proteomics database search software to identify extended protein ORFs from shotgun proteomics datasets. TAG sites in the extended ORFs are translated as X, or the 20 natural AAs, to populate the databases and enable peptide discovery and identification with custom filters. Protein or peptide level information can be culled from the extended proteome data and transferred into Skyline to develop MRM methods for quantitative proteomics.
We first generated custom E. coli databases that contain in silico predicted E. coli protein sequences in which all amber codons were reassigned to each of the 20 natural AAs (Figure 1B). The 321 annotated UAG-containing ORFs in the E. coli genome sequence (6,11) were then extended to the next in-frame non-TAG stop codon. These databases were then combined into a single database and used in conjunction with a conventional E. coli protein database for mining of the shotgun proteomic data. We then implemented an automated script to generate a list of tryptic peptides encoding all possible AAs at TAG loci.
We first tested our method by analyzing global UAG codon readthrough in three E. coli strains with demonstrated utility in directing Sep incorporation at UAG codons (13,21). Genetically encoding Sep uses a Sep-OTS, which includes a phosphoseryl-tRNA synthetase, an amber codon decoding tRNASep and a variant of elongation factor Tu (EF-Sep)(21). In proteomic analysis of these strains, over 1000 proteins were identified, including 83 TAG containing ORFs (see Supplementary Table S2). We examined a wild-type BL21 strain (BL21.WT.SEP.OTS2.1) and detected no significant translation of UAG codons with one exception (see Figure 2A and Supplementary Tables S4 and S5). We then compared the same BL21 strain encoding an established Sep-OTS (21) (BL21.L11C.SEP.OTS2.1; see Supplementary Table S1). In this strain, we identified two native ORFs (ilvA and luxS) in which a terminal amber codon directed Sep insertion and extension of the native protein to the next non-amber stop codon (Figure 2A). Finally, we examined the RF1 deletion strain EcAR7 (EcAR7.SEP.OTS2.1) (13), again with Sep-OTS2.1 and identified seven phosphopeptides (see Supplementary Table S3) all with Sep incorporation at native UAG codons (Figure 2A and B). The increased number of phosphopeptides identified in the RF1 deficient EcAR7strain indicates that our method readily detects enhanced readthrough of native UAGs resulting from RF1 deletion.
Figure 2.

Identification of natural AAs and Sep incorporated at native UAG codons in Escherichia coli. (A) Summary of peptides spanning UAG sites from RF1 + BL21 strains (BL21.WT.SEP.OTS2.1 (green) and BL21.L11C.SEP.OTS2.1 (violet)) and the RF1 deficient strain EcAR7.SEP.OTS2.1 (brown) discovered with the proteomics workflow. Peptides with Sep@UAG (red), Q@UAG (white or red underlined; *two Q in sucB) and V@UAG (black, italic) are listed. (B) Annotated tandem MS spectrum of a phosphopeptide showing incorporation of Sep at the native UAG codon in speG.
One of the goals of genetic code expansion is to produce proteins with an expanded AA repertoire while preserving the same fidelity as natural protein synthesis. In addition to on-target Sep incorporation, our method identified off-target incorporation of glutamine at native UAG codons (Figure 2A). These observations were more frequent in the RF1 deficient strain suggesting that misincorporation of AAs at UAG sites might be enhanced in this strain.
