


Abstract
In post-transcriptional regulation, an mRNA molecule is bound by many proteins and/or miRNAs to modulate its function. To enable combinatorial gene regulation, these binding partners of an RNA must communicate with each other, exhibiting cooperativity. Even in the absence of direct physical interactions between the binding partners, such cooperativity can be mediated through RNA secondary structures, since they affect the accessibility of the binding sites. Here we propose a quantitative measure of this structure-mediated cooperativity that can be numerically calculated for an arbitrary RNA sequence. Focusing on an RNA with two binding sites, we derive a characteristic difference of free energy differences, i.e. ΔΔG, as a measure of the effect of the occupancy of one binding site on the binding strength of another. We apply this measure to a large number of human and Caenorhabditis elegans mRNAs, and find that structure-mediated cooperativity is a generic feature. Interestingly, this cooperativity not only affects binding sites in close proximity along the sequence but also configurations in which one binding site is located in the 5′UTR and the other is located in the 3′UTR of the mRNA. Furthermore, we find that this end-to-end cooperativity is determined by the UTR sequences while the sequences of the coding regions are irrelevant.
INTRODUCTION
Post-transcriptional regulation of messenger RNAs (mRNAs) is an important component in cellular information processing. At the molecular level, this post-transcriptional regulation involves the binding of a multitude of proteins and other factors such as microRNAs incorporated into riboprotein complexes to an mRNA molecule (1,2). These proteins affect gene expression through regulation of mRNA stability, localization and translation. The importance of these RNA protein interactions can be gauged from the fact that there are close to 800 RNA binding proteins annotated in the proteome of the human embryonic kidney (3) and 860 in HeLa cells (4), and that a PubMed title search for the words ‘RNA’, ‘protein’ and ‘binding’ yields more than 2500 articles.
In order to obtain ‘combinatorial’ gene regulation, binding events of RNA binding proteins and other RNA binding factors have to be interdependent, or in biochemical terms ‘cooperative’. Since, most protein and microRNA binding sites are located in the untranslated regions (UTRs) at the ends of an mRNA (5), it is an interesting question how cooperativity between these distant regions of an RNA molecule is achieved, especially in light of recent evidence that the protein bridge between the 3′ and 5′UTRs in eukaryotes mediated by proteins that simultaneously bind the poly(A) tail and the cap may not be essential (6). For some transcripts the two UTRs might be brought together physically by binding of dimeric RNA binding proteins to one binding site in each UTR as recently hypothesized for the GLD-1 protein in Caenorhabditis elegans (7). Here, we present a natural mechanism for such cooperativity that does not require specific proteins but relies on RNA secondary structure alone.
Qualitatively the mechanism for this cooperativity is as follows: many RNA binding proteins bind to single-stranded regions of RNA (8–10). Therefore, a natural competition arises between the formation of intramolecular base pairs in the RNA and the binding of proteins to unpaired bases (here and in the following we will restrict our language to ‘RNA binding proteins’ even though in the context of this study any RNA binding factor that competes with the RNA for base pairing, such as, e.g. a microRNA, can play the role of what we call an ‘RNA binding protein’). We have recently discovered that this competition between binding to a single-stranded RNA binding protein and formation of intramolecular base pairs provides a possible mechanism for cooperativity among proteins binding an RNA molecule (11). A protein binding an RNA molecule will change the RNA's secondary structure by prohibiting the bound bases from pairing, which in turn affects the base pairing at the binding site of the second protein, and thus the ability of the second protein to bind (see Figure 1). In our previous work (11) we have focused on establishing that this RNA secondary structure mediated cooperativity is a long-range effect, and used simplified models of RNA folding to understand this behavior for random sequences. Here, we will show that this cooperativity (i) occurs for natural sequences, (ii) is of a biologically relevant order of magnitude of several kcal/mol, (iii) not only occurs between protein binding sites that are within 100 nt or so along the RNA but also between the 5′UTR and the 3′UTR and (iv) is determined by the UTR sequences with little effect from the coding regions.
Figure 1.
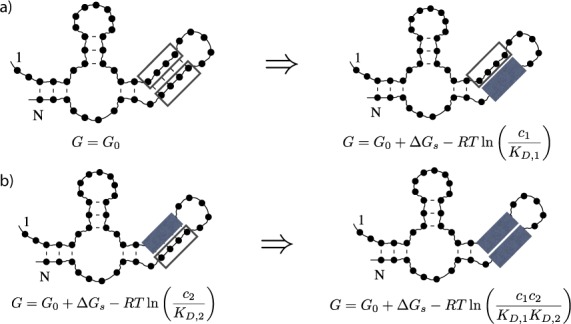
An example of structure-mediated cooperativity, where open boxes indicate unoccupied protein binding sites and filled boxes indicate occupied protein binding sites. G is the free energy of the RNA–protein complex in each step, G0 is the free energy of the RNA secondary structure alone, ΔGs is the free energy associated with breaking the base pairs in the stem containing the two protein binding sites, c1, c2, KD, 1 and KD, 2 are the concentrations and dissociation constants of the first and the second protein, respectively, R is the gas constant and T is the temperature in kelvin. (a) To have the first protein bind to bases in a stem of the structure in the absence of the second protein, the stem has to be broken, resulting in a free energy difference of ΔG0 → 1 = ΔGs − RTln (c1/KD, 1). (b) Once the second protein is bound to the opposing bases in the stem, the first protein can directly bind to its binding site without the loss of free energy by breaking additional base pairs, yielding ΔG2 → 12 = −RTln (c1/KD, 1) < ΔG0 → 1. Thus, in this case the two proteins exhibit positive cooperativity.
MATERIALS AND METHODS
RNA secondary structure prediction
A single-stranded RNA folds into different configurations by pairing its nucleotides and forming stable Watson–Crick (i.e. A–U or G–C) or wobble (i.e. G–U) base pairs. Stacks of these base pairs form stiff helices connected by flexible linkers of unpaired bases. In the case of structural RNAs these elements fold into a three-dimensional structure required for the biological function of the molecule, called the tertiary structure. However, given the generic propensity of nucleotides to base pair, also mRNAs, which are not primarily designed to fold into a specific structure, will form base pairs. Since here we are interested in mRNAs, we will focus on these secondary structures only. For computational convenience we follow the usual approach of the field and exclude the formation of pseudoknots because such structures contribute small amounts of free energy if they are short and become kinetically suppressed if they are long (12). More specifically, we use the Vienna package (13) to calculate the free energies of secondary structure folding, which implements the state-of-the-art nearest-neighbor free energy model of RNA secondary structure formation (14).
