

Abstract
The imprinted Kcnq1 domain contains a differentially methylated region (KvDMR) in intron 11 of Kcnq1. The Kcnq1ot1 non-coding RNA emerges from the unmethylated paternal KvDMR in antisense direction, resulting in cis-repression of neighboring genes. The KvDMR encompasses the Kcnq1ot1 promoter, CTCF sites and other DNA elements, whose individual contribution to regulation of the endogenous domain is unknown. We find that paternal inheritance of a deletion of the minimal Kcnq1ot1 promoter derepresses the upstream Cdkn1c gene. Surprisingly, Kcnq1ot1 transcripts continue to emerge from alternative sites, evidence that silencing depends, not on the ncRNA, but on the promoter sequence. Detailed analyses of Kcnq1ot during cardiogenesis show substantial chromatin reorganization coinciding with discontinuous RNA production in both wild-type and mutant mice, with loss of imprinting. We show that CTCF binds to both methylated and unmethylated alleles of the KvDMR. Furthermore, we report a multitude of enhancers within the Kcnq1ot1 region, and present conformational dynamics of a novel heart enhancer engaged in Kcnq1 expression. Our results have important implications on tissue-specific imprinting patterns and how transcriptional mechanisms compete to maximize the expression of vital genes, in addition to shifting our perception on the role of the long ncRNA in regulating this imprinted domain.
INTRODUCTION
Thousands of long non-coding (lnc) RNAs are produced by the mammalian genome, but few are larger than 10–20 kb. Kcnq1ot1 has been described historically as a lncRNA of ∼90 kb, emerging from intron 11 of Kcnq1 in the antisense direction (1,2). In the early embryo, Kcnq1ot1 is only expressed paternally and silences three upstream genes, Cdkn1c, Slc22a18 and Phlda2, acting in cis (reviewed in (3,4)) (Figure 1A). The mechanism by which the ncRNA regulates its neighboring genes has not been clearly established, although two hypotheses have been put forward: one proposing a direct action of the Kcnq1ot molecule by spreading and recruiting repressive factors (5); and the other, suggesting that regulatory DNA elements exposed by the act of transcription of the ncRNA are responsible for the silencing (6,7).
Figure 1.
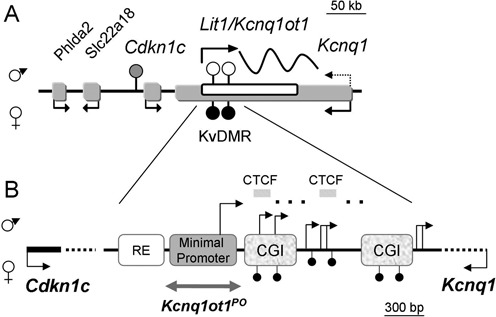
Kcnq1 domain: expression and imprinting features. (A) Overview of the genes within the Kcnq1 domain exhibiting monoallelic expression in the embryo. Paternal and maternal expressions are indicated with arrows above and below the sequences, respectively. Arrows show the direction of transcription. Dotted arrow emerging from Kcnq1 indicates transition to biallelic expression in mid-gestation. Filled and unfilled circles indicate methylated and unmethylated regions, respectively; hatched circle represents a methylation mark acquired after fertilization. Scale is indicated above the domain. (B) Schematic of the ∼4300 bp KvDMR and its regulatory features. RE, regulatory element; CGI, CpG islands; hatched bars, CTCF sites; broken arrows indicate the transcriptional start sites reported previously (16,19,24), some of which are tissue-specific. Gray double-headed arrow below the minimal promoter represents the region deleted in this report. Scale is indicated below the schematic.
Although it was initially assumed that Kcnq1ot1 also silenced its sense counterpart, the Kcnq1 gene, two observations led us to reassess how the ncRNA regulates Kcnq1: (i) studies in whole embryos and neonatal mouse tissues revealed that expression of Kcnq1 transitions to a biallelic mode after mid-gestation (8); (ii) detailed expression and conformational profiles of the developing heart showed that the loss of Kcnq1 imprinting coincides with activation of strong cardiac-specific enhancers that physically engage with the Kcnq1 promoter (9). Based on these observations, we suggested that enhancer-driven expression could successfully compete and override the silencing effects of Kcnq1ot1 transcription on the paternal Kcnq1 allele.
Defects in KCNQ1 are responsible for congenital long-QT syndrome, with cardiac phenotypes of different severities. Cdkn1c, the closest gene silenced on the paternal chromosome, is also important for regulating proliferation during heart development (10,11), specifically at E9.5–13.5, probably acting as a downstream target of Bmp10 signaling.
The main Kcnq1ot1 promoter is part of a region designated as the KvDMR (Figure 1B). Previous gene targeting studies have deleted the entire KvDMR, or in some cases, large segments that had regulatory activities in vitro (Supplementary Figure S1). This ∼3000 bp region includes elements that control the establishment of Kcnq1ot1 imprinting, although these have not yet been delimited. Two CG islands (CGIs) downstream of the promoter are methylated on the maternal chromosome. This constitutes a primary imprinting mark inherited from the oocyte that inhibits Kcnq1ot1 expression from that allele, allowing maternal expression of the neighboring upstream genes and the sense Kcnq1 gene (1,12,13). In addition, the KvDMR contains other potential regulatory elements, including a sequence with enhancer activity in vitro and two CTCF binding sites with unknown function (6,14,15).
To refine our understanding of the endogenous roles of the sequence elements in the KvDMR, we generated a mutant mouse in which a region previously defined as the Kcnq1ot1 minimal promoter (MP) in vitro (16) was ablated (designated as PO, or promoter-out allele). In contrast to previous studies, this deletion leaves intact the two CTCF binding sites as well as the two CGI (Supplementary Figure S1). Our data show that, in spite of the absence of the MP, alternative transcripts continued to emerge from alternative sites in the Kcnq1ot1 region. Surprisingly, although the deletion did not ablate Kcnq1ot1 expression, Cdkn1c was no longer monoallelic, suggesting that the residual Kcnq1ot1 transcripts had lost silencing capability or that imprinted Cdkn1c expression required the MP sequence itself.
Unexpected results from our studies raised the question of whether Kcnq1ot1 is a single entity or rather a series of overlapping transcripts. Complete elucidation of the transcript structure is required to understand the silencing mechanism attributed to it. Estimates of the length of the Kcnq1ot1 transcript came from reverse transcriptase-polymerase chain reaction (RT-PCR) scans and RNA-sequencing (17), but these technologies could not establish that Kcnq1ot1 is a single RNA molecule of 92 kb. By integrating publicly available conservation, strand-specific RNA-sequencing and chromatin immunoprecipitation (ChIP)-sequencing data from the Kcnq1ot1 region, we observed many discrete sequences that exhibited features of enhancers or promoters. We reasoned that if there are multiple independent RNAs emerging from the Kcnq1ot1 region, instead of (or in addition to) a 92 kb entity, the transcriptional start sites (TSS) could be identified by existing technologies. By performing targeted 5′ RACE and 3C assays, we show that several such regions are active transcriptionally and at least one is in contact with the Kcnq1 promoter in the heart. Our results completely change previous assumptions on Kcnq1ot1 and lead us to propose that the ncRNA is a by-product of the transcriptional process, which maintains an open chromatin structure allowing access to a variety of enhancers encoded in introns 10 and 11 of Kcnq1. Our experiments also show that deletion of a 200-bp region leads to loss of imprinting of Cdkn1c, strongly suggesting that this sequence is a long-distance silencer. These studies are valuable for understanding the mechanisms that govern imprinting in the context of other co-existing regulatory mechanisms.
MATERIALS AND METHODS
Targeting of embryonic stem cells
Targeting strategy is illustrated in Supplementary Figure S2A. pTV-Kcnq1ot1-MP was constructed by Genebridges (Heidelberg, Germany) and modified to add the negative selection marker DTA (Diphtheria toxin A fragment) (Taconic, Germantown, NY, USA). The targeting vector was linearized with SalI and electroporated into B6-3 ES cells. G418-resistant clones were isolated and restriction enzyme digestions and Southern blots were used to confirm accurate homologous recombination (Supplementary Figure S2B). Two confirmed targeted clones underwent a second round of electroporation with Flp to delete the neomycin-resistance (neoR) marker, and G418-sensitive clones were identified and confirmed with long-range PCR (Supplementary Figure S2C). Two confirmed neoR-deleted clones were injected into BALB/c mouse blastocysts and chimeric mice were generated.
Mice
All mouse protocols were approved by the Institutional Animal Care and Use Committee at Temple University Medical School. For each chimeric line, germline transmission of the targeted mutation was confirmed by PCR genotyping of progeny from chimeric mice crossed with C57BL/6 mice on DNA isolated from tail biopsies (see Supplementary Table S1 for primer sequences and Supplementary Figure S2 for schematic and results of genotyping PCR). Nomenclature indicates the maternal allele first. The Kcnq1ot1 MP was excised by crossing heterozygous mutant mice (Kcnq1ot1+/flox-MP) to mice expressing CMV-Cre recombinase (obtained from Jackson Lab, Bar Harbor, Maine, USA). Excision of the MP was confirmed in the progeny by PCR analysis (Supplementary Figure S2D). Positive heterozygous progeny, designated as Kcnq1ot1+/PO, were maintained by backcrossing to C57BL/6 or bred to homozygosity.
To distinguish the parental alleles for allelic assays (for RNA and methylation analyses, see below), mutant mice were crossed to C57BL/6(CAST7), a consomic strain homozygous for a Mus musculus castaneus chromosome 7 (18). In all experiments, mutant mice were compared to their wild-type (WT) littermates. All results were confirmed in two independent mutant lines.
To obtain embryonic tissues, reciprocal crosses were set up between C57BL/6 and C57BL/6(CAST7) mice, or between Kcnq1ot1+/PO and C57BL/6(CAST7) mice. Noon of the day of the plug was designated E0.5. Pregnant dams were killed at the appropriate day of gestation and fetal tissues were dissected and frozen for further analysis. Neonatal tissues were obtained from postnatal day 2 pups.