Optimization of Sep-OTS systems and quantitation of NSAA incorporation
We applied our workflow to quantify the fidelity of several Sep-OTS systems engineered for enhanced performance in RF1 deficient strains. An established GFP reporter (13) was used to simultaneously quantify on-target Sep incorporation and off-target AA incorporation (Figure 3A). We expressed this protein in EcAR7, with and without the Sep-OTS and performed a standard shotgun proteomic analysis of the purified proteins. We then analyzed the shotgun data with our bioinformatics pipeline as described above. Briefly, LC-MS/MS data was searched with MASCOT against an E. coli database and a custom database for GFP that considered all 20 canonical AAs at the UAG codon (see Figure 3A and Supplementary Table S6). Modified- or non-standard AAs can be specified via custom modifications in the database search engine. We note that the sequences of the peptides reporting suppression of UAG are unique in the peptide search results and reporter peptides reporting UAG readthrough can be identified in Microsoft Excel with a simple text filter containing the partial reporter peptide sequence. In addition to Sep, our method identified an unexpectedly large number of AAs incorporated at the UAG codon. We identified GFP peptides with off-target insertions of Q, Y, W, K, G, E, S and T (see Supplementary Table S6). We reasoned that the number of off-target AAs identified increased due to the fact that we were targeting a single protein which increases the sensitivity and dynamic range compared with our whole proteome shotgun experiments (Figure 2A). Off-target incorporation of Q, Y and W has been reported previously (9) and results from near-cognate recognition of amber codons by the respective native aminoacyl-tRNAs (see Supplementary Figure S2). Our method uniquely identified near cognate incorporation of E and K in addition to misincorporation of G, S and T. Lysine insertion at UAG was verified in our proteomics data with an unbiased database and trypsin cleavage rules where lysine incorporation at UAG would produce novel tryptic cleavage sites.
Figure 3.
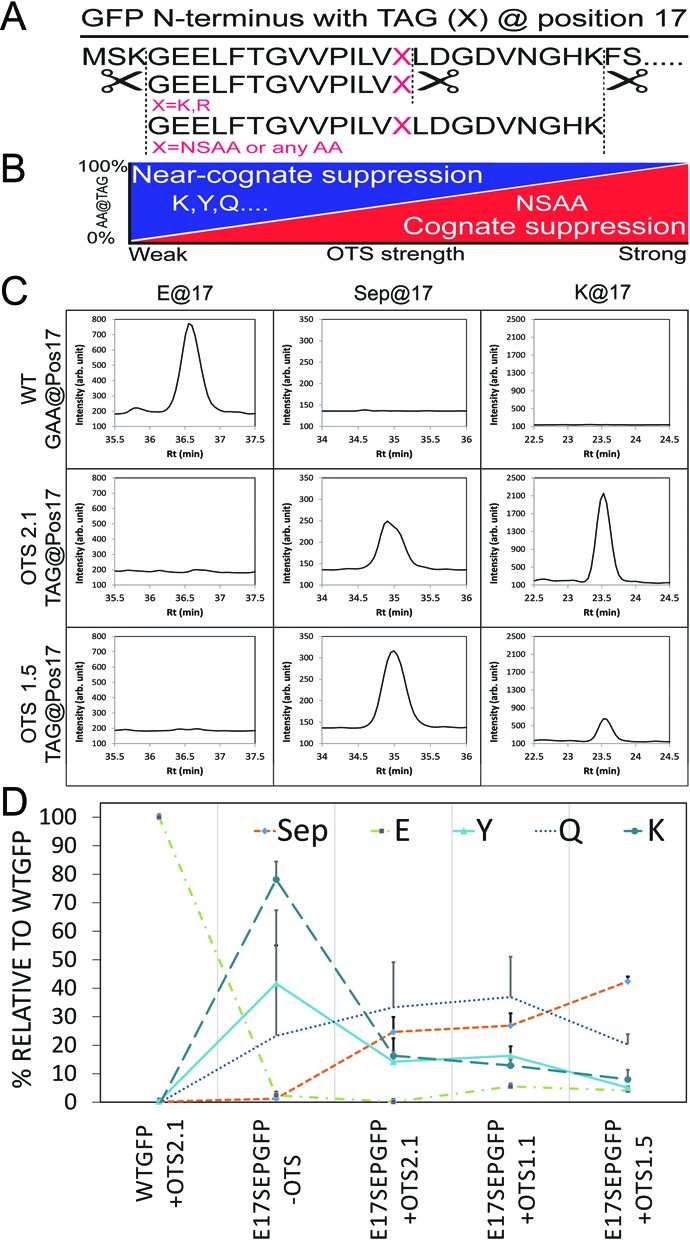
Label-free MRM quantitation of AA incorporation at UAG codons. (A) The N-terminus of the GFP reporter with the E17 codon replaced with a TAG. Trypsin cuts the reporter at K or R residues and generates peptides that report the incorporation of non-standard or natural AAs (red). (B) Increasing the strength of an OTS for NSAA incorporation is predicted to outcompete near-cognate suppression. (C) Extracted MRM traces from experiments with the GFP reporter and a GAA codon (E) or with TAG and two different Sep-OTSs. OTS2.1 was our original two plasmid system containing one tRNASep (21). We merged the two plasmids (OTS2.1) to obtain a single plasmid containing 1 (OTS1.1) and 5 (OTS1.5) tRNASep respectively while leaving the copy number for EF-Sep and the SepRS constant. MRMs for Sep, E, Y and K are shown. (D) Quantitation of Sep and near-cognate AA insertion across a series of Sep-OTSs showing elevated Sep incorporation with increased strength of the Sep-OTS and reduced misincorporation of Y, K and Q.