RNA–protein binding
To discuss RNA–protein binding events, we consider only the simplest effects of the binding of the protein on the RNA secondary structure (11,15): (i) a bound protein prevents all nucleotides in its footprint from base pairing and (ii) binding of a protein to the RNA results in a free energy gain of RTln (c/KD), where c is the concentration of the protein in solution, KD is the dissociation constant of the protein from the specific site in the RNA, which contains all the details of the interaction between the protein and the RNA, R is the gas constant and T is the temperature in kelvin (16). More sophisticated effects of RNA–protein binding, for example the increased geometrical distance of the bases adjacent to the protein binding site upon binding of the protein or any other modifications to the binding propensities of nucleotides not within the footprint of the protein, are not considered in our minimal model and will be subject of future research.
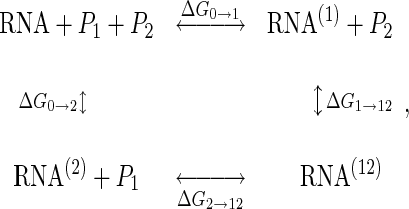
We will further constrain ourselves to the case that there are only two proteins, P1 and P2, binding one binding site each on a given RNA molecule, which is the simplest system to investigate protein–protein cooperativity in (11). Consequently, there are four possible RNA–protein complexes, distinguished by whether the first and/or the second protein binding site is occupied or not, which we label as RNA, RNA(1), RNA(2) and RNA(12) as illustrated in Figure 2. For each complex, different sets of base pairs can participate in base pairing interactions. We calculate the free energies G(0), G(1), G(2) and G(12) of the secondary structures for each of the four complexes using the constraint folding capabilities of the Vienna package to exclude the nucleotides ‘hidden’ in the footprints of bound proteins from base pairing. To obtain the free energies of the entire complexes the protein binding free energy RTln (c1/KD, 1) has to be subtracted from G(1) and G(12) and the protein binding free energy RTln (c2/KD, 2) of the second protein has to be subtracted from G(2) and G(12).
Figure 2.
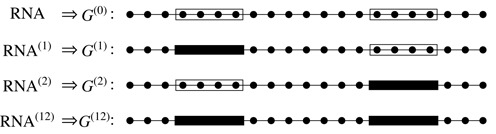
The four possible RNA–protein complexes to consider. Lines are RNA backbones and dots are bases. Transparent and black blocks represent the unoccupied and occupied protein binding sites, respectively. Only the bases that are not in the occupied binding regions participate in base pairing.
Quantitating protein–protein cooperativity
With the two proteins P1 and P2, all RNA–protein binding reactions can be expressed as
 |
where the free energy differences ΔG of the four reactions are given by
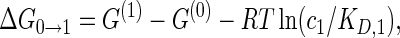 |
(1a) |
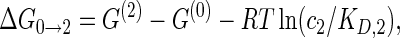 |
(1b) |
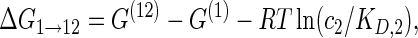 |
(1c) |
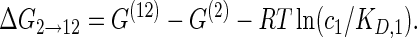 |
(1d) |
The cooperativity between the two binding sites can be quantified by comparing the binding free energies ΔG of the reactions on opposite sides of this diagram, e.g. the difference in binding free energy ΔG0 → 1 for the binding of protein P1 in the absence of protein P2 and the binding free energy ΔG2 → 12 associated with binding of protein P1 in the presence of protein P2 (or vice versa with the two proteins interchanged which leads to the same result). This yields the cooperativity free energy
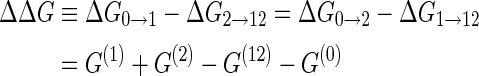 |
(2) |
which becomes independent of the protein concentrations c1 and c2. Therefore, for an arbitrary RNA sequence, once the two protein binding sites are assigned, the Vienna package can be used to calculate the structural free energies G(0), G(1), G(2) and G(12), from which the free energy difference ΔΔG quantifying the cooperativity can be deduced via Equation (2). For a positive cooperativity, a protein already bound helps the other protein bind, leading to ΔG2 → 12 < ΔG0 → 1 and thus a positive ΔΔG; for a negative cooperativity, the situation is reversed and ΔΔG is negative.
Locations of protein binding sites
We are especially interested in the cooperativity between the proteins bound on the 5′ and 3′UTRs close to the 5′ and 3′ ends of the mRNAs, respectively. Therefore, we assign the two protein binding sites of each mRNA in their 5′ and 3′ ends, and vary their distances from the ends of the RNAs (note that in our minimal model the binding site of each protein is specified explicitly, i.e. that we assume perfect sequence specificity for the sequence at whatever binding site we choose without actually specifying that sequence). The relative location of the two binding sites is quantified by the outside distance, Dout, which is defined in Figure 3 as the sum Dout = n1 + n2 of the distances n1 and n2 of the two binding sites from their respective sequence ends. In order to avoid the very significant computational cost of varying the two distances n1 and n2 independently, we consider only the situation that the two binding sites are symmetric, n1 = n2, when we calculate the ΔΔG(Dout) of an ensemble of mRNAs or of several extremely long sequences; we do not expect any qualitative differences for asymmetric situations. In our studies involving microRNA binding sites described below, we choose the known microRNA binding site in the 3′UTR and vary the location of the putative protein binding site in the 5′UTR. The footprint of the proteins is set to be l = 6 nt in agreement with typical footprint sizes of RNA binding proteins.
Figure 3.
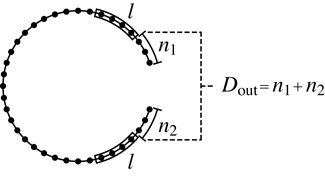
Definition of the outside distance, Dout, as the sum of the distances from each of the two protein binding sites to their corresponding ends in units of nt. Black dots represent bases and the two blocks represent the protein binding sites with footprint length l (in this figure l = 4 nt). In our numerical calculation we restrict ourselves to the symmetric case n1 = n2 and use a footprint size of l = 6 nt.