Gene-specific-primed cDNA synthesis
RNA was extracted from snap frozen 10.5 dpc, 12.5 dpc, neonatal and adult tissues using the Roche High Pure RNA Kit (Nutley, NJ, USA). An additional DNase step was performed using Invitrogen Turbo DNase (Grand Island, NY, USA). GSP cDNA was generated for each specific qPCR analysis using Invitrogen SuperScript III at 65°C incubation for 50 min, with controls including minus reverse transcriptase and minus primers indicating no contaminating DNA nor non-specific cDNA synthesis. Samples were either used immediately for qPCR or frozen at −20°C. Primers are listed in Supplementary Table S1.
Quantitative RT-qPCR
Immediately following GSP cDNA synthesis, RT-qPCR was performed using 1x Power Sybr Green (Invitrogen) according to manufacturer's protocol with primer-optimized reaction conditions (primers and efficiencies listed in Supplementary Table S1) and according to MIQE guidelines. PCR products were analyzed both by melt curve analysis and polyacrylamide gel electrophoresis. Mean Ct values for each triplicate reaction were normalized to geometric mean of expression values of β-actin, Ppia and Rplp1, three housekeeping genes that show no expression variation across cardiac developmental stages nor between heart and brain. Statistical analyses were performed using Student's t-test with GraphPad Prism software v6. Results are the average of three independent biological replicates (one mouse from each of three different litters), each performed in triplicate.
5′ and 3′ rapid amplification of cDNA
5′ and 3′ rapid amplification of cDNA ends (RACE) was performed with the 5′ and 3′ RACE systems (Invitrogen). For 5′ RACE, primers were located downstream of the predicted transcription factor binding sites. For 3′ RACE, primers were anchored at 100, 500 and 1000 bp away from the TSS found by 5′RACE. PCR products amplified according to the manufacturer's recommendation were run on 3% agarose gels and bands were extracted, cloned and sequenced. Sequences were aligned with BLAT in the UCSC genome browser. Primers for 5′ and 3′ RACE assays are listed in Supplementary Table S1.
Protein expression analysis
For western blot analyses, total proteins were isolated from mouse neonatal hearts using RIPA buffer [20 mM Tris (pH 8.0), 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% NP-40, 0.5% sodium deoxycholate]. Protein samples (30 μg) were separated on 10% SDS–polyacrylamide gels and subjected to a standard western procedure. Antibodies for Kcnq1 and β-actin were purchased from Santa Cruz (SC-20816 and SC-47778, respectively), and both were used at a 1:400 dilution. Secondary antibody was horseradish peroxidase conjugated and used at a 1/5000 dilution. Blot was visualized on an Alpha Innotech imaging system and quantified with the Alpha FluorChem software.
Chromatin immunoprecipitation
Tissues were extracted from postnatal day 2 (P2) mice and cross-linked in 0.5% formaldehyde in phosphate buffered saline (PBS) and protease inhibitors for 5 min (heart) or 1% formaldehyde for 10 min (brain) and quenched with 0.125 M glycine. Intact nuclei were isolated from tissue digestions and split so that each subsequent reaction had 5 × 106 nuclei. Micrococcal nuclease digestion in 1 mM CaCl2 was performed [20 U for 1 min] and the reaction was terminated with 2 mM ethylene glycol tetraacetic acid (EGTA), followed immediately by sonication in a 0.1% SDS–PBS solution. Direct ChIP was performed on the soluble nuclear extract at 4°C overnight using either CTCF antibody (Active Motif, Carlsbad, CA, USA) or IgG control antibody (Vector Laboratories, Burlingame, CA, USA). Substrates were pulled down with Invitrogen Protein A Dynabeads. After washing and elution, samples were incubated with Proteinase K overnight at 65°C to reverse cross-links and enriched DNA was purified with phenol–chloroform. Ethanol precipitated DNA was resuspended in 10 mM Tris–HCl solution and analyzed by qPCR using the ActiveMotif qPCR Analysis Kit. Each ChIP assay was performed three times and qPCR results represent the mean ± SEM. ChIP primers are listed in Supplementary Table S1.
Bisulfite mutagenesis sequencing
1 ug of DNA was mutagenized using the EZ-DNA Methylation Gold Kit (Zymo Research #5005) following the manufacturer's protocol. Amplification was performed with nested primers (listed in Supplementary Table S1) and PCR products cloned into the pCR2.1-TOPO vector using a TOPO-TA Cloning Kit (Life Technologies#K4500-01). The bacterial colonies were cultured and plasmids purified using a Spin Mini Prep Kit (QIAGEN #30020). Sequencing of the plasmid inserts was done by Eurofins MWG Operon. Sequences were analyzed with Geneious Pro 6.5. Data were obtained from three independent biological samples, with at least 10 clones sequenced from each.
Pyrosequencing
Biotin-labeled PCR products were amplified from either cDNA or bisulfite-treated genomic DNA using Qiagen PyroMark Custom Assay Primers and Hot Start Taq Polymerase (Qiagen Catalogue #203203). The list of PCR primers and sequencing primers is provided in Supplementary Table S1. Pyrosequencing was carried out in duplicate on a PSQ96 system with a Pyro-Gold reagent Kit (Qiagen Catalogue #972804) and the results were analyzed by PyroMark Q96 ID software version 1.0 (Qiagen). Known polymorphisms (http://phenome.jax.org/) between M. musculus castaneus and C57BL/6 genomes were used to determine maternal/paternal contributions with Allele Quantification (AQ) assays. For methylation assays, the ratio of C-to-T at cytosine residues within CpG islands was quantified.
Methylated DNA immunoprecipitation (MeDIP)
Genomic DNA was extracted from snap-frozen hybrid neonatal (P2) tissues using the Quick-gDNA MiniPrep Kit (Zymo Research #D3024). Using the EpiSeeker MeDIP Kit (Abcam #ab117133), 1 ug of the extracted gDNA was then subject to sonication and immunoprecipitation according to manufacturer's protocol. PCR was performed on the methylated DNA enriched samples alongside IgG and Input controls with the Hot Start Taq Polymerase Kit (Qiagen #203203) and with PyroMark Custom Assay Primers (listed in Supplementary Table S1). The resulting biotinylated PCR products were bound to Strepavidin Sepharose beads (GE Healthcare #17-5113-01) and pyrosequenced in duplicated on the PSQ96 Pyrosequencing Platform with the Pyro-Gold reagent Kit (Qiagen #972804). Resulting Allele Quantification (AQ) data were analyzed by PyroMark Q96 ID software version 1.0 (Qiagen) and percent maternal/paternal contribution was determined by comparing known polymorphisms between M. musculus castaneus and C57BL/6 mouse strains. Successful enrichment of methylated DNA was confirmed by comparing AQ data from immunoprecipitated samples against comparable IgG and Input controls.
Transgenic embryo assays
A 500-bp region spanning 5 sequences, including the Tbx5 candidate binding site was amplified from C57BL/6J genomic DNA and subcloned into the Hsp68-LacZ vector. For microinjection, constructs were linearized, gel purified by electroelution and run through the Wizard DNA Clean Up columns (Promega). DNA was injected into male pronuclei of C57BL/6J embryos, and pseudopregnant Swiss Webster females (Taconic) were used as recipients. Transgenic embryos were collected at 11.5 dpc and stained with X-gal (Sigma) for 20 min to detect β-galactosidase activity. Images were obtained with an Olympus MVX10 stereoscope, cropped and levels adjusted with Adobe Photoshop.
Chromosome conformation capture (3C)
Chromosome conformation capture (3C) assays were conducted as previously reported (19) with the appropriate controls (20). Cross-linked chromatin substrates were digested with AflIII and NcoI. Purified DNA samples were analyzed on 0.8% agarose gels to estimate concentration. The linear range of amplification was determined for each sample by serial dilution. An appropriate amount of DNA within the linear range (typically 500 ng of DNA) was subsequently used for the experiments. The control template was prepared from BACs spanning the Kcnq1, Kcnq1ot1 and Cdkn1c genes. The linear range of the control template was determined with a serial dilution of the random ligation mix made in the same amount (500 ng) of digested and ligated BAC DNA. Primer sequences are listed in Supplementary Table S1. All PCR products were cross-linking and ligation-dependent. PCR products were run on 7% polyacrylamide gels. Intensity of the fragments was quantified on a Typhoon 9200 imager (Molecular Dynamics) and normalized between gels. All data points were generated from three different biological replicates, each performed in triplicate. Relative cross-linking frequency for a fragment X was calculated as: γ(x) = [I(A-X)3C/I(A-X)C]/[I(A-NF)3C/I (A-NF)C, where I(A-X)3C is the intensity of PCR products from ligations between the anchor (A) and another fragment in the Kcnq1ot1 region in the 3C substrate; I(A-X)C is the same for the BAC control template; I(A-NF)3C is the intensity of PCR products from ligations between the anchor and the nearest fragment to the anchor in the 3C substrate; I(A-NF) is the same for the BAC control template. Data were expressed as mean ± SD.
Bioinformatics analysis
RNA-seq data were downloaded from the Genome Browser and imported into the Integrative Genomics Viewer (IGV) (21,22). Annotation for Mouse Genome (mm9) was selected and the RefSeq gene table visualized. After choosing the correct region (Kcnq1ot1), spliced reads from RNA-seq data were used to predict putative spliced isoforms and a Sashimi plot was generated with the Sashimi tool in the Integrative Genomics viewer (22).
RESULTS
Generation of the Kcnq1ot1PO allele
To determine the effect of deleting the previously defined MP for Kcnq1ot1, mice carrying a mutant allele at the endogenous locus were generated by homologous recombination in embryonic stem (ES) cells (Supplementary Figure S2). This allele, designated as Kcnq1ot1PO (Promoter-Out), removes 564 bp of sequence and leaves two CTCF sites and two CG-islands (CGIs) intact. A comparison of the Kcnq1ot1PO allele with previously generated deletions is shown in Supplementary Figure S1, illustrating that CTCF sites were either eliminated or partially truncated in those studies. Mutant progeny inheriting the deletion maternally or paternally were assayed for defects in Kcnq1ot1, Cdkn1c and Kcnq1 expression and imprinting, CTCF binding and alterations in chromosome conformation. Parental origin of the alleles was distinguished by crossing heterozygous mutant mice with B6(CAST7) mice, a consomic strain in which chromosome 7 is derived from M. musculus castaneus (23).