We speculated that UAG readthrough by near-cognate aminoacyl-tRNAs would be the most abundant off-target events. Therefore, we quantitatively assessed the impact of RF1 deletion on UAG translation fidelity and Sep insertion efficiency quantified. Our methods included a pathway to build quantitative MRM assays from shotgun discovery data via Skyline (19) (Figure 1B). This enables a method to simultaneously quantify both Sep and natural AA insertion at the UAG codon. To test this, we used a series of Sep-OTSs with increasing copy numbers of tRNASep to better compete against near-cognate readthrough. We used our established GFP reporter (13) and new MRM assay (see Supplementary Tables S7 and S8) to perform label-free quantitation of Sep and natural AA incorporation at a single UAG codon. Importantly, these experiments were carried out in the RF1 deficient EcAR7 strain (13). We used two reference peptides located downstream of the TAG-locus to normalize our samples for total GFP (see Supplementary Figure S3). This enabled a direct comparison of relative abundances for each reporter peptide across experiments. The high reproducibility of this MRM assay is demonstrated by the low standard deviation for the detection of a spike-in stable isotope labeled versions of one of the reference peptides (see Supplementary Figure S3). We choose not to perform a comparison of absolute intensities between peptides reporting the insertion of different AAs because the peptide length, AA composition and charge are known to alter ionization properties. This methodology could be easily extended for absolute quantitation of the reporter peptides if stable isotope labeled peptides (e.g. AQUA peptides (22)) were included for each analyte in the assay.
We first quantified AA incorporation in our GFP reporter with a native glutamate (E) codon GAA at position 17 (Figure 3C and D). As expected, only on-target E incorporation was detected at this native sense codon. We then simultaneously quantified the incorporation of Sep, Q, E, Y and K in a GFP construct with TAG at position 17 co-expressed with Sep-OTSs of varying strengths (Figure 3D). Introduction of the Sep-OTS showed robust and abundant on-target Sep incorporation at UAG sites. A striking diversity of amber codon readthrough was readily quantified. Our assay directly measured aminoacyl-tRNA competition taking place on the ribosome. We observed that Q, Y and K near-cognate readthrough decreases with increasing strength of the Sep-OTS directed by increasing the copy number of the cognate tRNASep. These results not only demonstrate the utility of quantitative assessment of Sep-OTS efficiency and purity of the phosphoprotein product, they also point to further improvements in E. coli strains, such as a completely recoded cell (6), that might improve fitness of the host strain and the performance of the Sep-OTS.