Sequences
We select human mRNA sequences in NCBI's RefSeq database (17) to study cooperativity between 5′ and 3′UTRs. As a comparison to gauge the influence of the length of the UTRs, we also investigate C. elegans mRNAs from the WormBase database (18), in which the UTRs are much shorter than those in human mRNAs.
Some of the sequences are deposited in these databases including their poly(A) tails. Since these tails, as homopolymers, contribute very little to the RNA secondary structure, and in order to treat all mRNAs consistently no matter if they were submitted to the databases with or without poly(A) tails, we discard all poly(A) tails. Specifically, we remove any runs of three or more consecutive adenines at the 3′ ends of the sequences in the database. We are aware that in eliminating the poly(A) tails, we lose the ability of applying our results to the important class of poly(A) binding proteins (PABPs) (19–21); however, since the poly(A) tails should not form secondary structures, PABPs are not expected to be subject to structure-mediated cooperativity, and thus are irrelevant in this study.
Since RNA secondary structure prediction scales as the third power of sequence length, computational resources constrain us to focus on the shorter mRNAs in the database. Dictated by computational feasibility, we thus choose the range from 500 to 1500 bases (counted after dropping the poly(A) tails). For human sequences, we select those sequences in which both 5′ and 3′UTRs are longer than 110 bases to ensure that all our binding sites are always in the UTRs even for the greatest outside distance, max (Dout) = 200 nt, yielding a total of 2282 sequences. For C. elegans sequences, we select the sequences with short UTRs by constraining the UTR lengths between 30 and 150 bases, yielding a total of 1277 sequences.
Since we want to identify which, if any, regions of the human mRNAs might have evolved to generate cooperativity, we also generate several sets of artificial RNA sequences based on our set of human sequences: (i) we shuffle each of the 2282 selected human sequences, generating an ensemble of random sequences with equal dinucleotide frequencies (22). (ii) We shuffle the UTRs of each of the 2282 human sequences to generate an ensemble of random sequences with equal dinucleotide frequencies in the UTRs but with unchanged coding sequences (CDSs). (iii) We select sequences that have UTR lengths in the range from 110 to 400 nt and replace their CDSs by the CDS of the single sequence NM004242 (300 nt), rendering an ensemble of totally 1377 artificial sequences with a fixed CDS.
In order to show that our results are not limited to short mRNA sequences, we also selected four individual mRNA sequences with more than 2000 bases, namely PTP4A1, MARCKS, MYC and PPP1R15B for verification purposes (it is computationally feasibly to study a few individual long mRNAs, but averaging over the entirety or even just a large number of these is not possible).
Human microRNA binding sites
Above, we describe symmetric protein binding sites, which are designed to have n1 = n2 in Dout = n1 + n2. However, in addition to these ‘assigned’ binding sites, we also investigate natural microRNA binding sites on 3′UTRs (23), and specifically their cooperativity with proteins binding on the 5′UTR.
We fetch microRNA binding sites from microRNA.org (24) using a strict mirSVR score cutoff of −1 (25). Again, we select only the mRNA sequences with total lengths from 500 to 1500 nt and UTR lengths above 110 nt for computational reasons. We limit the distance between the microRNA binding site and the 3′ end of the mRNA to be less than 100 bases, i.e. n2 ≤ 100, because we want to focus on the cooperativity between microRNAs bound on the end and proteins bound at the beginning of the same mRNA sequence. The above-mentioned conditions yield 1173 microRNA–mRNA binding pairs, in which some of the mRNA sequences correspond to more than one microRNA binding site. We investigate each such microRNA–mRNA pair independently, i.e. we consider only one specific microRNA interacting with its bound mRNA at a time. The cooperativity is calculated between the microRNA and a protein binding site around the end of the 5′UTR. We set the protein footprint to be l = 6 nt and investigate all possible protein binding sites from exactly at the end (n1 = 0 nt) to 100 bases from the end of 5′UTR (n1 = 100 nt). For each microRNA–mRNA pair we find the 5′UTR protein binding site that has the greatest ΔΔG with respect to the microRNA, recording it as the strongest cooperativity of the microRNA–mRNA pair.
As a comparison, multiple random ensembles of microRNA–mRNA binding pairs are generated. In each random ensemble the microRNA binding sites, which are located by their n2 on their naturally bound mRNA, are randomly assigned to a different mRNA. The strongest cooperativities of the reassigned microRNA–mRNA pairs are recorded as a reference, to which the cooperativities of the natural microRNA–mRNA pairs can be compared.
Artificial evolution of highly cooperative sequences
We use an evolutionary algorithm, in order to explore how high structure-mediate cooperativities can become. We choose five natural sequences, namely NM_006308, NM_022645, NM_001104548, NM_001244390 and NM_001272086, as starting sequences. We calculate the maximum over all |ΔΔG| for all possible pairs of protein binding sites. Considering the massive computational resources required for a search through all possible protein binding site pairs (on the order of N3 × N2 ∼ N5 in which N is the length of sequence), we compromise on merely searching protein binding sites in the region of 5 nt ≤ n1, n2 ≤ 25 nt, as we are more interested in the cooperativity between the ends of the two UTRs. In each evolutionary step, we randomly choose and mutate a nucleotide of the sequence and recalculate the maximum of |ΔΔG| for all protein binding pairs. If the maximum is greater than that of the previous sequence, we keep the new sequence; otherwise we continue with the previous sequence. This evolutionary step is repeated 1000 times.
RESULTS
Distance dependence of structure-mediated cooperativity
We quantify RNA secondary structure mediated protein binding cooperativity by the cooperativity free energy ΔΔG, which is defined as the difference between the free energies of binding of the first protein in the presence and absence of the second protein (see the Materials and Methods section). If it is positive, the presence of the second protein promotes binding of the first while if it is negative, the presence of the second protein prevents binding of the first. In order to demonstrate that RNA secondary structure mediated protein binding cooperativity is a generic effect providing cooperativity between proteins binding at the two ends of an RNA molecule, we select 2282 human mRNA sequences (see the Materials and Methods section). We choose one protein binding site to be close to the 5′ end of each molecule and one close to the 3′ end and systematically vary their distance from their respective ends such that the sum Dout of the two distances from the ends ranges from 0 to 200 nt (see Figure 3). We calculate the cooperativity free energy ΔΔG for each ‘outside’ distance Dout and each of the 2282 sequences.
