

Abstract
Controllable gene delivery via vector-based systems remains a formidable challenge in mammalian synthetic biology and a desirable asset in gene therapy applications. Here, we introduce a methodology to control the copies and residence time of a gene product delivered in host human cells but also selectively disrupt fragments of the delivery vehicle. A crucial element of the proposed system is the CRISPR protein Cas9. Upon delivery, Cas9 guided by a custom RNA sequence cleaves the delivery vector at strategically placed targets thereby inactivating a co-expressed gene of interest. Importantly, using experiments in human embryonic kidney cells, we show that specific parameters of the system can be adjusted to fine-tune the delivery properties. We envision future applications in complex synthetic biology architectures, gene therapy and trace-free delivery.
INTRODUCTION
We employ the well-established nuclease activity of Cas9 (1–6), a type II CRISPR associated protein, which has been codon-optimized for expression in mammalian systems (1). As an RNA-guided system, Cas9 requires a guide RNA (gRNA) to direct the binding of the nuclease to a 17–21 bp DNA protospacer target (7). Cas9 preferentially interrogates the protospacer adjacent motif (PAM) sequence (8) and upon encountering the gRNA target sequence it creates a double stranded break (DSB) in close proximity to the PAM (Figure 1a).
Figure 1.
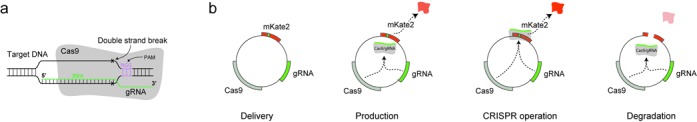
CRISPR-based self-cleaving delivery mechanism. (a) The gRNA–Cas9 complex targets template DNA through complementarity with the gRNA sequence. (b) The concept of self-cleaving vehicle for delivery.
Our proposed strategy for precise inactivation and fine-tuned gene delivery is based on a single vehicle that packages all the necessary components for the CRISPR operation in addition to the desired gene product cassette. A top-level operation of the system is illustrated in Figure 1b. Upon delivery, the Cas9 and gRNA modules, together with the desired gene product (e.g. a fluorescent protein mKate2), are produced by their corresponding promoters. The Cas9 and gRNA then form a complex and scan for potential targets. If the Cas9–gRNA complex recognizes a specific sequence in the open reading frame (ORF) of mKate2, it will cleave the vector and thus inhibit the protein production, triggering the vector degradation. In this case, the outcome in a single-cell will be the disruption of the delivery vehicle and the gradual degradation of the products. As a result, the system can be adopted in order to control the transient expression of transgenes in viral and non-viral delivery techniques (9,10).
MATERIALS AND METHODS
Recombinant DNA constructs
A complete list of plasmids constructed appears in Supplementary Table S1. The human codon optimized Cas9 containing nuclear localization signals and an empty gRNA backbone were obtained from Addgene (1). Q5 Polymerase (NEB) was utilized for all polymerase chain reaction (PCR) product amplifications according to the manufacturer's protocol. Oligos were ordered form Sigma and are listed in Supplementary Table S2. The plasmids were constructed utilizing PCR amplification, restriction digest and ligation with T4 ligase (NEB). Gel purification and PCR purification were performed with QIAquick kits (Qiagen). All plasmids over 9 kb were transformed in XL10 Gold Ultracompetent Cells (Agilent) and those <9 kb were transformed in NEB 5-alpha high efficiency Escherichia coli. Colonies were screened by PCR amplification using Taq (NEB) and verified by Sanger sequencing (Genewiz). DNA isolation was performed with the QIAprep miniprep kit (Qiagen). Translated ORFs appear in Supplementary Figure S1 and specific cloning steps in the Supplementary Material.