DISCUSSION
It is a common misconception that engineered or expanded genetic codes have the same level of fidelity as the natural code. Approaches that can rapidly assess the fidelity of protein synthesis in a whole proteome were not available. For example, we found that the standard proteomics approaches were unable to identify multiple AA insertion at the same codon in an unbiased fashion. Here we introduce a strategy that uniquely identifies and quantifies AA diversity, or reduced translational fidelity, during translation of the amber codon. The method permits proteome level characterization of organisms with expanded genetic codes. A key innovation in our approach was a set of databases that contain C-terminally extended protein sequences of all known UAG terminated ORFs in E. coli (11). The databases contain 20 natural AAs at the UAG sites to produce a comprehensive set of extended UAG ORFs. We then successfully used this approach to mine large-scale shotgun proteomics data to identify proteins resulting from stop codon readthrough. We applied these databases with the search engine MASCOT but we note that our databases are compatible with other search engines including Andromeda (MaxQuant) (23) and Myrimatch (24). Our method allowed us to identify 10 extended ORFs (Figure 2A) that are potentially useful native reporters for further OTS and E. coli engineering (6). We imagine that this workflow can be easily adapted to monitor stop codon readthrough or alternate codon assignment in any organism with a sequenced genome.
Within this framework, NSAA incorporation can be detected via custom modifications of standard AAs. Because we are using a peptide based (bottom-up) approach, the site-specific incorporation of the NSAA can often be directly confirmed from the fragmentation spectrum of a peptide (Figure 2B). Using the Sep OTS (21), we were able to easily identify peptides reporting incorporation of Sep at off-target UAGs in native E. coli ORFs (Figure 2A). Not only did we detect increased Sep incorporation in the RF1 deficient strain, but we also found evidence for near-cognate misincorporation of glutamine (Q) in the RF1 deficient strain (Figure 2A). While the number of detected extended ORFs was limited in this dataset, we envision that the coverage of the method can be extended by applying multidimensional protein identification technology (MudPIT) (25,26) prior to MS analysis which should extend proteome coverage.
One of our goals is to optimize the purity and quality of NSAA containing proteins expressed in organism with UAG reassigned as a sense codon. The characterization of translation at this ‘new’ sense codon is important as it can guide further strain- and OTS engineering efforts leading to increased protein yields and purity. Toward this goal, we applied our workflow to characterize translation of an amber codon in GFP permissible for Sep incorporation (Figure 3A). Using a custom database for our reporter that considered any of the 20 natural AAs at the suppressor site we identified site-specific incorporation of the NSAA Sep at UAG. In addition, we confirmed near-cognate misincorporation of Q, Y, W, E and K but the high sensitivity of our method allowed us to detect evidence for misincorporation of G and T. Future work will establish the absolute levels of these events and could possibly compare translation at these UAG codons to the natural fidelity of protein synthesis at reassigned sense codons (27).
Toward our goal to define the purity of NSAA containing proteins, we used our workflow (Figure 1B) to generate a label-free MRM assay that permitted in-depth quantitation of phosphoprotein purity resulting from our new Sep-OTSs (Figure 3C and D). As expected, off-target misincorporation of Y and Q was reduced when we increased the copy number of tRNASep in our system. Our best Sep-OTS system (OTS1.5) not only increased the yield of Sep containing protein (see Supplementary Figure S1) but also increased the purity of the phosphoprotein (Figure 3D). The data suggests that further improvements for this OTS will be necessary to increase phosphoprotein purity. Furthermore, this supports our idea that new OTS systems should be characterized in greater detail than previously recognized. Clearly, further optimization of the Sep-OTS system will be necessary before this technology can be applied for medical applications or bio manufacturing. Our data suggest that other existing OTS systems should be re-examined to quantify their ability to produce pure NSAA-containing recombinant protein and also to assess their impact on the proteome resulting from off-target UAG translation.
We are aware that this method has potential applications beyond the characterization of NSAA incorporation. Elevated misincorporation rates of natural AAs or readthrough of natural stop codons in bacteria subjected to stress (e.g. antibiotics) is well recognized. The high sensitivity of our workflow suggests that it may be a useful tool for the discovery and quantitation of such events.