CTCF exhibits biallelic binding to the KvDMR in the neonatal heart in both wild-type and Kcnq1otPO mice
Two CTCF sites located immediately downstream of the MP have been proposed to play a role in Kcnq1ot1 expression or as insulators regulating access of neighboring genes to tissue-specific enhancers (24) (Figure 2A). Allele-specific binding of CTCF to the unmethylated paternal sites has also been reported, suggesting a role in establishing or maintaining differential methylation at the KvDMR. We performed ChIP assays using anti-CTCF antibodies on neonatal hearts of WT mice. Quantitative PCR with primers for CTCF binding sites 1 and 2 (CTCF-1, CTCF-2) showed positive occupancy of CTCF (Figure 2B, WT). To determine allele-specificity of binding, we performed allele-specific restriction digests of the PCR products from immunoprecipitated DNA. Surprisingly, we observed biallelic CTCF binding at the KvDMR in the heart (Figure 2C).
Figure 2.
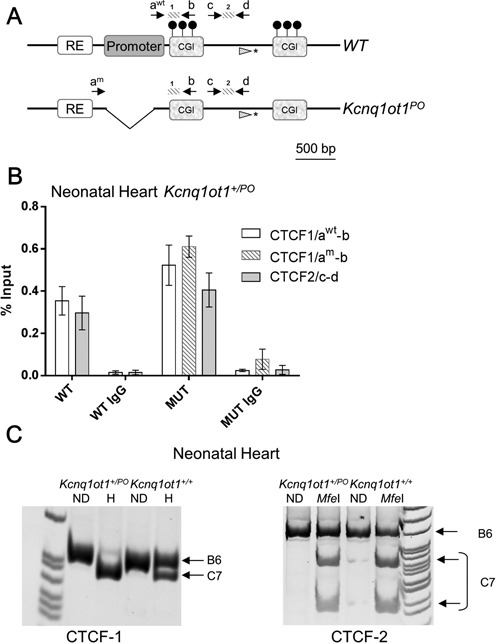
Chromatin immunoprecipitation analysis for CTCF at the KvDMR. (A) Schematic of the wild-type (WT) and Kcnq1ot1PO alleles. Primers awt and b amplify CTCF binding site 1 (CTCF-1) in the WT allele only, since awt anneals to a sequence in the promoter, which is absent in the mutant allele; primers am and b amplify CTCF binding site 1 in the mutant allele (PO). Primers c and d amplify CTCF binding site 2 (CTCF-2) of both the WT and Kcnq1otPO allele. Asterisk denotes SNP analyzed with the primer depicted with the hatched arrowhead. (B) Quantitative PCR of immunoprecipitated substrates from neonatal hearts of WT mice and Kcnq1ot1+/PO littermates. Assays were performed on mice from three litters each, in triplicate, and results expressed as percentage input normalized to H19 R1 (repeat 1 of the differentially methylated domain). Error bars indicate SEM. CTCF-1 shows occupancy of CTCF in both parental alleles of in both the WT mouse and the Kcnq1ot1+/PO mouse. For the Kcnq1ot1+/PO mouse (MUT), the white bar represents the data for the WT maternal (primers awt and b) and the hatched bar represents the mutant paternal allele (primers am and b) CTCF-2 shows occupancy of CTCF for both WT and mutant alleles. (C) Allele-specific restriction digests of ChIP-PCR products for primer pairs for CTCF-1 (left panel) and CTCF-2 (right panel). ND, non-digested; H, Hpy1881 digest. For CTCF-1, Hpy1881 cuts only the castaneus allele; for CTCF-2, MfeI cuts only the castaneus allele.
To determine if deletion of the MP affected CTCF occupancy, ChIP was performed with chromatin purified from neonatal hearts of Kcnq1ot1+/PO mice (Figure 2B, MUT). Quantitative PCR with the primers specific for the WT (maternal) and mutant (paternal) alleles for binding site 1 yielded a positive signal for both. To confirm that CTCF was occupying the maternal allele at binding site 1, we performed allele-specific restriction digests on the PCR product amplified with WT-specific primers. The PCR product was the expected size, and digestion revealed CTCF binding to the maternal B6(CAST7) allele. Primers for CTCF-2, which detect both WT and mutant alleles, were also positive for CTCF occupancy and allele-specific restriction digests of the PCR products showed binding to both alleles (Figure 2C).
To confirm this result with a different assay, we quantified allelic presence of CTCF by pyrosequencing immunoprecipitated substrates (primers and the polymorphism analyzed indicated in Figure 2A). Although CTCF occupancy was higher on the paternal allele, we observed significant enrichment at the maternal allele relative to input samples (for Kcnq1ot1+/PO, P = 0.004, n = 6; for Kcnq1ot1+/+ P < 0.0001, n = 4). Thus, methylation of the maternal allele does not affect CTCF binding to its cognate sites at the KvDMR. Importantly, CTCF binding is not perturbed by the absence of the MP, making it a good model for testing the role of CTCF in the Kcnq1ot1 region.
Cdkn1c loses imprinted expression in the absence of the Kcnq1ot1 minimal promoter
To determine the effect of deleting the Kcnq1ot1 MP on imprinted expression of Cdkn1c, we performed quantitative pyrosequencing of expressed strain-specific single nucleotide polymorphisms (SNPs). Although Cdkn1c was monoallelic in all WT tissues, Kcnq1ot1+/PO mice showed biallelic expression of Cdkn1c in every tissue tested (Figure 3A). Paternal expression of Cdkn1c was significantly higher in Kcnq1ot1+/PO than in WT mice (for all stages, P < 0.0001), indicating that absence of the MP releases the repression of the paternal Cdkn1c allele. However, the maternally biased expression was maintained, suggesting that repressive epigenetic marks were not completely removed. Surprisingly, despite the biallelic expression pattern, total RNA abundance of Cdkn1c does not increase significantly in the Kcnq1ot1+/PO heart and brain, as assayed by qPCR. This implies the existence of a feedback mechanism to limit Cdkn1c levels in those tissues. Cdkn1c maintained a monoallelic pattern in all neonatal tissues from progeny of the reciprocal cross (Kcnq1ot1PO/+) (Supplementary Figure S3).
Figure 3.
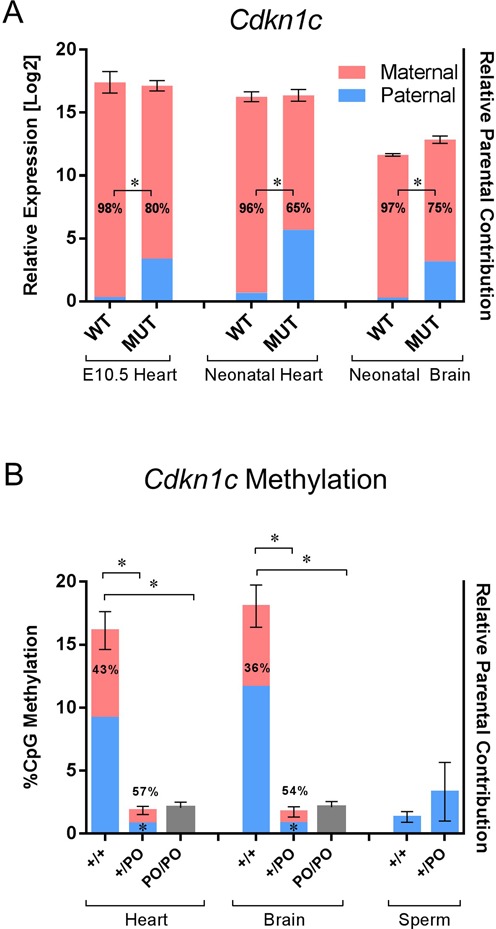
Effects of the paternal inheritance of the Kcnq1ot1PO allele on expression, imprinting and methylation of Cdkn1c. (A) Combined analysis of cardiac and brain Cdkn1c expression and imprinting in wild-type and Kcnq1ot1+/PO mice as assayed by qPCR and quantitative pyrosequencing, respectively. For qPCR, mean CT values were normalized to the geometric mean of β-actin, Ppia and Rplp1. Error bars indicate SEM. Numbers inside the bars are the percent maternal allele abundance relative to total Cdkn1c expression. Asterisks between bars indicate significant difference in parental contribution (P < 0.0001). (B) Methylation analysis of the Cdkn1c promoter in wild-type (+/+), Kcnq1ot1+/PO (+/PO) and Kcnq1ot1PO/PO (PO/PO) tissues by pyrosequencing. Bisulfite-treated genomic DNA was subjected to PCR with biotin-labeled primers; pyrosequencing results were analyzed for ratio of C to T transitions. Asterisks above the bars indicate a significant difference in methylation levels (P < 0.0001). Numbers inside the bars are percent maternal allelic abundance relative to total Cdkn1c expression and asterisks indicate a significant difference in parental contribution compared to wild-type (P = 0.002 for heart, P = 0.0003 for brain, n = 4).
The Cdkn1c gene lies in a CG-rich region that harbors a secondary methylation mark on the paternal allele (25), and it has been suggested that the Kcnq1ot1 ncRNA is directly responsible for recruiting DNA-methyltransferases to establish this modification (26,27). We tested the methylation status of the promoter in WT, Kcnq1ot1+/PO and Kcnq1ot1PO/PO mice by bisulfite mutagenesis and pyrosequencing. The Cdkn1c promoter is unmethylated in sperm from WT and mutant (heterozygous or homozygous) mice. In neonatal heart and brain tissues of the Kcnq1ot1+/PO and Kcnq1ot1PO/PO mice, the Cdkn1c promoter exhibits complete lack of parental methylation compared to WT mice (Figure 3B). This is in agreement with the biallelic expression in the mutants, indicating that transcription from or physical presence of the Kcnq1ot1 MP is required for acquisition of the secondary mark at the paternal Cdkn1c allele.