Figure 4 shows the mean and standard deviation of the cooperativity free energy ΔΔG over all 2282 sequences as a function of the outside distance Dout of the two protein binding sites. The mean cooperativity free energy remains within about 0.1 kcal/mol of zero, more or less independent of the outside distance between the protein binding sites. That in principle leaves two possibilities, namely that either RNA secondary structure mediated cooperativity is a very small effect or that it is a significant effect but roughly equal numbers of sequences show positive as negative cooperativity. The standard deviation allows us to differentiate between these two scenarios: the fact that it starts out above 1 kcal/mol when the two binding sites are very close to their respective ends and then falls to about 0.5 kcal/mol for outside distances of 30 nt and stays in this range for binding sites up to 100 nt away from their respective ends (yielding a total outside distance of 200 nt) implies that many sequences must have cooperativity free energies with absolute values of this order of magnitude.
Figure 4.
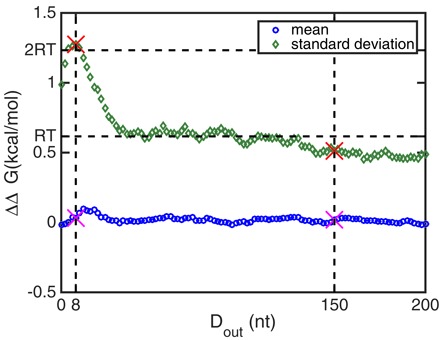
The mean (circles) and standard deviation (diamonds) of the cooperativity free energy ΔΔG of 2282 human sequences, as a function of the outside distance Dout between the two protein binding sites. The mean of the cooperativity free energy ΔΔG is close to zero for all outside distances, while its standard deviation is on the order of or greater than the thermal energy scale of RT =0.6 kcal/mol. The dashed vertical lines label two different outside distances Dout for which the whole distribution of the cooperativity free energies is compared in Figure 5. The maximum of the standard deviation of the cooperativity free energy occurs at Dout = 8 nt.
Structure-mediated cooperativity is significant for large numbers of mRNA molecules
In order to classify cooperativity free energies as significant or not, it is convenient to compare them to the thermal energy of RT, which is approximately 0.6 kcal/mol at temperatures of 37°C. Free energy differences below this magnitude will not have a measurable effect on protein binding in the thermal environment of a cell. Free energy differences larger than RT do result in significant protein binding cooperativity. The horizontal dashed lines in Figure 4 indicate cooperativity free energy levels of RT and 2RT and show that the standard deviation of the cooperativity free energy is on the order of RT in the distance range of 30 nt ≲ Dout ≲ 150 nt. Thus a good fraction of molecules must have cooperativity free energies larger than RT even at these outside distances. This is illustrated in Figure 5 for two specific outside distances, namely Dout = 8 nt where both proteins are very close to their respective ends and the standard deviation of the cooperativity free energy has its maximum, and Dout = 150 nt, where the standard deviation of the cooperativity free energy starts to fall below RT. The figure illustrates how many sequences have a given cooperativity free energy in bin sizes of 0.2 kcal/mol. As suggested by the small mean, the distributions of cooperativity free energies are symmetric with equal numbers of molecules showing positive as negative cooperativity. It can also be seen that the distribution at Dout = 8 nt is much wider than the distribution at Dout = 150 nt as expected from their standard deviations shown in Figure 4. The inset of Figure 5 shows the same distributions as the main figure but on a logarithmic scale that emphasizes the long tails of the distribution indicating some sequences with very large cooperativity free energies.
Figure 5.
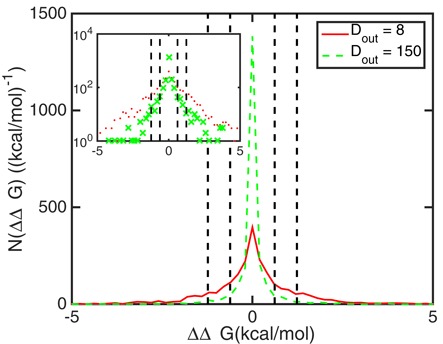
The number of mRNA molecules with respect to given cooperativity free energies ΔΔG (in bin sizes of 0.2 kcal/mol) at outside distances between the protein binding sites of Dout = 8 (solid line) and Dout = 150 (dashed line). The inset shows the same data on a logarithmic scale. The two distributions are both centered at 0. The distribution for Dout = 8 (when the protein binding sites are close to their respective ends of the molecule) extends much wider than that for Dout = 150 when the protein binding sites are further away from the ends of the molecule. Vertical black dashed and dash-dotted lines label |ΔΔG| = RT and |ΔΔG| = 2 RT, respectively.
An additional way to quantify the significance of the cooperativity free energies is to count the number of molecules out of the 2282 tested that have a cooperativity free energy above a certain threshold for each outside distance Dout. Figure 6 shows these data for thresholds of RT and 2RT. The data show that when the proteins bind very close to their respective ends of the RNA molecule, about half of the totally 2282 investigated sequences have |ΔΔG| > RT and a quarter have |ΔΔG| > 2RT (at Dout= 6 nt), implying that a significant cooperativity between two protein binding sites, of which one is at the 5′ end of a molecule and the other is at the 3′ end of a molecule, is a generic property among mRNAs. In addition, even at an outside distance of 200 nt (each protein binds 100 nt from its respective end of the RNA) still 10% of the 2282 mRNAs studied show a cooperativity free energy of at least RT and more than a hundred show a cooperativity of at least 2RT.
Figure 6.