Our detailed and quantitative examination of protein synthesis at an ‘open’ UAG codon provided exciting new experimental evidence for a long-standing theory in the protein synthesis field. These experiments recapitulate a scenario for the evolution of the genetic code that was envisioned by Woese in the 1960′s (28). He argued that the genetic code must have evolved through stages, the more primitive of which likely produced ‘statistical proteins’ that would result from an inaccurate genetic code with many ambiguously decoded codons. Using our methodology, we found that opening UAG leads to a codon that is read by several aminoacyl-tRNAs. By expressing the Sep-OTS at increasing strengths, we were able to substantially shift the meaning of UAG toward Sep. It is conceivable that over the course of evolution, selection could produce a more clearly defined meaning for UAG in a similar way that might involve altering aminoacyl-tRNA concentrations in the cell or modulating competition for UAG decoding on the ribosome.
UAG readthrough is not confined to the field of protein engineering and genetic code expansion. Metagenomics studies have unveiled thousands of natural reassignments of the UAG codon from stop to sense (8) and ribosome profiling data indicate that in Drosophila UAG and other stop codons are read at significant rates in certain genes as part of normal protein synthesis (29). We suggest that our method will provide quantitative insight into engineered proteomes, improve OTS development and advance discoveries into the unexpected diversity of natural proteomes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Farren Isaacs, Miriam Amiram, and Jiqiang ‘Lanny’ Ling for critical review of our manuscript. Agilent Technologies, Jim Lynch, Doug Postl, Sanika Kulkarni and Gerry Alexis for support of the MRM platform. Chris Colangelo and Thomas Abbott for operation of the 5600 triple TOF instrument at the W.M. Keck Foundation Biotechnology Resource Laboratory at Yale. Terence Wu and Brandon Gassaway for help with the Orbitrap Velos instrument.
FUNDING
National Institutes of Health [NIDDK-K01DK089006 to J.R.]; Defense Advanced Research Projects Agency [N66001-12-C-4211 to J.R.]; Natural Sciences and Engineering Research Council of Canada [RGPIN 04282-2014 to P.O'D.]. Funding for open access charge: Defense Advanced Research Projects Agency [N66001-12-C-4211 to J.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Crick F.H.C. The origin of the genetic code. J. Mol. Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 2.Nirenberg M., Leder P., Bernfield M., Brimacombe R., Trupin J., Rottman F., O'Neal C. RNA codewords and protein synthesis, VII. On the general nature of the RNA code. Proc. Natl. Acad. Sci. U.S.A. 1965;53:1161–1168. doi: 10.1073/pnas.53.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soll D., Ohtsuka E., Jones D.S., Lohrmann R., Hayatsu H., Nishimura S., Khorana H.G. Studies on polynucleotides, XLIX. Stimulation of the binding of aminoacyl-sRNA's to ribosomes by ribotrinucleotides and a survey of codon assignments for 20 amino acids. Proc. Natl. Acad. Sci. U.S.A. 1965;54:1378–1385. doi: 10.1073/pnas.54.5.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu C.C., Schultz P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 5.O'Donoghue P., Ling J., Wang Y.S., Soll D. Upgrading protein synthesis for synthetic biology. Nat. Chem. Biol. 2013;9:594–598. doi: 10.1038/nchembio.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lajoie M.J., Rovner A.J., Goodman D.B., Aerni H.R., Haimovich A.D., Kuznetsov G., Mercer J.A., Wang H.H., Carr P.A., Mosberg J.A., et al. Genomically recoded organisms expand biological functions. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bröcker M.J., Ho Joanne M.L., Church George M., Söll Dieter, O'Donoghue Patrick. Recoding the genetic code with selenocysteine. Angew. Chem. Int. Ed. Engl. 2014;53:319–323. doi: 10.1002/anie.201308584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanova N.N., Schwientek P., Tripp H.J., Rinke C., Pati A., Huntemann M., Visel A., Woyke T., Kyrpides N.C., Rubin E.M. Stop codon reassignments in the wild. Science. 2014;344:909–913. doi: 10.1126/science.1250691. [DOI] [PubMed] [Google Scholar]