The mono- to biallelic transition for Kcnq1 is maintained in Kcnq1ot1+/PO cardiogenesis
In WT mice, Kcnq1 transitions from mono- to biallelic mode at 13.5 dpc in all the expressing tissues (8). This pattern is disrupted in brain of the Kcnq1ot1+/PO mice, with full biallelic expression of Kcnq1 in early stage embryos (not shown). In the fetal heart, however, absence of the MP results in only partial loss of imprinted expression, with paternal levels contributing less than 20% of the total RNA abundance in 10.5 dpc hearts, as assayed by pyrosequencing (Figure 4). A transition to full biallelic expression is still noticeable, with maternal and paternal contributions to Kcnq1 expression equalizing in Kcnq1ot1+/PO neonatal hearts. Together with results from previous studies (9), these data underscore that developmentally regulated imprinting patterns of the Kcnq1 region in the early stage heart are from other tissues.
Figure 4.
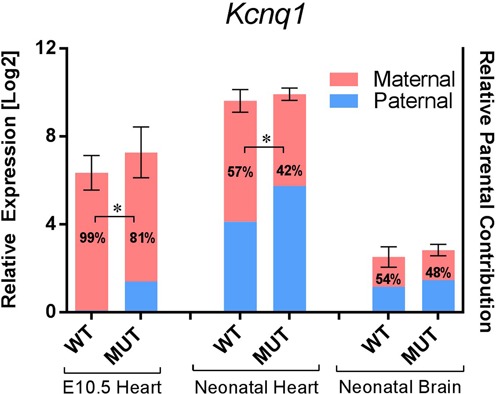
Effects of the paternal inheritance of the Kcnq1ot1PO allele on expression and imprinting Kcnq1. Combined analysis of cardiac and brain Kcnq1 expression and imprinting in wild-type and Kcnq1ot1+/PO mice (assays as described in Figure 3A). Error bars indicate SEM. Numbers inside the bars are percent maternal allelic abundance relative to total Kcnq1 expression. Asterisks between the bars indicate a significant difference in parental contribution compared to wild-type (P = 0.0011 for 10.5 dpc heart, P < 0.0001 for neonatal heart, n = 4).
To determine if the Kcnq1ot1 MP sequence per se contributed to the RNA levels of Kcnq1, we performed RT-qPCR for Kcnq1 from 10.5 dpc and neonatal heart and neonatal brain in Kcnq1ot1+/PO mice. Absence of the Kcnq1ot1 MP had no effect on levels of Kcnq1, even though both maternal and paternal alleles were now expressed (Figure 4). Protein levels of Kcnq1 were also invariant in in Kcnq1ot1+/PO neonatal hearts (Supplementary Figure S4), suggesting that transcription from the MP or the MP sequence itself is not required for full activation of Kcnq1.
The Kcnq1ot1 ncRNA is still expressed in the absence of the minimal promoter
Loss of imprinted Cdkn1c expression in the Kcnq1ot1+/PO mouse could be due to absence of the Kcnq1ot1 ncRNA or to an intrinsic regulatory activity of the MP. To distinguish between these two possibilities, we compared the levels of Kcnq1ot1 transcript in neonatal hearts and brains of WT and Kcnq1ot1+/PO mice, by performing RT-qPCR with sets of primers that spanned the entire 92 kb length of the RNA (Figure 5). Contrary to expectations, deletion of the MP did not lead to absence of the Kcnq1ot1 transcript in any tissue. The RNA levels in the Kcnq1ot1+/PO hearts were significantly lower with primer sets at 2 and 33 kb downstream of the canonical TSS (P = 0.0021 and P = 0.0045, respectively), but they were similar to WT levels with primer sets further downstream (44, 60 and 90 kb) (Figure 5B). On the other hand, Kcnq1ot1 ncRNA levels in Kcnq1ot1+/PO brains showed significant decreases with primer sets 2, 33 and 44 kb (P = 0.0478, 0.024 and 0.0162, respectively), and a trend toward lower expression with the remaining primer sets further downstream (Figure 5C). These results show (i) that WT levels of RNA resulting from transcription throughout the Kcnq1ot region are not uniform; (ii) that the sequence previously defined as the MP is not essential for transcription throughout the Kcnq1ot1 region and (iii) that in the heart, there are tissue-specific alternative TSS more than 33 kb downstream of the MP that act independently of it.
Figure 5.
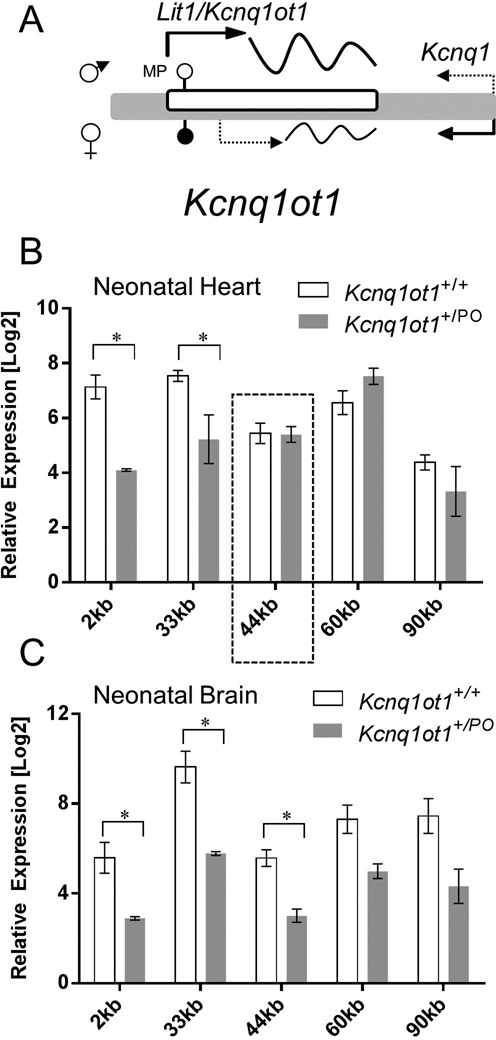
Analysis of cardiac and brain Kcnq1ot1 ncRNA expression. (A) Schematic of Kcnq1ot1 region (not to scale). Filled and unfilled circles are methylated and unmethylated CGIs, respectively and dotted arrow represents maternal transcription of the ncRNA in the heart. (B) Comparison of Kcnq1ot1 levels in neonatal hearts of wild-type and Kcnq1ot1+/PO mice with primers located 2, 33, 44, 60 and 90 kb downstream from the canonical transcriptional start site. For qPCR, mean CT values were normalized to the geometric mean of β-actin, Ppia and Rplp1. Error bars indicate SEM. Asterisks indicate significant difference between wild-type and Kcnq1ot1+/PO (for 2 kb, P = 0.005, n = 4; for 33 kb, P = 0.0245, n = 4). The dotted box indicates the region in which expression is consistently diminished, coinciding with a candidate enhancer region (see Figure 7, main text). (C) Comparison of Kcnq1ot1 levels in neonatal brains of wild-type and Kcnq1ot1+/PO mice with primers located 2, 33, 44, 60 and 90 kb downstream from the canonical TSS. For qPCR, mean CT values were normalized to the geometric mean of β-actin, Ppia and Rplp1. Error bars indicate SEM. Asterisks indicate significant difference between wild-type and Kcnq1ot1+/PO (for 2 kb, P = 0.0478, n = 4; for 33 kb, P = 0.0024, n = 4; for 44 kb, P = 0.0162, n = 4).
To determine the imprinting status of Kcnq1ot1 in Kcnq1ot1+/PO neonatal hearts and brains compared to WT, and to test whether parental contributions were similar throughout the locus, we performed pyrosequencing analysis at 2, 33, 44, 60 and 90 kb downstream of the MP. In the heart, loss of imprinting of Kcnq1ot1 was not observed at 2 kb, but was evident with primers from 33 kb onward. This was true in both in both WT and Kcnq1ot1+/PO mice in the cardiac tissue (Supplementary Figure S5B and SC). In contrast, paternal-specific expression was maintained in brain independently of the presence or absence of the MP (Supplementary Figure S5D and SE). Thus, the loss of imprinting of Kcnq1ot is a tissue-specific phenomenon. Activation of maternal allele suggests the presence of alternative cardiac promoters not subject to the imprinted KvDMR.
We quantified the contributions of the maternal and paternal alleles throughout the Kcnq1ot1 locus in neonatal hearts. In the WT mouse (Supplementary Figure S5B), a significantly greater contribution of maternal expression is seen with primers located at 44 kb downstream of the MP, relative to that observed at 2 kb (P < 0.0001). In the Kcnq1ot1+/PO hearts (Supplementary Figure S5C), all primer sets showed a higher maternal proportion of total expression relative to 2 kb (for 33 kb, P = 0.0018; for 44, 60 and 90 kb, P < 0.0001).
In the WT heart, maternal Kcnq1ot1 transcripts emerge from alternative start sites (TSS) that bypass the methylated CGIs downstream of the MP. We performed 5′ RACE assays to determine if these sites were active on the maternal allele in Kcnq1ot1+/PO neonatal hearts. Our results confirm that the maternal-specific TSS are activated normally in the mutant mice (Supplementary Figure S6), suggesting that the maternal contributions to Kcnq1ot expression are not captured equally with all primer sets, but depend on their location relative to the alternative TSS.
The minimal promoter is required for maintenance but not establishment of the hypomethylated paternal KvDMR
Figure 1 shows the KvDMR, a CG-rich region that is hypermethylated maternally and methylation-free paternally. To determine if the MP is required to prevent establishment of paternal methylation, we assayed DNA methylation by pyrosequencing in Kcnq1ot1PO/PO sperm and Kcnq1ot1+/PO neonatal heart and brain at the two CG islands (CGI 1 and 2) (Figure 6A). Although the KvDMR in sperm from mutant mice completely lacked methylation (not shown), the absence of the MP led to an increase in methylation of the region in neonatal tissues. The higher methylation levels were contributed by an increase in methylation of the paternal allele. Thus, the region remains refractive to de novo but not maintenance DNA methylation.
Figure 6.