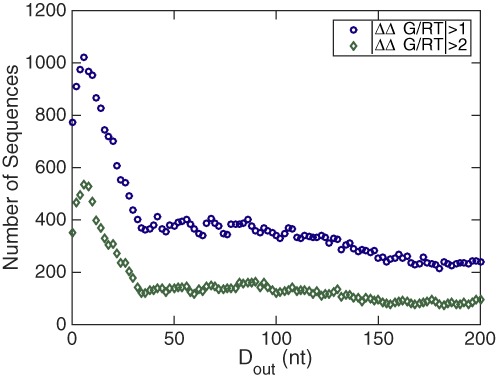
The number of mRNA sequences with biochemically relevant protein binding cooperativity among the 2282 investigated human mRNA sequences. The two data sets correspond to |ΔΔG|/RT > 1 (circles) and |ΔΔG|/RT > 2 (diamonds). The data show that at any given distance Dout a significant fraction of molecules has a significant cooperativity free energy.
Cooperativities in human mRNAs and random RNA sequences have different statistical characteristics
In our previous work we verified that structure-mediated cooperativity is a generic property of all RNA molecules, stating that cooperativity exists in an ensemble of random RNA sequences (11). In this study we investigate the cooperativity in human, and thus evolved, mRNAs. Thus, we are curious about what the similarities and differences between the cooperativities for a ‘random’ and a ‘natural’ sequence ensemble are.
To compare with the human mRNA sequences, we thus randomly permute nucleotides in each of the 2282 selected human sequences and generate two ensembles of 2282 random sequences (see the Materials and Methods section): (i) random sequences generated by shuffling the entire sequence of each human mRNA such that the dinucleotide frequencies are identical to those of the original human mRNAs, and (ii) random sequences generated by shuffling only the 5′ and 3′UTR sequences of each human mRNA while leaving the CDS unaffected. We investigate cooperativity in these two random sequence ensembles and compare their statistical properties with those of the natural human sequences.
The standard deviations of ΔΔG(Dout) of the two random ensembles and the human mRNA ensemble are shown in Figure 7. We notice that all these three ensembles comprise many sequences with biologically relevant cooperativities, i.e. ΔΔG is on the order of or greater than 0.6 kcal/mol. However, the standard deviation of the human mRNA ensemble has a large peak at small Dout and then sharply decreases to merely around RT at Dout ≥ 50 nt. Instead, the standard deviation of the completely shuffled random sequences smoothly decays in the entire investigated region, 0 nt ≤ Dout ≤ 200 nt. The standard deviation of the UTR-shuffled sequences is in between those of the human mRNA and fully shuffled sequence ensembles.
Figure 7.
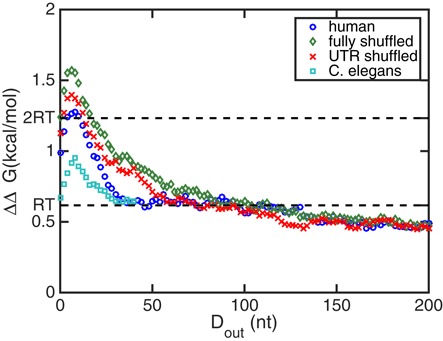
The standard deviations of the cooperativity free energy ΔΔG for different mRNA ensembles including 2282 sequences each, as functions of the distance Dout between the protein binding sites. Statistics of four sequence ensembles are shown: natural human mRNAs (circles), dinucleotide-shuffled human sequences (diamonds), sequences with randomized UTRs (crosses) and natural C. elegans mRNAs (squares). All four sequence ensembles have standard deviations on the order of or greater than the thermal energy scale of RT =0.6 kcal/mol, implying that a biologically relevant ΔΔG is a general property for mRNA sequence ensembles. However, different sequence ensembles show different statistical characteristics.
The fraction of sequences that have |ΔΔG| > 2RT in the human mRNA and the two random sequence ensembles are shown in Figure 8. Similar to the standard deviations, for the two random sequence ensembles, the fractions of sequences with strong cooperativities decrease smoothly as Dout increases, whereas for human mRNAs the fraction decreases sharply once Dout becomes greater than that for the maximum at Dout = 8 nt. Taken together, a human mRNA is likely to have strong cooperativity between the ends of the 5′ and 3′UTRs, while this cooperativity decreases rapidly as the two UTR binding sites are farther from their respective ends. On the other hand, random sequences perform power-law like, smoothly decaying cooperativity with respect to Dout. We thus conclude that human RNAs are different from random RNAs in terms of their cooperativity properties. While one might have expected stronger cooperativity in the evolved human sequences than in their randomly shuffled counterparts, we would like to point out that we, as discussed above, for computational reasons only look at symmetrically located binding sites of the two proteins. These will in general not correspond to the true protein binding sites on those natural mRNAs and it is conceivable that a possible design for cooperativity at true binding sites in fact leads to the suppression of cooperativity at the symmetrically located positions we interrogate.
Figure 8.
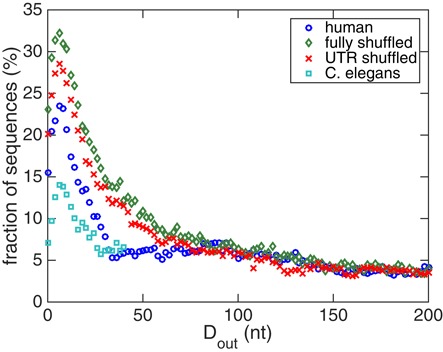
The fractions of sequences that have |ΔΔG(Dout)| > 2RT in different sequence ensembles: natural human mRNAs (circles), dinucleotide-shuffled human sequences (diamonds), sequences with randomized UTRs (crosses) and natural C. elegans mRNAs (squares). All four ensembles show maxima of the fraction at small Dout. However, their distance dependences are different.
Cooperativity is driven by the UTRs and not by the coding sequences
Since we have shown in the previous section that replacing the UTRs with shuffled sequences yields similar cooperativities as shuffling the entire sequence, we here ask the inverse question and retain the UTRs while replacing the CDSs of the ensemble of human mRNAs by one fixed CDS (see the Materials and Methods section). Figure 9 shows the comparison of the standard deviations of ΔΔG(Dout) of human mRNAs and the CDS-replaced sequences. The two ensembles of sequences show very similar profiles. This similarity suggests that the cooperativity between the 5′ and 3′UTR binding sites is determined by the two UTR sequences, while the coding region has little effect.
Figure 9.