Cell culture and transfection
HEK293 and Tet-On HEK293 were maintained in Dulbecco's modified Eagle's medium (DMEM) (ThermoFisher) supplemented with 9% Fetal Bovine Serum (FBS) (Atlanta Biologicals), 0.9% MEM NEAA (MEM Non-Essential Amino Acids) 100× (ThermoFisher) and 0.4% Penicillin Streptomycin (ThermoFisher). Cells were incubated at 37°C at 5% CO2 and 95% humidity. All transfections were performed with JetPRIME (Polyplus) in 12-well plates (Greiner Bio One) at a plating density of 250 000 cells 24 h before transfection according to the manufacturer's protocol. Each well received 100 ng of plasmid unless otherwise noted and 1.75 μl of JetPRIME was used per well. Co-transfection DNA was added to create a total mass of 500 ng per well with the exception of the experiments shown in Figures 2 and 4a where a total mass of 750 ng was used. For the experiments in Tet-On HEK293 cells, doxycycline was added immediately following transfection.
Figure 2.
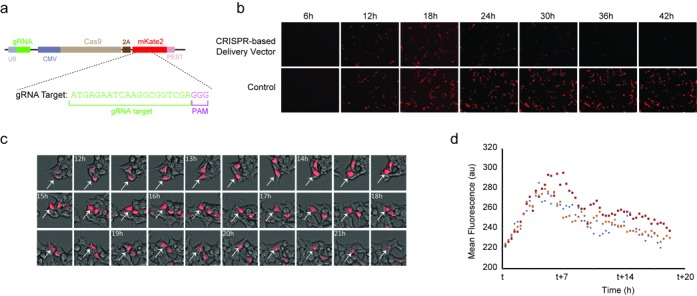
Initial validation experiments. (a) Representation of the single plasmid expression system with U6-gRNA combined with CMV-Cas9–2A-mKate2-PEST (pCas9–mKate2ps–T1gRNA). (b) Fluorescence microscopy time-lapse experiment showing the mKate2 dynamics for the CRISPR-based delivery vector (pCas9–mKate2ps–T1gRNA) and the control (pCas9–mKate2ps–CgRNA). Two hundred and fifty nanograms of each plasmid were transiently transfected in HEK293 cells. (c) Cropped images of the microscopy time-lapse showing the mKate2 dynamics in cells at 20 min intervals with post-transfection times shown. White arrow tracks a single cell. (d) Single-cell tracks showing mean fluorescence signal. The parameter t is the time after transfection of the CRISPR-based delivery vector pCas9–mKate2ps–T1gRNA (t = 6 h for blue, t = 10 h for red and t = 19 h for orange).
Figure 4.
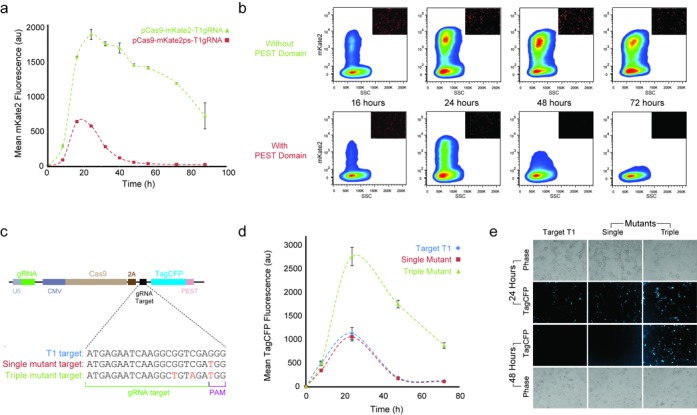
Protein degradation and Cas9–gRNA affinity impact on delivery properties. (a) Removal of the PEST domain (pCas9–mKate2–T1gRNA) from the T1gRNA plasmid (pCas9–mKate2ps–T1gRNA) yields a pulse of larger amplitude and residence time at equal mass transfection (100 ng of each plasmid). Error bars correspond to the standard deviation between three biological replicate experiments. (b) Representative flow cytometry measurements showing smoothed density plots of mKate2 intensity versus side scatter (SSC). Representative microscopy snapshots at each time point overlaid at top right corner. (c) Nucleotide sequences of mutant targets used to test different Cas9–gRNA affinities. (d) Single mutation at the first position of the PAM sequence of the target T1 shows same behavior to the T1 target while triple mutant has larger amplitude and longer residence time. Mean of three biological replicate flow cytometry measurements at 8, 24, 48 and 72 h post-transfection shown with standard deviation. (e) Representative fluorescence and phase microscopy of unmodified T1 target (pCas9–t1–CFPps–T1gRNA), single mutant (pCas9–mut1–CFPps–T1gRNA) and triple mutant (pCas9–mut3–CFPps–T1gRNA), at 24 and 48 h.