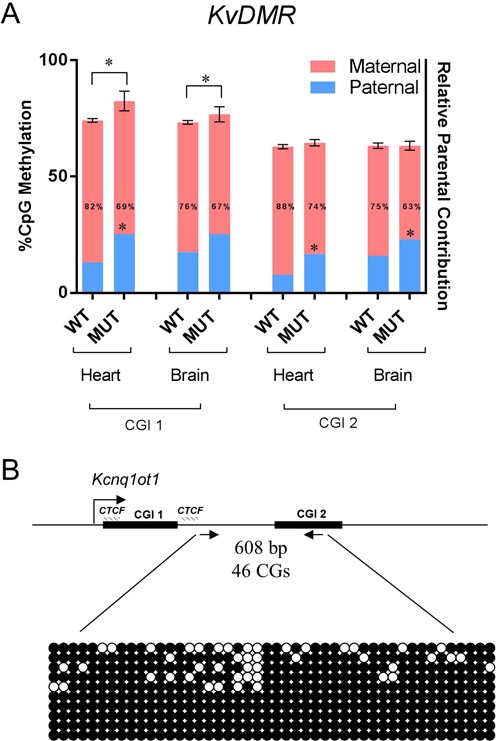
Methylation analyses of the KvDMR in the absence of the Kcnq1ot minimal promoter. (A) Combined analysis of percent total methylation (as assayed by pyrosequencing) and allelic distribution of the methylation (as assayed by MeDIP) between Kcnq1ot+/+ and Kcnq1ot1−/PO neonatal heart and brain at the two CG islands downstream of the Kcnq1ot1 promoter (CGI 1 and 2). Asterisks above the bars indicate a significant difference in methylation levels (P = 0.008 for heart, P = 0.0017 for brain at CG1). Numbers inside the bars are percent maternal methylation relative to total and asterisks indicate a significant difference compared to wild-type (P = 0.0012 for heart at CGI 1, P < 0.0001 for heart, P = 0.0005 for brain at CGI 2, n = 4). (B) Bisulfite mutagenesis sequencing of oocytes from Kcnq1ot1−/− females. Arrows indicate the primers that amplify a 608 bp sequence including CGI 2, with a total of 46 CGs (primers in Supplementary Table S1). Top, schematic of the amplified region, indicating the CGIs, the CTCF binding sites and the direction of transcription of Kcnq1ot. Below, representation of the methylation status of 10 individual DNA strands. Filled circles, methylated cytosines.
To determine if, on the other hand, the MP is required for normal acquisition of maternal methylation, bisulfite mutagenesis sequencing was performed at the KvDMR in Kcnq1ot1PO/PO oocytes (Figure 6B). Methylation was acquired normally in the KvDMR region, indicating that the minimal Kcnq1ot1 promoter is not required for DNA methylation establishment during oogenesis.
Kcnq1ot1 levels are inversely correlated with Kcnq1 expression in many tissues
Kcnq1ot1 exhibits tissue-specific transcriptional regulation, with down-regulation in the heart, but maintains high levels in the brain. Because Kcnq1, the sense gene, is highly expressed in heart but not easily detectable in whole brain, we hypothesized that Kcnq1ot1 levels might be inversely correlated to those of Kcnq1. Supporting this, previous results in which a major form of Kcnq1ot1 is prematurely terminated showed that Kcnq1 exhibited increased RNA levels in the heart (19). We examined strand-specific RNA-sequencing data in a range of tissues available from the ENCODE consortium; this confirmed that Kcnq1ot1 levels are inversely correlated with Kcnq1 in many tissues, suggesting that their expression is mutually exclusive (28) (Figure 7A). We also observed a trend toward higher expression of Kcnq1ot1 in embryonic compared to adult tissues.
Figure 7.
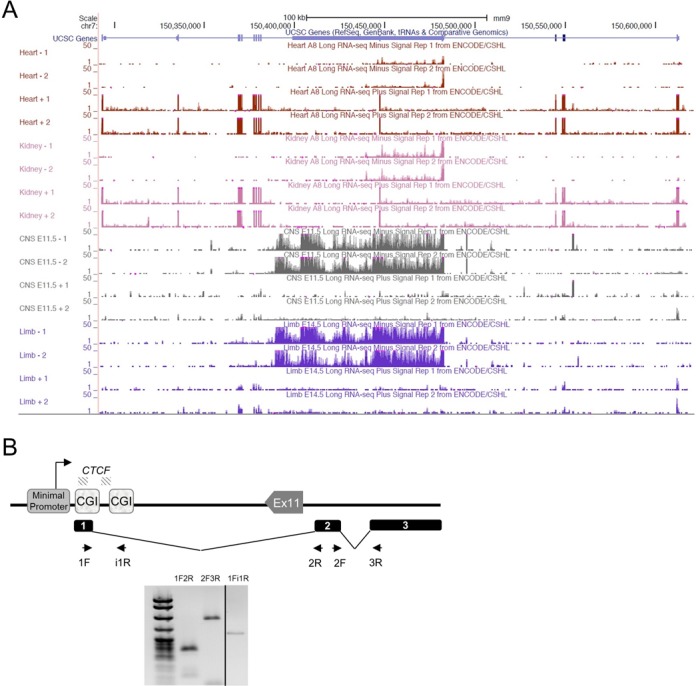
The Kcnq1ot1 ncRNA is spliced. (A) Long RNA-sequencing data showing strand-specific expression from ENCODE, visualized in the UCSC genome browser. Minus strand codes for the Kcnq1ot1 lncRNA; the plus strand codes for Kcnq1. Higher Kcnq1 expression correlates with lower and more discontinuous Kcnq1ot1 expression. (B) Confirmation of predicted splicing events for the Kcnq1ot1 lncRNA. Top, schematic of the Kcnq1ot coding region and the splicing variants (not to scale). cDNA from neonatal heart was used as substrates for qPCR. Shown is a 2% agarose gel with PCR products from neonatal heart. Primer sets 1F-2R and 2F-3R are designed to detect the junctions between exon 1 and 2 (190 bp), and 2 and 3 (369 bp), respectively. Primers 1F-i1R detect the unspliced version (297 bp). Exons 1 and 2 are 247 and 1043 bp, respectively.
The Kcnq1ot1 ncRNA is not a single entity
Several mechanisms have been proposed for regulation by antisense transcription, including direct effects of the non-coding RNAs (26) and/or the act of transcription itself as a source of transcriptional interference (29). To explore these possibilities, we turned our attention to fully characterizing the Kcnq1ot1 transcript.
When scanning the abundance of the Kcnq1ot1 transcript with five primer sets, we noticed that RNA levels were unequal, with a drop in abundance at 44 kb downstream of the major TSS that was reproducible in all our WT heart samples (Figure 5B). A consistent drop in RNA levels across a locus can be due to a splicing event or an alternative TSS. We explored both these possibilities.
We downloaded long strand-specific RNA-sequencing data from ENCODE/CSHL and used Sashimi plots to explore if there were alternatively spliced regions in the Kcnq1ot1 molecule. Two possible splicing events were detected by this analysis (Supplementary Figure S7). To validate them, we designed primers spanning the predicted introns and sequenced the PCR products. Reverse transcription and PCR of liver, brain and heart RNAs produced expected product sizes, and sequencing confirmed the presence of three exons (Figure 7B).
An alternative (though not exclusive) hypothesis to explain the variable levels of the Kcnq1ot1 transcript across the locus is that there are additional TSS contributing alternative RNAs to the total expression in the region. Several regions within introns 10 and 11 of Kcnq1 exhibit epigenetic features consistent with enhancer or promoter activity, harbor bioinformatically predicted transcription factor binding motifs (30) and/or exhibit transcription factor binding by ChIP-sequencing. We performed targeted 5′ RACE on several of these sites to determine if they were active in neonatal heart and brain (Figure 8A). Some candidate sites did not yield results in the 5′ RACE assays, at least for heart and brain RNA. However, assays for two of the candidate regulatory regions, positive for binding of Tbx5 in cultured heart cells (31) and Oct4 in mouse ES cells (32), respectively, indicated that there were novel RNA species emanating from them in both neonatal heart and brain (Figure 8A). To test whether these RNAs were enhancer RNAs, we performed 3′ RACE with primers located 500, 1000 and 1500 bp from the 5′ end; no products were detected, suggesting that the RNAs produced are longer than 1500 bp or are not polyadenylated.
Figure 8.
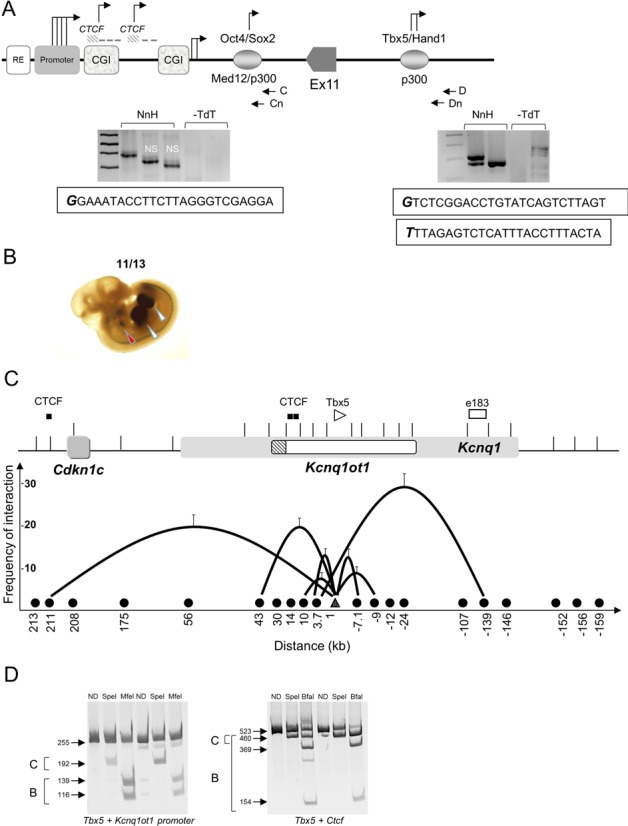
A novel heart enhancer activates Kcnq1. (A) Schematic showing the Kcnq1ot1 MP and part of the coding region, including the Oct4 and Tbx5 binding sites (not to scale). The Oct4 site is 7 kb downstream of the MP and the Tbx5 site is 10 kb downstream of Exon 11 and 45 kb downstream of the MP. 5′ RACE analyses were performed with nested primers, C and Cn for Oct4 site, and D and Dn for Tbx5 site. Below, gels showing the fragments obtained and the first 25 bases of the RNA transcribed from each site. For Oct4, the positive fragment was 326 bp; NS, non-specific; NnH, neonatal heart; -dTd, minus terminal deoxynucleotidyl transferase. For Tbx5, the positive fragments were 282 and 247 bp. (B) 11.5 dpc transgenic embryo analysis with the Tbx5 element. Representative LacZ-stained embryo exhibiting in vivo tissue-specific enhancer activity. Red arrowheads indicate the heart; white arrowheads point to the limb buds. Numbers show the reproducibility of LacZ reporter staining (11 of 13 total embryos). (C) Chromatin loop formation by 3C in wild-type neonatal heart. Top, schematic of the Kcnq1 region, with the Kcnq1ot1 promoter as a hatched box. CTCF sites and enhancer element 183 indicated. Bottom, graphic representation of 3C results. Black circles on the X-axis indicate sites tested; interactions between Tbx5 binding site (white triangle) and other restriction fragments depicted by black lines, with height of the curve determined by cross-linking frequency and the X-axis representing the distance from the anchor fragment in kilobases. Error bars represent SD. (D) Allele-specificity of the chromatin loops in the neonatal heart. Restriction digests of the 3C products between the Tbx5 binding site and the Kcnq1ot1 promoter and CTCF site. Digestion of the 255 bp Tbx5–Kcnq1ot promoter loop with SpeI cuts the castaneus allele into two fragments of 192 and 63 bp (the latter not shown); MfeI digests the C57BL/6 allele into fragments of 139 and 116 bp. Digestion of the 523 bp Tbx5–CTCF loop with SpeI cuts the castaneus allele into 460 and 63 bp fragments (the latter not shown); BfaI digests the castaneus allele into fragments of 458 and 65 bp and the C57BL/6 allele into fragments of 369 and 154 bp. ND, not digested; B, C57BL/6 and C, castaneus.