- 9.Johnson D.B., Xu J., Shen Z., Takimoto J.K., Schultz M.D., Schmitz R.J., Xiang Z., Ecker J.R., Briggs S.P., Wang L. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 2011;7:779–786. doi: 10.1038/nchembio.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukai T., Hayashi A., Iraha F., Sato A., Ohtake K., Yokoyama S., Sakamoto K. Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 2010;38:8188–8195. doi: 10.1093/nar/gkq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isaacs F.J., Carr P.A., Wang H.H., Lajoie M.J., Sterling B., Kraal L., Tolonen A.C., Gianoulis T.A., Goodman D.B., Reppas N.B., et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Donoghue P., Prat L., Heinemann I.U., Ling J., Odoi K., Liu W.R., Söll D. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012;586:3931–3937. doi: 10.1016/j.febslet.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinemann I.U., Rovner A.J., Aerni H.R., Rogulina S., Cheng L., Olds W., Fischer J.T., Soll D., Isaacs F.J., Rinehart J. Enhanced phosphoserine insertion during Escherichia coli protein synthesis via partial UAG codon reassignment and release factor 1 deletion. FEBS Lett. 2012;586:3716–3722. doi: 10.1016/j.febslet.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Odoi K.A., Huang Y., Rezenom Y.H., Liu W.R. Nonsense and sense suppression abilities of original and derivative Methanosarcina mazei pyrrolysyl-tRNA synthetase-tRNA(Pyl) pairs in the Escherichia coli BL21(DE3) cell strain. PloS One. 2013;8:e57035. doi: 10.1371/journal.pone.0057035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markowitz V.M., Chen I.M., Palaniappan K., Chu K., Szeto E., Pillay M., Ratner A., Huang J., Woyke T., Huntemann M., et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2007;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 17.Link A.J., Jennings J.L., Washburn M.P. Current Protocols in Protein Science. New York: John Wiley & Sons, Inc.; 2001. [Google Scholar]
- 18.Keseler I.M., Collado-Vides J., Santos-Zavaleta A., Peralta-Gil M., Gama-Castro S., Muniz-Rascado L., Bonavides-Martinez C., Paley S., Krummenacker M., Altman T., et al. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2011;39:D583–D590. doi: 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maclean B., Tomazela D.M., Abbatiello S.E., Zhang S., Whiteaker J.R., Paulovich A.G., Carr S.A., Maccoss M.J. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal. Chem. 2010;82:10116–10124. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohtake K., Sato A., Mukai T., Hino N., Yokoyama S., Sakamoto K. Efficient decoding of the UAG triplet as a full-fledged sense codon enhances the growth of a prfA-deficient strain of Escherichia coli. J. Bacteriol. 2012;194:2606–2613. doi: 10.1128/JB.00195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park H.S., Hohn M.J., Umehara T., Guo L.T., Osborne E.M., Benner J., Noren C.J., Rinehart J., Söll D. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kettenbach A.N., Rush J., Gerber S.A. Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat. Protoc. 2011;6:175–186. doi: 10.1038/nprot.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neuhauser N., Michalski A., Cox J., Mann M. Expert system for computer-assisted annotation of MS/MS spectra. Mol. Cell. Proteomics. 2012;11:1500–1509. doi: 10.1074/mcp.M112.020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tabb D.L., Fernando C.G., Chambers M.C. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007;6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Link A.J., Eng J., Schieltz D.M., Carmack E., Mize G.J., Morris D.R., Garvik B.M., Yates J.R. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 26.Washburn M.P., Wolters D., Yates J.R., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 27.Lajoie M.J., Kosuri S., Mosberg J.A., Gregg C.J., Zhang D., Church G.M. Probing the limits of genetic recoding in essential genes. Science. 2013;342:361–363. doi: 10.1126/science.1241460. [DOI] [PubMed] [Google Scholar]
- 28.Woese C.R. On the evolution of the genetic code. Proc. Natl. Acad. Sci. U.S.A. 1965;54:1546–1552. doi: 10.1073/pnas.54.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunn J.G., Foo C.K., Belletier N.G., Gavis E.R., Weissman J.S. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife. 2013;2:e01179. doi: 10.7554/eLife.01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.