Microscopy
A Hamamatsu camera attached to an Olympus IX81 microscope with a 10× objective was used to capture images with the following filters (Chroma): ET560/40x (excitation) and ET630/75m (emission) for mKate2 and ET436/20x (excitation) and ET480/40m (emission) for Tag-CFP. The phase images were captured at 10 ms exposure and Tag-CFP at 150 ms exposure. The exposure for mKate2 was 15 ms in Figures 2 and 4 and 250 ms in Figure 6. For all experiments, image normalization and processing were consistent. All raw images were captured with SlideBook 5.0 and all time-lapse images were captured at 20 min intervals. Microscopy Supplementary Videos 1 and 2 were captured for 40 h starting five hours post-transfection with each frame representing a 20-min interval. The final frame in the videos represents 45 h post-transfection. All time-lapse images were captured at 15 ms exposure for mKate2.
Figure 6.
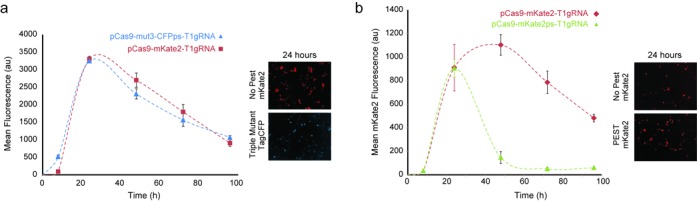
Fine-tuned delivery. (a) Transient transfection of the triple mutant (100 ng of pCas9–mut3–CFPps–T1gRNA) and the system without the PEST domain (10 ng of pCas9–mKate2–T1gRNA) yield the same maximum output levels and a similar trajectory. Error bars correspond to the standard deviation between three biological replicate experiments. We include representative microscopy snapshots, taken at 24 h. (b) Transient transfection of pCas9–mKate2ps–T1gRNA (45 ng) and the pCas9–mKate2–T1gRNA vector lacking the PEST domain (10 ng). The output lacking the PEST domain remains stable approximately twice as long as the output carrying the fast degradation domain. Error bars correspond to the standard deviation between three biological replicate experiments. We include representative microscopy snapshots, taken at 24 h.
Single-cell analysis
Multipoint 16 bit intensity TIFF files were exported from SlideBook and processed with ImageJ. The TIFF files were exported into individual images and pre-processed with the software package Circadian Gene Expression (CGE). To quantify fluorescence, the center of fluorescence at individual cells was marked for each frame of the pulse. CGE then generated a mean intensity value for tracked cells over each frame and individual tracks were combined in Excel for analysis of amplitude and residence time.
Flow cytometry
The cells were prepared for flow cytometry by trypsinizing each well with 0.2 ml 0.25% trypsin–EDTA followed by adding 0.7 ml of DMEM after 2 min. The resulting cell suspension was collected and centrifuged at 2000 rpm for 1 min. The supernatant was removed and the pellet resuspended by pipetting in 0.3 ml Dulbecco's Phosphate-Buffered Saline (DPBS) buffer (Invitrogen). Cells were run on a LSR Fortessa (BD Biosciences) flow cytometer equipped with the FACSDiva software program. Excitation occurred at 561 nm with a 610/20 band-pass (mKate2) and 445 nm with a 470/20 band-pass (Tag-CFP). Voltages were 280 V for FSC, 290 V for SSC, 375 V for Tag-CFP and 490 V for mKate2 with the exception of Figure 4 which was 375 V for mKate2. Acquisition was performed with BD FACSDiva and a global elliptical FSC/SSC gate was used to stop recording at a threshold of 50 000 gated events with all events saved and processed in FlowJo. Background fluorescence calculated from untransfected cells was subtracted from each recording.