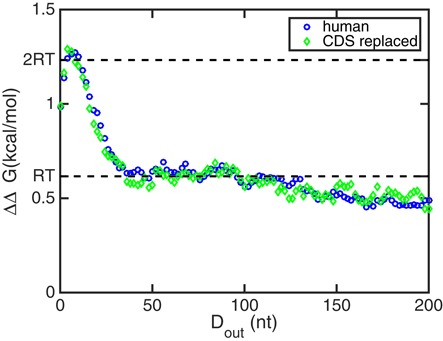
The standard deviations of the cooperativity free energy ΔΔG of 2282 human mRNAs and 1377 CDS-replaced sequences. The two standard deviations are very similar to each other, implying that the cooperativity depends on UTRs instead of CDSs.
Cooperativities in C. elegans mRNAs are weaker than those in human mRNAs
Compared to human mRNA sequences, C. elegans mRNAs comprise much shorter UTRs (17,18). As we have confirmed in the last section that the cooperativity is driven by the UTRs, it is natural to investigate the cooperativities of an ensemble of sequences that have short UTRs. Thus, we choose an ensemble of 1277 C. elegans mRNAs, both UTRs of which are in the range from 30 to 150 bases and the total sequence lengths are between 500 and 1500 bases. Their standard deviation of ΔΔG(Dout) and the number of sequences having significant ΔΔG(Dout) are plotted in Figures 7 and 8, respectively. The two figures show two consistent consequences. First, the cooperativities in C. elegans mRNAs are also biologically relevant, i.e. |ΔΔG| ≳ RT. Second, although cooperativities in C. elegans mRNAs are biologically relevant, they are significantly weaker than the cooperativities in human sequences. The latter result is additional evidence for the strong relationship between cooperativity and UTR sequences.
RNA structure mediated cooperativity between the sequence ends persists in long mRNA molecules
While computational complexity does not allow us to include mRNA molecules longer than 1500 nt in our systematic study above, we selected four individual mRNA molecules with more than 2000 nt to determine if our findings of significant cooperativity between 5′ and 3′ ends of an mRNA hold for longer molecules as well. Figure 10 shows the cooperativity free energy of these four molecules as a function of outside distance Dout. It can be seen that all four chosen molecules have some outside distances Dout at which |ΔΔG| > 2RT and even though for two of them (MYC and PPP1R15B) the binding sites with such large cooperativity free energy are very close to the ends, all molecules also have cooperativity free energies in excess of 1RT at some outside distance between 50 and 150 nt.
Figure 10.
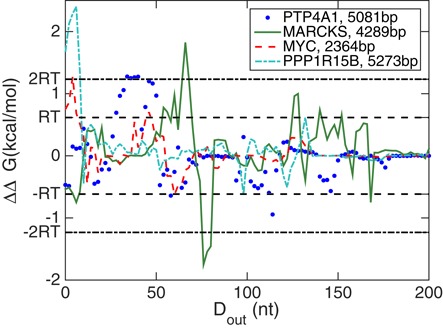
The cooperativity free energy of four randomly selected mRNA molecules longer than 2000 nt as a function of outside distance ΔΔG(Dout). Horizontal lines label the scales of ±RT and ±2RT. For all four molecules, ΔΔG exceeds these energy scales and thus develops significant cooperativity at specific values of the outside distance Dout.
Human microRNA binding sites show significant negative cooperativities
A large number of microRNAs bind to the 3′UTRs of mRNA sequences to regulate their abundance. At the same time, proteins are bound to the 5′UTRs, e.g. see (7,26). Thus, in addition to the symmetric binding sites with n1 = n2, studied so far, we also investigate the cooperativities between these microRNAs binding at their experimentally verified sites in the 3′UTR and putative 5′UTR binding proteins. We select 1173 microRNA–mRNA binding pairs from the microRNA.org database (see the Materials and Methods section). For each microRNA binding site in a 3′UTR, we search through putative protein binding sites in the first 100 bases in the 5′UTR of the corresponding mRNA and record the cooperativity free energy ΔΔG of the binding site with the greatest absolute value of the cooperativity free energy ΔΔG, termed ΔΔGmax. We note that although the position of the putative protein binding site in the 5′UTR is selected by maximizing the absolute value of the cooperativity free energy, the chosen cooperativity free energy still can be either positive or negative. As a comparison, we randomly shuffle the positions of the microRNA binding sites, matching them with different mRNAs. The ΔΔGmax calculated from these randomly shuffled microRNA–mRNA binding pairs helps us evaluate the significance of the characteristics discovered in the natural microRNA–mRNA pairs.
Figure 11 shows the distribution of the ΔΔGmax in the 1173 microRNA–mRNA binding pairs in bin sizes of 0.2 kcal/mol, with the gray regions representing the distribution of 10 ensembles of randomly shuffled binding pairs. The dark gray region for each cooperativity free energy bin designates the range of frequencies observed among the 10 ensembles while the light gray region indicates the regime of four standard deviations. The figure illustrates that for most cooperativity free energies, the ensemble of natural binding pairs does not show significant differences from the ensembles of randomly shuffled binding pairs. However, in the region of negative ΔΔGmax, the natural binding pairs exhibit a characteristic difference from random fluctuations, namely a peak at ΔΔGmax ≈ −3.8 kcal/mol ≈ −6RT labeled in Figure 11. The number of microRNAs with cooperativities of this magnitude is greater than its expected frequency based on the 10 random sequence ensembles by more than four standard deviations. We conclude that this peak is statistically significant, suggesting that a group of microRNAs results in strongly negative cooperativities with respect to proteins bound to the 5′UTRs. Considering the role of microRNAs in silencing and suppression of gene regulation (23), the negative cooperativity we discover in the statistics of human microRNA–mRNA binding pairs is reasonable and, furthermore, implies that a microRNA can participate in combinatorial regulation with proteins binding far from its binding site.
Figure 11.
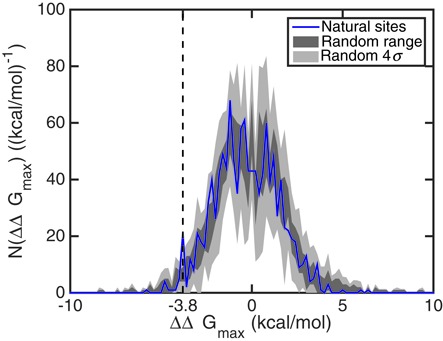
The number of microRNA–mRNA binding pairs, for which the strongest cooperativity with respect to a binding site in the 5′UTR corresponds to a given value of ΔΔGmax, in bins of 0.2 kcal/mol. While the number distribution of human binding pairs does not show significant differences compared to randomly shuffled binding pairs for most values of ΔΔGmax, a peak at ΔΔGmax ≈ −3.8 kcal/mol suggests that there is a group of human sequences with strongly negative cooperativities between the 3′UTR-bound microRNA and putative 5′UTR-bound proteins.