Thus, the Kcnq1ot1 region produces multiple transcripts of varying sizes, emerging from a previously defined MP and at least two additional TSS. Transcripts produced from the MP can undergo splicing, with at least two introns removed from the ncRNA.
An enhancer in the Kcnq1ot1 region directs expression to the heart and limb buds
Enhancers can direct the production of ncRNAs that are required for activation of their corresponding promoters (33). We reasoned that RNAs emanating from the Tbx5 and Oct4 sites could be indicative of enhancer activity. We performed transgenic mouse assays with the Tbx5 region. At E11.5, the Tbx5 sequence exhibited positive β-galactosidase staining in heart and limb buds, consistent with the known regulatory activities of Tbx5 (34–36) (Figure 8B, Supplementary Figure S8).
To determine whether the Tbx5 binding site makes contact with the Kcnq1ot1, Kcnq1 or Cdkn1c promoters to regulate their expression, we used 3C assays, which identify DNA sequences that interact in vivo. Cross-linked chromatin from WT neonatal hearts was analyzed by semi-quantitative PCR using an invariant primer anchor at the Tbx5 sequence. As shown in Figure 8C, the Tbx5 site loops to a previously identified heart enhancer (‘element 183′) (37) in intron 1 of Kcnq1, that in turn was shown to interact with the Kcnq1 promoter in the heart (9). This interaction is already present in the heart at E12.5, and is absent in the brain. The Tbx5 fragment also contacts the Kcnq1ot1 promoter and a CTCF binding site 211 kb upstream that lies between the Cdkn1c and Slc22a1 genes. Allele-specific restriction digests of the 3C products (Figure 8D) and sequencing of the ligated fragments indicates that the Tbx5 loops occur on both maternal and paternal alleles.
DISCUSSION
In this report, we provide strong evidence that DNA sequence elements and their relative location in the Kcnq1ot1 region are responsible for imprinted expression of Cdkn1c and, to some degree, of early embryonic Kcnq1. This undermines the current paradigm whereby the Kcnq1ot1 ncRNA is a single molecule that spreads and directly silences its neighboring genes. In the absence of the MP, a sequence that contains the main Kcnq1ot1 promoter, Cdkn1c loses imprinted expression, but the ncRNA continues to be transcribed across the Kcnq1ot1 region from multiple initiation sites. We show that Kcnq1ot1 is not a single molecular entity, but rather encodes a complex population of overlapping RNAs, at least one of which is spliced. We propose that the transcription throughout the Kcnq1ot1 locus in the early stage embryo serves to make regulatory elements available to their protein binding partners, setting the stage for their use later in development. In fact, we identify tissue-specific usage of a cardiac enhancer involving chromatin looping to allow physical proximity to other regulatory elements. This conformation is not present in the early embryo, suggesting it requires specific transcriptional factors to be established. We also propose that as the Kcnq1 promoter and/or other genes contact elements within the Kcnq1ot1 locus, alternative TSS are activated, driving expression of several overlapping ncRNAs.
The Kcnq1ot1 minimal promoter is required for imprinting of Cdkn1c
The KvDMR was initially defined as a region of differential methylation between parental alleles in intron 11 of Kcnq1, with a ncRNA emerging from the unmethylated paternal copy in antisense direction to Kcnq1. Several experiments in vivo and in vitro identified a MP, an enhancer-like activity, a silencing element, two CTCF binding sites and several repetitive sequences (16,38,39). Targeted deletions in the KvDMR ranging from 200 to 4000 bp as well as insertions to engineer the premature termination of the Kcnq1ot1 ncRNA all disrupted imprinted expression of the Cdkn1c gene (summarized in Supplementary Figure S1) (14,27,40), even though ncRNA transcripts continue to emerge from the region from alternative TSS that were identified later (19). Together, our data and those of other labs (7) suggest that maintenance of repression of the paternal Cdkn1c gene in the embryo is independent of the Kcnq1ot1 transcript per se, but rather requires one or more DNA elements within the KvDMR.
Initial repression of Cdkn1c accompanied by secondary methylation at its promoter may still be dependent on the Kcnq1ot1 transcript, as posited by several labs (27). We observed a decrease in the ncRNA abundance with primers spanning the initial 33 kb of the Kcnq1ot1 locus in the absence of the MP. This indicates that expression of this region, though not of downstream sequence, depends on the MP. In addition, it could suggest that the first kilobases of the ncRNA have repressive activity and that below a certain threshold level, it cannot silence Cdkn1c.
In the WT heart, down-regulation of Kcnq1ot1 and its loss of imprinting does not affect Cdkn1c imprinted expression, whereas upon removal of certain sequences within the KvDMR, as in the Kcnq1ot1+/PO mice, Cdkn1c loses imprinting in every tissue. This strongly supports the hypothesis that establishment and maintenance of Cdkn1c imprinting are independent events. While either the ncRNA or the act of transcription at Kcnq1ot1 is required for the establishment of methylation at the Cdkn1c promoter, maintenance of monoallelic Cdkn1c expression thereafter depends on the continued presence of a specific DNA element localized at the Kcnq1ot1 promoter. This is in partial agreement with previous reports ablating Kcnq1ot1 post-transcriptionally (7).
Surprisingly, although Cdkn1c expression is biallelic in Kcnq1ot1+/PO mice, total RNA levels in the heart are not significantly increased. Possibly a negative feedback mechanism operates above a certain threshold of Cdkn1c RNA or protein. More detailed profiling of Cdkn1c expression in the developing heart and additional mutations are necessary to understand the regulatory mechanisms at this locus.
The effect of removing the Kcnq1ot1 MP on Kcnq1 imprinting is especially interesting. Full biallelic expression of Kcnq1 is seen in the mutant mice, even at early stages which exhibit monoallelic expression in WT mice. However, in the heart, maternally biased expression is conserved and a transition to full biallelic mode is still observed after 13.5 dpc, with the same dynamics as in the WT. This suggests that the paternal Kcnq1 allele is repressed in the heart through a unique mechanism, a question that needs to be further explored. It is interesting to note that heart-specific chromatin remodeling complexes have been identified, underscoring that proteins involved in epigenetic regulation can be diverse and tissue-specific (41). The biology of the binuclear cardiomyocytes may also require exceptional regulatory mechanisms. Single-cell technology to visualize transcriptional events should provide more detailed information on the activities in this domain. Because Kcnq1 is an important gene for cardiac function, this region is a model for the co-existence of opposing regulatory mechanisms and how they interact, compete and prevail during development.
Revisiting the relationship between DNA methylation, CTCF binding and transcription of DMRs
We were interested in seeing if removal of the Kcnq1ot1 MP affected the regulatory elements within the KvDMR in Kcnq1ot1+/PO mice. Two main observations are particularly revealing: (i) in neonatal heart, CTCF exhibited binding to the maternal allele despite its hypermethylated status; (ii) the Kcnq1ot1 region was transcribed in spite of the lack of the main promoter. These results led us to re-examine the assumptions on the structure and function of the KvDMR and the ncRNA itself in the WT mouse.
Previous studies reported allele-specific binding of CTCF to the KvDMR, and it was hypothesized that maternal methylation impeded CTCF binding, allowing Cdkn1c access to an upstream enhancer element (24,39). Conversely, it was suggested that CTCF occupancy of its sites on the paternal allele could have a role in protecting the region from methylation. Our results show that in the heart, CTCF is capable of occupying sites 1 and 2 (four and one CpGs, respectively) on both maternal and paternal alleles. This agrees with a report of biallelic CTCF presence at the KvDMR in mouse embryonic fibroblasts (42), and contrasts with previous reports, perhaps because of the differences in tissues and developmental stages. Although CTCF binding is usually assumed to be methylation-sensitive, even in the classic model of the H19 DMD (differentially methylated domain), mutations in specific CpGs allowed binding of CTCF despite the presence of methylation (43). Thus, it seems that CTCF methylation-sensitivity may depend on the location of the CGs within its binding site and is dependent on context. Our results prove that CTCF does not function as an enhancer-blocking element at the endogenous locus, at least for Cdkn1c. Removal of the MP is sufficient to activate the paternal Cdkn1c, as noted above, despite the occupancy of CTCF on that allele. This result also suggests that the elements required for Cdkn1c expression in heart and brain lie downstream of the MP.