RESULTS
To explore the feasibility and properties of the proposed intriguing mechanism, we commenced experiments to test the efficiency of CRISPR/Cas9 in targeting the ORF of mKate2 using transient transfections in human embryonic kidney cells (HEK293). Published results show that Cas9 has high nuclease activity while allowing for mismatches in the target sequence (11,12). In order to circumvent potential off-target effects we screened the mKate2 coding sequence targets against the human genome (11). Subsequently, using Gibson assembly (13), we constructed the two gRNAs (T1gRNA and T2gRNA) with the lowest off-target effect likelihood (Supplementary Methods and Supplementary Tables S3 and S4) and cloned them under the control of a U6 type III RNA polymerase promoter. As a negative control, we constructed a third gRNA (CgRNA) with no targets in the mKate2 ORF (Supplementary Table S3). Finally, we cloned Cas9 and mKate2 under the control of a CMV promoter, separated by a self-cleaving Thosea asigna peptide sequence (2A-like). This peptide allows the expression of more than one gene product under the same promoter (14).
We then combined the gRNA, Cas9, mKate2 and their corresponding promoters into a single plasmid. This delivery vehicle utilizes a CMV promoter to express Cas9 and mKate2 separated by the 2A sequence and the U6 promoter for the gRNA (Figure 2a). Considering the stability and corresponding long half-life of the fluorescent protein mKate2 (15), we introduced a PEST amino acid sequence into the C-terminus of our reporter to catalyze its faster degradation (16). The particular PEST domain has been shown to reduce the protein half-life to ∼2 h in mammalian cells (16). Finally, we verified that both single plasmids with T1gRNA and T2gRNA degraded mKate2 while the non-targeting plasmid (carrying CgRNA) did not (Supplementary Figure S2).
To characterize the behavior of the proposed delivery vehicle we transiently transfected HEK293 cells and performed time-lapse microscopy. On a population level (Figure 2b), we observed a pulse behavior in our pCas9–mKate2ps–T1gRNA CRISPR-based delivery vehicle (i.e. T1gRNA, Supplementary Video 1) while the control (non-targeting CgRNA, Supplementary Video 2) showed sustained mKate2 production. Single-cell analysis confirms the mKate2 rapid accumulation and subsequent degradation phase (Figure 2c). Additional microscopy time-lapse experiments show the mKate2 production by both systems between 6 and 30 h (Supplementary Figure S3) as well as 6 and 42 h (Supplementary Figure S4). Next, we processed the raw microscopy files (17) and retrieved the mean fluorescence value for each time frame in single cells. Representative tracks of three cells further validated the pulse behavior, and point to the peak fluorescence at ∼7 h after the mKate2 appearance (Figure 2d).
To probe the plasmid degradation after CRIPR-based cleavage, we cloned a second reporter cassette UbC-CFP-PEST into our pCas9–mKate2ps–T1gRNA and pCas9–mKate2ps–CgRNA plasmids. Indeed, we observe that the Tag–CFP levels decrease after Cas9–T1gRNA cleavage, albeit at a reduced rate (Supplementary Figure S5).
Prior to additional experimentation, we performed in silico experiments to probe the transient behavior of the system. We used ordinary differential equations (ODE) to model our system (Supplementary Tables S5 and S6) and utilized MATLAB to simulate the effects of parametric changes at the transcription, translation, and post-translational level. Our first observation is the ability to control the amplitude of the delivered product by adjusting the vector mass (Figure 3a). The model predicted that an increase in the DNA template would predominately increase the amplitude while having a mild effect on the residence time of the protein output (Figure 3a). We then investigated the effect of the output protein half-life to the response of the system. Specifically, we performed simulations gradually decreasing the degradation rate of mKate2. Both the residence time and amplitude of the pulse increased as a result of greater protein stability (Figure 3b). Finally, we performed simulations gradually decreasing the affinity of the Cas9–gRNA complex for its target DNA (Figure 3c). The simulations predicted that lowering the Cas9–gRNA activity would increase both the residence time and amplitude of the pulse. Motivated by these observations, we engineered customized versions of the delivery system to probe the ability to control the residence time and amplitude of the delivered gene.
Figure 3.