Cooperativities of artificially evolved RNA sequences can be large
In order to find out how strong cooperativities between the 3′UTR and the 5′UTR can possibly become, we do not restrict ourselves to natural sequences but rather search for the maximum of the cooperativity free energy ΔΔG among a huge number of possible sequences using an evolutionary algorithm (see the Materials and Methods section). In Figure 12 we show evolution trajectories starting from five human sequences over 1000 mutation steps. At the end of the artificial evolution trajectories all five human mRNAs mutate to artificial sequences comprising very strong cooperativities. Three sequences NM_001104548, NM_006308 and NM_022645 eventually mutate to sequences with |ΔΔGmax| ≈ 20RT, and NM_001244390 and NM_001272086 have even higher final cooperativities of |ΔΔGmax| ≈ 30RT. This result shows that appropriately designed RNA molecules can support cooperativities much stronger than typically found in natural mRNA sequences. However, considering that most biochemical reactions occur at energy scales of a few RT, such large ΔΔG are actually unrealistic and inappropriate for biological functions as the strong interactions they would confer would not be reversible thus rendering regulation impossible.
Figure 12.
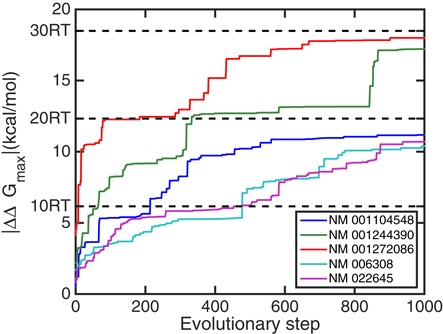
Cooperativities along artificial evolutionary trajectories starting from five human sequences seeking to maximize the cooperativity free energy. Compared to the cooperativities in natural human sequences, which are on the order of RT, the artificial sequences generated by mutations support much stronger cooperativities of |ΔΔG| > 10RT. The original sequences for initiating the evolutions are (from top to bottom at the final evolutionary step of 1000): NM_001272086, NM_001244390, NM_001104548, NM_022645 and NM_006308.
DISCUSSION
In this study, we have introduced a quantitative measure of RNA secondary structure based protein binding cooperativity and we have demonstrated that significant cooperativity between protein binding sites at the two ends of an RNA molecule is a generic feature of RNA secondary structure formation. We have come to this conclusion by systematically studying the ensembles of human mRNAs, C. elegans mRNAs, and random sequences generated from human mRNAs, in which the sequence lengths are between 500 and 1500 nt, and verified the effect in selected longer human mRNAs.
The cooperativity free energy ΔΔG rises up to a biologically relevant value (i.e. |ΔΔG| > RT ≈ 0.6 kcal/mol) in more than 1000 among the 2282 human mRNAs we investigated. Our statistical investigation of these sequences shows that this cooperativity gets stronger as the two binding sites get closer to their respective ends, suggesting a significant ‘end-to-end’ cooperativity between two proteins or microRNAs that bind on the opposing ends of an mRNA molecule. We interpret this observed cooperativity to imply that the two binding partners, albeit far from each other, when measured in terms of nucleotides along the mRNA sequence, can actually be close to each other in physical and secondary structure space due to the intricate secondary structures of the mRNA molecule. In consequence, the binding of the two partners becomes interdependent, and this effect allows for combinatorial post-transcriptional regulation.
We compared the cooperativity free energies ΔΔG(Dout) of human mRNAs and two ensembles of the random sequences generated from the human mRNAs. We discovered that all three sequence ensembles support biologically relevant cooperativity, |ΔΔG| > RT and have maximal cooperativities when the binding sites are relatively close to the ends of the UTRs. However, in human mRNAs the cooperativity ΔΔG decreases rapidly as the distance Dout increases; on the other hand, the cooperativity ΔΔG(Dout) of random sequences decays smoothly, as shown in Figure 7. We view the difference as an evidence that human mRNAs have distinguishable cooperativity properties compared to random sequences and note that the fact that human sequences appear to show a weaker cooperativity than their random counterparts can be a consequence of our only looking at symmetrically located putative binding sites that likely do not coincide with the true pairs of binding sites in human UTRs. Consistently with this, when we investigated experimentally annotated microRNA binding sites, we did find a group of microRNA binding sites with a strong cooperativity free energy around ΔΔG = −3.8 kcal/mol ≈ −6RT that is statistically over-represented compared to randomly permuted sites.
An intriguing phenomenon about RNA secondary structure induced cooperativity is its symmetry in terms of the sign of the cooperativity. The symmetry of the distribution of cooperativity free energies shown in Figure 5 implies that the number of mRNAs with positive cooperativity is roughly equal to the number of those with negative cooperativity. Whether this symmetry is a generic property of RNA or if it is specific to the ensembles of mRNAs studied here is an interesting topic for future investigations.
Another interesting fact we find about the cooperativity between the two ends of an RNA is that it appears exclusively determined by the 5′ and 3′UTR sequences. Our investigation of C. elegans mRNAs shows a positive relationship between UTR length and cooperativity strength. Moreover, an ensemble of sequences generated by substituting a fixed CDS for the CDSs of human mRNAs shows very similar cooperativities to the ensemble of original human mRNAs. These two results support the picture that the 5′ and 3′UTRs ‘communicate’ with each other with little interference from the CDS.
In our study we first chose the protein binding sites to be symmetrically arranged, i.e. at equal distances from their respective ends of the mRNA molecule. Based on the properties of RNA secondary structure, we would expect that statistically asymmetric configurations of the protein binding site will behave the same as the symmetric configurations with the outside distance Dout being the only relevant parameter. However, when studying the cooperativity free energy of a specific molecule, being able to vary the positions of both binding sites independently should dramatically increase the search space and thus enable even higher cooperativity free energies. Unfortunately, the computational cost of varying the two positions independently is prohibitive since each combination of binding sites requires a new folding of the entire molecule.