It has been hypothesized that transcription across differentially methylated regions in the oocyte is required for establishment of maternal methylation marks (44). Absence of the MP does not lead to hypomethylation when the mutation is transmitted maternally, suggesting that either that the KvDMR can be methylated regardless of its transcriptional status, or alternatively, that the additional TSS, if active in the oocyte, are sufficient for the methylation machinery to access the region. Alternatively, transcription across the KvDMR emanating from the sense Kcnq1 in oocytes may be sufficient to afford access to the methylation machinery.
Kcnq1ot1 is not a single entity
Antisense transcripts often exhibit low fidelity of transcription initiation and for Kcnq1ot1, we had identified alternative TSS up to 1.3 kb downstream of the MP, some of which were tissue-specific (19). Surprisingly, removal of the MP did not affect activation of these sites in the Kcnq1ot1+/PO mice, with RNAs emerging from both parental alleles in the heart. This suggests that activity of alternative TSS relies on tissue-specific factors.
In our studies and in publicly available expression data, we noticed that abundance of the Kcnq1ot1 transcript was not uniform across the locus. This was not due to difference in primer efficiencies nor unusually repetitive sequences affecting the RNA-seq data. Previous studies had not tested along the full length of the transcript, thus overlooking this uneven abundance. A possible explanation is that the Kcnq1ot1 either has unannotated TSS or is spliced, or both. In fact, our analysis of RNA-seq data and subsequent experiments revealed two splicing events. Even more striking was the fact that there were several sites that initiated transcripts at locations that also bear features of regulatory regions. One of these harbors a motif recognized by Tbx5, an important factor in cardiac and limb development (34,45). Tbx5 is part of a network of transcriptional factors that activate heart-specific genes (46). We speculated that Tbx5 and its binding site could be important for activation of Kcnq1 and could help determine the domain-wide conformation of the region in the heart.
The Kcnq1ot1 region is an enhancer jungle
Tissue-specific activation of the Kcnq1 gene is dependent on interactions between the promoter and at least two cardiac enhancers, one of which lies within the region encoding Kcnq1ot1. We previously reported physical proximity between the Kcnq1 promoter and a region in intron 1, already present in embryonic stages, with a contact established specifically in the heart at an upstream element. Here, we find an additional interaction between the intron 1 element (e183) (30) with a sequence that harbors many hallmarks of a cardiac/limb bud enhancer. Our assays to determine if these are eRNAs were inconclusive, and expression from the overlapping Kcnq1 gene complicate the analyses. However, transgenic embryo assays revealed that one candidate, with a recognition motif for Tbx5, exhibits tissue-specific expression in heart and limb.
One pending question is whether Kcnq1 expression matches the Tbx5 pattern in the limb bud, or if this particular enhancer is used in the heart for Kcnq1 and engages other distant genes in the limb bud. The expression domain suggested by the conformational assays includes the Cdkn1c gene, raising the question of how cell-cycle inhibition is coordinated with activation of cardiac-specific genes within this domain. Furthermore, it will be interesting to refine the timeline for these regulatory events, since there may be temporal differences between the maternal and paternal alleles due to their distinct chromatin status.
Overall, our data suggest that the products of the Kcnq1ot1 region vary in molecular structure, abundance and complexity as development ensues. The ∼90 kb Kcnq1ot1 molecule differs from other macroRNAs that act directly by silencing other genes, either by a spreading mechanism (47) or by transcriptional overlap (29). In fact, our results strongly suggest that the MP, or sequence elements within it, rather than the RNA molecule itself, are required for Cdkn1c repression, and that methylation on the maternal allele precludes this activity. Conformational analyses in an in vitro muscle differentiation model support this hypothesis, since they show that the MP is in close proximity to the repressed Cdkn1c promoter and this interaction is released upon differentiation (48).
The human Kcnq1 domain is conserved in gene order, expression patterns and imprinting control with the mouse. Based on sequence conservation and histone modifications associated with enhancers (ENCODE), we identified a region in Kcnq1 intron 10 overlapping with the Kcnq1ot1 ncRNA that, upon analysis with rVISTA (49), was predicted to bind Tbx5, Nkx2–5 and MyoD, among other cardiac transcription factors (Figure 9A), strongly pointing to heart-specific activity. Since Kcnq1 is associated with long QT syndrome, it will be interesting to determine whether there are mutations in the non-coding portions of the gene that could be responsible for the cardiac phenotypes that have not been associated with mutations in the protein-coding regions.
Figure 9.
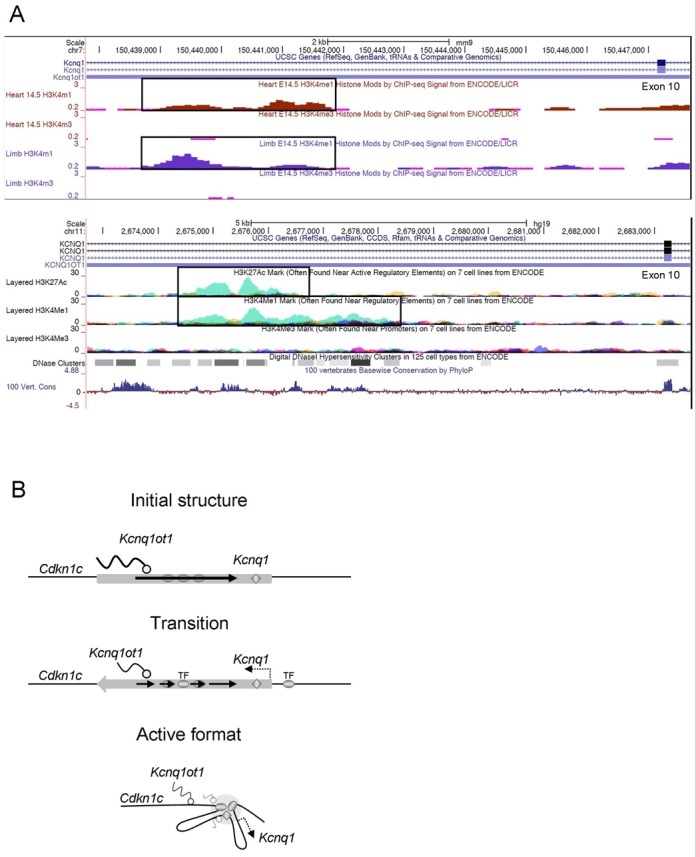
The Kcnq1ot1 region encompasses multiple enhancers and may adopt distinct conformations to accommodate specific enhancer-gene interactions in different tissues. (A) Top, H3K4Me1 and H3K4Me3 profiles of the Tbx5 binding site in mouse tissues as shown in the UCSC browser. Bottom, identification of a candidate enhancer in intron 10 of the human Kcnq1 gene, highly homologous to the novel enhancer reported here. The image shows a partial region of the Kcnq1 and Kcnq1ot1 genes, with the H3K27Ac, H3K4Me1 and H3K4Me3 profiles of the candidate Tbx5 enhancer. H3K4Me1 and H3K27Ac with low H3K4Me3 levels are frequently found at enhancer elements. (B) Model of heart-specific conformation as suggested by the 3C data. Ovals represent enhancers, diamond is the regulatory element active in early embryogenesis, wavy lines indicate RNAs. Width of lines represent levels of expression. Initially, Kcnq1ot1 is highly expressed and tissue-specific enhancers are inactive. Between 11.5 and 14.5 dpc, the Kcnq1ot1 and Kcnq1 genes become biallelic, with Kcnq1 levels increasing as transcription factors are expressed and bound to their cognate sites. Kcnq1ot1 levels start to decrease and become discontinuous as, finally, the domain is compressed and restructured when enhancers are brought into proximity with the Kcnq1 promoter.
There are a multitude of sequences within the Kcnq1ot1 region exhibiting features of regulatory elements that need to be tested. We propose that the Kcnq1ot1 region downstream of the KvDMR is a repository of enhancers and that the discontinuous levels of the ncRNA are due to activation of these enhancers by transcription factors. This leads to reorganization of the chromatin fiber, favoring induction of genes necessary for lineage determination at later stages. In the heart, for example, enhancers involved in cardiogenesis are engaged by the Kcnq1 promoter, and this coincides with a maximum decrease in the abundance of Kcnq1ot1 at 12.5 dpc, an up-regulation and loss of imprinting of Kcnq1 and the compression of the whole domain (50,51) (Figure 9B). We also envision that some enhancers may not be functional in vivo, and that this region may be an evolutionary nursery for regulatory elements, as suggested by others (52). In a region as complex as the Kcnq1 domain, with many genes active at different stages and in different tissues, insulators are likely to be important to ensure that genes are activated by the right enhancer. Ultimately we need to identify all the regulatory sequences, but also their relative locations, their tissue specificity and their three-dimensional architecture to fully understand their developmental role in vivo. The Kcnq1 region is an excellent model to understand mechanistically how enhancers, insulators, ncRNAs and the target genes are deployed to generate the appropriate expression outputs in the endogenous context.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Lisa Korostowski for establishment of the Kcnq1ot1+/PO mice and Lina Abouh Saleh, Dale Haines, Sijia Yu and Brian Barnette for technical assistance; also, we are grateful to Keith Latham, Marisa Bartolomei and Joanne Thorvaldsen for comments on the manuscript.
FUNDING
National Institutes of Health [R01 GM093066]. Funding for open access charge: National Institutes of Health, USA.
Conflict of interest statement. None declared.