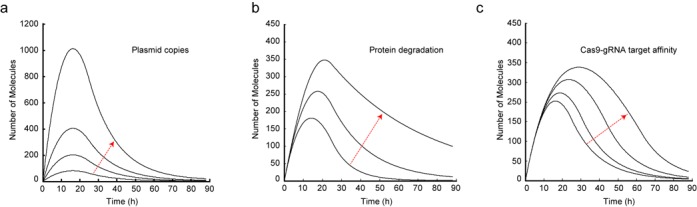
Parametric simulations of the self-cleaving delivery mechanism. (a) Increasing the plasmid copies (arrow direction) yields larger response amplitude without a significant impact in residence time. (b) Increasing protein stability (arrow direction) results in larger amplitude and longer pulse duration. (c) Decreasing the gRNA–Cas9 complex targeting affinity (arrow direction) increases the pulse amplitude and duration.
We first removed the PEST domain from our system and performed multipoint flow cytometry measurements. The population-based results show that the delivery vehicle with the PEST fast-degradation tag yields a pulse of protein production with duration of ∼48 h (Figure 4a). The simulations show that a more stable mKate2 protein would lead to increased duration and residence time. Indeed, the removal of the PEST domain caused a marked increase in the duration of the pulse. For the system lacking PEST, the output signal remains present at 88 h, yet in the degradation phase (Figure 4a). We verified the sustained expression of the non-targeting system throughout 88 h (Supplementary Figure S6). For both delivery vehicles, we plotted the mKate2 intensity versus the side scatter (SSC) (Figure 4b). The system without PEST shows a significant population of cells with high signal at 24, 48 and 72 h, with the number of cells and mean fluorescence intensity gradually decreasing (Figure 4b). Representative microscopy snapshots (Figure 4b) demonstrate the decrease in signal to near background in the mKate2–PEST fusion over the period from 24 to 48 h post-transfection.
Subsequently, we designed new mutant protospacers to reduce the affinity of Cas9–gRNA complex to the targeted DNA. To circumvent using the target within the ORF of the mKate2 gene, we first replaced our reporter with Tag–CFP fused with the PEST domain. We screened the Tag–CFP ORF against the T1gRNA target and verified that there is no protospacer of sufficient similarity to trigger the CRISPR cleavage (Supplementary Figure S7) (12). We proceeded to create three Tag–CFP versions of our delivery system with two mutant targets and the original T1 target sequence immediately preceding the start codon of Tag–CFP (Figure 4c). All three constructs contain the unmodified T1gRNA expressed with the U6 promoter.
The Cas9–gRNA complex crystal structure revealed that the PAM interacting domain interrogates DNA separately from the region that presents the gRNA to the DNA (18). The transcripts for gRNAs do not contain the PAM sequence and the Cas9–gRNA complex should tolerate any nucleotide in the N position of the PAM sequence (8). Our results are indeed aligned with these findings, indicating a mutation at the N position (G to T) yields similar pulse behavior to the original T1 target control (Figure 4d). Our triple mutant contains the single mutation with an additional two mutated nucleotides proximal to the PAM (Figure 4c). This mutant exhibits increased residence time and amplitude versus the original target control experiment (Figure 4d and e). The results of Figure 4d are also presented as raw flow cytometry data and show the population of cells at different time points (Supplementary Figure S8). Representative microscopy snapshots demonstrate the pulse behavior of the original target and single mutant while the triple mutant maintains high fluorescence at 48 h (Figure 4e).
Furthermore, based on our simulation results (Figure 3a), we directed our attention to controlling the output amplitude. These simulations show that the plasmid mass can be adjusted to control the product amplitude. Accordingly, we performed transfections at 25, 100 and 250 ng of plasmid. We observed that the maximum fluorescent intensity is directly correlated to the transfection mass (Figure 5a and b). Additionally, the average mKate2 intensity values of the pCas9–mKate2ps–T1gRNA vector, as recorded by flow cytometry, returned to near background levels for all the concentrations at 48 h (Figure 5, Supplementary Figure S9). The control plasmid (i.e. with the CgRNA) again demonstrates sustained high mKate2 expression at 24 and 48 h (Supplementary Figure S9). Therefore, the ability to control the amplitude while maintaining similar residence time provides an effective means for controlling protein delivery dosage. We were also interested in probing our system using an inducible promoter driving the production of the Cas9 and mKate2 transcript. To accomplish this, we replaced our CMV promoter with the TRE3G system and tested various levels of doxycycline (Dox) in Tet-on cells (HEK293 cells that stably produce the transcription factor rtTA). We observed that we can indeed control the amplitude of the output protein and maintain the same residence time (Figure 5c). The control experiments with no Dox as well as the CMV-driven vector pCas9–mKate2ps–T1gRNA are included in Supplementary Figures S10 and S11.