However, we were still able to investigate asymmetric configurations in some limited cases. In our study of microRNAs, the 3′UTR binding sites are fixed, and thus we were able to tune the 5′UTR binding sites and search through a range of different n1 with respect to the asymmetric, fixed n2. Also, when we looked for the maxima of cooperativity in artificially evolved RNA sequences, we constrained the calculation of ΔΔG in a small range of asymmetric n1 and n2, as a compromise given the limitations imposed by computational cost. In this case we found very significant cooperativity free energies up to |ΔΔGmax| ≈ 30RT.
An important aspect for future investigations is the inclusion of sequence specificity. For the purpose of this work, we assumed perfect sequence specificity and allowed the protein (or microRNA) to only bind at one pre-defined location along the mRNA. With techniques like RNAcompete (9,10) systematically measuring the affinities of many important RNA binding proteins to all possible binding sites, it should be possible to allow sequence-dependent binding of two proteins to be incorporated into RNA secondary structure prediction and we will pursue this avenue in order to identify mRNAs and pairs of proteins that actually employ this mechanism of post-transcriptional regulation and could be verified experimentally. Lastly, while our study entirely focused on thermodynamical quantities, studying the kinetics of the interplay of RNA structure and protein binding will be important as well.
Acknowledgments
We thank Nikolaus Rajewsky and Michael Poirier for fruitful discussions initiating this work and the anonymous reviewers for suggesting interesting additional directions incorporated into this work.
FUNDING
National Science Foundation [DMR-01105458, DMR-01410172]. Funding for open access charge: National Science Foundation [DMR-01105458].
Conflict of interest statement. None declared.
REFERENCES
- 1.Burd C.G., Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 2.Glisovic T., Bachorik J.L., Yong J., Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltz A.G., Munschauer M., Schwanhäusser B., Vasile A., Murakawa Y., Schueler M., Youngs N., Penfold-Brown D., Drew K., Milek M., et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 4.Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M., et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. (2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Mignone F., Gissi C., Liuni S., Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3:REVIEWS0004. doi: 10.1186/gb-2002-3-3-reviews0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park E.-H., Walker S.E., Lee J.M., Rothenburg S., Lorsch J.R., Hinnebusch A.G. Multiple elements in the eIF4G1 N-terminus promote assembly of eIF4G1·PABP mRNPs in vivo. EMBO J. 2011;30:302–316. doi: 10.1038/emboj.2010.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jungkamp A.C., Stoeckius M., Mecenas D., Grun D., Mastrobuoni G., Kempa S., Rajewsky N. In vivo and transcriptome-wide identification of RNA binding protein target sites. Mol. Cell. 2011;44:828–840. doi: 10.1016/j.molcel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antson A.A. Single stranded RNA binding proteins. Curr. Opin. Struct. Biol. 2000;10:87–94. doi: 10.1016/s0959-440x(99)00054-8. [DOI] [PubMed] [Google Scholar]
- 9.Ray D., Kazan H., Chan E.T., Castillo L.P., Chaudhry S., Talukder S., Blencowe B.J., Morris Q., Hughes T.R. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009;27:667–670. doi: 10.1038/nbt.1550. [DOI] [PubMed] [Google Scholar]
- 10.Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A., et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin Y.-H., Bundschuh R. Interplay between single-stranded binding proteins on RNA secondary structure. Phys. Rev. E. 2013;88:052707. doi: 10.1103/PhysRevE.88.052707. [DOI] [PubMed] [Google Scholar]
- 12.Higgs P.G. RNA secondary structure: physical and computational aspects. Q. Rev. Biophys. 2000;33:199–253. doi: 10.1017/s0033583500003620. [DOI] [PubMed] [Google Scholar]
- 13.Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- 14.Mathews D.H., Sabina J., Zuker M., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 15.Forties R.A., Bundschuh R. Modeling the interplay of single-stranded binding proteins and nucleic acid secondary structure. Bioinformatics. 2010;26:61–67. doi: 10.1093/bioinformatics/btp627. [DOI] [PubMed] [Google Scholar]
- 16.Schroeder D.V. An Introduction to Thermal Physics. San Francisco, CA: Addison Wesley Longman; 2000. [Google Scholar]
- 17.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yook K., Harris T.W., Bieri T., Cabunoc A., Chan J., Chen W.J., Davis P., de la Cruz N., Duong A., Fang R., et al. WormBase 2012: more genomes, more data, new website. Nucleic Acids Res. 2012;40:D735–D741. doi: 10.1093/nar/gkr954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Görlach M., Burd C.G., Dreyfuss G. The mRNA poly(A)-binding protein: localization, abundance, and RNA-binding specificity. Exp. Cell. Res. 1994;211:400–407. doi: 10.1006/excr.1994.1104. [DOI] [PubMed] [Google Scholar]
- 20.Kahvejian A., Roy G., Sonenberg N. The mRNA closed-loop model: the function of PABP and PABP-interacting proteins in mRNA translation. Cold Spring Harb. Symp. Quant. Biol. 2001;66:293–300. doi: 10.1101/sqb.2001.66.293. [DOI] [PubMed] [Google Scholar]
- 21.Kahvejian A., Svitkin Y.V., Sukarieh R., M'Boutchou M.-N., Sonenberg N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005;19:104–113. doi: 10.1101/gad.1262905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschul S.F., Erickson B.W. Significance of nucleotide sequence alignments: a method for random sequence permutation that preserves dinucleotide and codon usage. Mol. Biol. Evol. 1985;2:526–538. doi: 10.1093/oxfordjournals.molbev.a040370. [DOI] [PubMed] [Google Scholar]
- 23.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Betel D., Wilson M., Gabow A., Marks D.S., Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Betel D., Koppal A., Agius P., Sanderm C., Leslie C. The mirSVR regression method for predicting likelihood of target mRNA down-regulation from sequence and structure features in microRNA/mRNA predicted target sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medenbach J., Seiler M., Hentze M.W. Translational control via protein-regulated upstream open reading frames. Cell. 2011;145:902–913. doi: 10.1016/j.cell.2011.05.005. [DOI] [PubMed] [Google Scholar]