REFERENCES
- 1.Smilinich N.J., Day C.D., Fitzpatrick G.V., Caldwell G.M., Lossie A.C., Cooper P.R., Smallwood A.C., Joyce J.A., Schofield P.N., Reik W., et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl. Acad. Sci. U.S.A. 1999;96:8064–8069. doi: 10.1073/pnas.96.14.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee M.P., Hu R., Johnson L.A., Feinberg A.P. Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat. Genet. 1997;15:181–185. doi: 10.1038/ng0297-181. [DOI] [PubMed] [Google Scholar]
- 3.Verona R.I., Mann M.R., Bartolomei M.S. Genomic imprinting: intricacies of epigenetic regulation in clusters. Annu. Rev. Cell Dev. Biol. 2003;19:237–259. doi: 10.1146/annurev.cellbio.19.111401.092717. [DOI] [PubMed] [Google Scholar]
- 4.Barlow D.P. Genomic imprinting: a mammalian epigenetic discovery model. Annu. Rev. Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- 5.Thakur N., Tiwari V.K., Thomassin H., Pandey R.R., Kanduri M., Gondor A., Grange T., Ohlsson R., Kanduri C. An antisense RNA regulates the bidirectional silencing property of the kcnq1 imprinting control region. Mol. Cell. Biol. 2004;24:7855–7862. doi: 10.1128/MCB.24.18.7855-7862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin J.Y., Fitzpatrick G.V., Higgins M.J. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J. 2008;27:168–178. doi: 10.1038/sj.emboj.7601960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golding M.C., Magri L.S., Zhang L., Lalone S.A., Higgins M.J., Mann M.R. Depletion of Kcnq1ot1 non-coding RNA does not affect imprinting maintenance in stem cells. Development. 2011;138:3667–3678. doi: 10.1242/dev.057778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gould T.D., Pfeifer K. Imprinting of mouse Kvlqt1 is developmentally regulated. Hum. Mol. Genet. 1998;7:483–487. doi: 10.1093/hmg/7.3.483. [DOI] [PubMed] [Google Scholar]
- 9.Korostowski L., Raval A., Breuer G., Engel N. Enhancer-driven chromatin interactions during development promote escape from silencing by a long non-coding RNA. Epigenet. Chromatin. 2011;4:21–32. doi: 10.1186/1756-8935-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochilas L.K., Li J., Jin F., Buck C.A., Epstein J.A. p57Kip2 expression is enhanced during mid-cardiac murine development and is restricted to trabecular myocardium. Pediatr. Res. 1999;45:635–642. doi: 10.1203/00006450-199905010-00004. [DOI] [PubMed] [Google Scholar]
- 11.Chen H., Shi S., Acosta L., Li W., Lu J., Bao S., Chen Z., Yang Z., Schneider M.D., Chien K.R., et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–2231. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engemann S., Strodicke M., Paulsen M., Franck O., Reinhardt R., Lane N., Reik W., Walter J. Sequence and functional comparison in the Beckwith-Wiedemann region: implications for a novel imprinting centre and extended imprinting. Hum. Mol. Genet. 2000;9:2691–2706. doi: 10.1093/hmg/9.18.2691. [DOI] [PubMed] [Google Scholar]
- 13.Lee M.P., DeBaun M.R., Mitsuya K., Galonek H.L., Brandenburg S., Oshimura M., Feinberg A.P. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5203–5208. doi: 10.1073/pnas.96.9.5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mancini-Dinardo D., Steele S.J., Levorse J.M., Ingram R.S., Tilghman S.M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006;20:1268–1282. doi: 10.1101/gad.1416906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guseva N., Mondal T., Kanduri C. Antisense noncoding RNA promoter regulates the timing of de novo methylation of an imprinting control region. Dev. Biol. 2012;361:403–411. doi: 10.1016/j.ydbio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Mancini-DiNardo D., Steele S.J., Ingram R.S., Tilghman S.M. A differentially methylated region within the gene Kcnq1 functions as an imprinted promoter and silencer. Hum. Mol. Genet. 2003;12:283–294. doi: 10.1093/hmg/ddg024. [DOI] [PubMed] [Google Scholar]
- 17.Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-Dinardo D., Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 18.Mann M.R., Chung Y.G., Nolen L.D., Verona R.I., Latham K.E., Bartolomei M.S. Disruption of imprinted gene methylation and expression in cloned preimplantation stage mouse embryos. Biol. Reprod. 2003;69:902–914. doi: 10.1095/biolreprod.103.017293. [DOI] [PubMed] [Google Scholar]
- 19.Korostowski L., Sedlak N., Engel N. The kcnq1ot1 long non-coding RNA affects chromatin conformation and expression of kcnq1, but does not regulate its imprinting in the developing heart. PLoS Genet. 2012;8:e1002956. doi: 10.1371/journal.pgen.1002956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dekker J. The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat. Methods. 2006;3:17–21. doi: 10.1038/nmeth823. [DOI] [PubMed] [Google Scholar]
- 21.Thorvaldsdottir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2012;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mann M.R., Lee S.S., Doherty A.S., Verona R.I., Nolen L.D., Schultz R.M., Bartolomei M.S. Selective loss of imprinting in the placenta following preimplantation development in culture. Development. 2004;131:3727–3735. doi: 10.1242/dev.01241. [DOI] [PubMed] [Google Scholar]
- 24.Fitzpatrick G.V., Pugacheva E.M., Shin J.Y., Abdullaev Z., Yang Y., Khatod K., Lobanenkov V.V., Higgins M.J. Allele-specific binding of CTCF to the multipartite imprinting control region KvDMR1. Mol. Cell. Biol. 2007;27:2636–2647. doi: 10.1128/MCB.02036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhogal B., Arnaudo A., Dymkowski A., Best A., Davis T.L. Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics. 2004;84:961–970. doi: 10.1016/j.ygeno.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 26.Mohammad F., Mondal T., Guseva N., Pandey G.K., Kanduri C. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development. 2010;137:2493–2499. doi: 10.1242/dev.048181. [DOI] [PubMed] [Google Scholar]
- 27.Mohammad F., Pandey G.K., Mondal T., Enroth S., Redrup L., Gyllensten U., Kanduri C. Long noncoding RNA-mediated maintenance of DNA methylation and transcriptional gene silencing. Development. 2012;139:2792–2803. doi: 10.1242/dev.079566. [DOI] [PubMed] [Google Scholar]
- 28.Consortium ENCODE. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latos P.A., Pauler F.M., Koerner M.V., Senergin H.B., Hudson Q.J., Stocsits R.R., Allhoff W., Stricker S.H., Klement R.M., Warczok K.E., et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. 2013;338:1469–1472. doi: 10.1126/science.1228110. [DOI] [PubMed] [Google Scholar]
- 30.Visel A., Minovitsky S., Dubchak I., Pennacchio L.A. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He A., Kong S.W., Ma Q., Pu W.T. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. U.S.A. 2011;108:5632–5637. doi: 10.1073/pnas.1016959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S., et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orom U.A., Shiekhattar R. Long non-coding RNAs and enhancers. Curr. Opin. Genet. Dev. 2011;21:194–198. doi: 10.1016/j.gde.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q.Y., Newbury-Ecob R.A., Terrett J.A., Wilson D.I., Curtis A.R., Yi C.H., Gebuhr T., Bullen P.J., Robson S.C., Strachan T., et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 1997;15:21–29. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- 35.Basson C.T., Huang T., Lin R.C., Bachinsky D.R., Weremowicz S., Vaglio A., Bruzzone R., Quadrelli R., Lerone M., Romeo G., et al. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc. Natl. Acad. Sci. U.S.A. 1999;96:2919–2924. doi: 10.1073/pnas.96.6.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rallis C., Bruneau B.G., Del Buono J., Seidman C.E., Seidman J.G., Nissim S., Tabin C.J., Logan M.P. Tbx5 is required for forelimb bud formation and continued outgrowth. Development. 2003;130:2741–2751. doi: 10.1242/dev.00473. [DOI] [PubMed] [Google Scholar]
- 37.Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F., et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thakur N., Kanduri M., Holmgren C., Mukhopadhyay R., Kanduri C. Bidirectional silencing and DNA methylation-sensitive methylation-spreading properties of the Kcnq1 imprinting control region map to the same regions. J. Biol. Chem. 2003;278:9514–9519. doi: 10.1074/jbc.M212203200. [DOI] [PubMed] [Google Scholar]
- 39.Kanduri C., Fitzpatrick G., Mukhopadhyay R., Kanduri M., Lobanenkov V., Higgins M., Ohlsson R. A differentially methylated imprinting control region within the Kcnq1 locus harbors a methylation-sensitive chromatin insulator. J. Biol. Chem. 2002;277:18106–18110. doi: 10.1074/jbc.M200031200. [DOI] [PubMed] [Google Scholar]
- 40.Fitzpatrick G.V., Soloway P.D., Higgins M.J. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 2002;32:426–431. doi: 10.1038/ng988. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi J.K., Lou X., Alexander J.M., Sugizaki H., Delgado-Olguin P., Holloway A.K., Mori A.D., Wylie J.N., Munson C., Zhu Y., et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat. Commun. 2011;2:187. doi: 10.1038/ncomms1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin S., Ferguson-Smith A.C., Schultz R.M., Bartolomei M.S. Nonallelic transcriptional roles of CTCF and cohesins at imprinted loci. Mol. Cell. Biol. 2011;31:3094–3104. doi: 10.1128/MCB.01449-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engel N., West A.G., Felsenfeld G., Bartolomei M.S. Antagonism between DNA hypermethylation and enhancer-blocking activity at the H19 DMD is uncovered by CpG mutations. Nat. Genet. 2004;36:883–888. doi: 10.1038/ng1399. [DOI] [PubMed] [Google Scholar]
- 44.Chotalia M., Smallwood S.A., Ruf N., Dawson C., Lucifero D., Frontera M., James K., Dean W., Kelsey G. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009;23:105–117. doi: 10.1101/gad.495809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smemo S., Campos L.C., Moskowitz I.P., Krieger J.E., Pereira A.C., Nobrega M.A. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 2012;21:3255–3263. doi: 10.1093/hmg/dds165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–1048. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Lee J.T. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–1842. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Busanello A., Battistelli C., Carbone M., Mostocotto C., Maione R. MyoD regulates p57kip2 expression by interacting with a distant cis-element and modifying a higher order chromatin structure. Nucleic Acids Res. 2012;40:8266–8275. doi: 10.1093/nar/gks619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loots G.G., Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004;32:W217–W221. doi: 10.1093/nar/gkh383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terranova R., Yokobayashi S., Stadler M.B., Otte A.P., van Lohuizen M., Orkin S.H., Peters A.H. Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev. Cell. 2008;15:668–679. doi: 10.1016/j.devcel.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 51.Fedoriw A.M., Calabrese J.M., Mu W., Yee D., Magnuson T. Differentiation-driven nucleolar association of the mouse imprinted Kcnq1 locus. G3. 2012;2:1521–1528. doi: 10.1534/g3.112.004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hong J.W., Hendrix D.A., Levine M.S. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.