Figure 5.
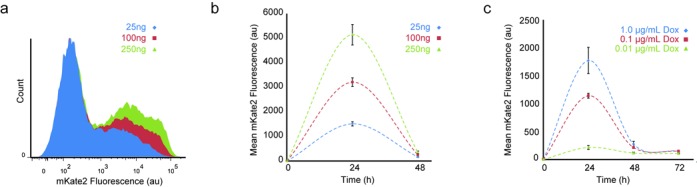
Plasmid concentration impact on delivery properties. (a) Transfection mass titration in overlaid flow cytometry histograms (pCas9-–mKate2ps–T1gRNA), measured at 24 h. (b) Mean mKate2 fluorescence intensity from triplicate biological replicates at 24 and 48 h indicate control of amplitude by mass titration (pCas9–mKate2ps–T1gRNA). (c) Titrations of doxycycline show change in amplitude for fixed mass transfections at 24, 48 and 72 h (ptreCas9–mKate2–T1gRNA). Error bars correspond to the standard deviation between three biological replicate experiments.
Considering the capacity to fine-tune the amplitude of our delivered protein, our last objective was to compare the dynamics of the three delivery vehicles (i.e. PEST, no-PEST and triple mutant) by adjusting the transfection mass in order to achieve the same maximum amplitude. We discovered that the 10:1 mass ratio (Supplementary Figure S12) between the triple mutant and the system without the PEST domain yields the same maximum output levels (Figure 6a). Lowering the activity of the Cas9–gRNA complex by mutating the targets yields a similar pulse trajectory to that of the mKate2 lacking a PEST domain (Figure 6a). Finally, we transfected the pCas9–mKate2ps–T1gRNA plasmid and the pCas9–mKate2–T1gRNA vector lacking the PEST domain after identifying a ratio in the range of 6:1 and again showed that we can achieve the same maximum output at 24 h yet control the residence time of delivery (Figure 6b, Supplementary Figures S13 and S14).
DISCUSSION
Here we present a ‘self-destructing message’, in the form of plasmid DNA that can only be read a finite number of times by the cell. As the messenger RNAs are transcribed and then translated the protein Cas9 in complex with the gRNA returns to the plasmid and creates DSB, effectively disrupting its function and destroying the delivery mechanism. Additionally, the proposed mechanism can be adopted to produce trace-free delivery for potential applications in protection of genetic material intellectual property. Introducing multiple, strategically located targets in the delivery vehicle can result in short fragments that randomize the original code, rendering it unrecoverable.
The CRISPR off-target effects remain a significant challenge to applying Cas9 for therapeutic use. Here, we introduce a mechanism which is independent of the actual nucleotide content of the gRNA targets. Accordingly, our system can be updated with gRNAs proven to have minimal off-target impact. Alternatively, in order to eliminate potential double strand breaks, a user can replace the Cas9 with a double nicking mechanism (4,19).
To conclude, the proposed methodology complements current tools (20–24) to produce genetic switches in synthetic architectures (25–27). Moreover, we show that specific parameters of the system can be used to fine-tune the response of the output protein dynamics. CRISPR Cas9 systems have the potential to build complex gene regulatory modules (22) and we envision future applications where our delivery vehicle is used in synthetic architectures and for controlled therapeutic gene delivery (28,29).
AVAILABILITY
Correspondence and requests for materials should be addressed to L.B. (bleris@utdallas.edu).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Kristina Ehrhardt and Yiling Bao for technical assistance.
Author contributions: L.B. and R.M. designed the experiments. R.M., A.S., M.L, S.P., Y.L. and T.K. performed the experiments. R.M. and L.B. analyzed the data. R.M. and L.B. prepared the manuscript. L.B. conceived and supervised the project.
FUNDING
US National Institutes of Health (NIH) [GM098984, GM096271, CA17001801]; US National Science Foundation (NSF) [CBNET-1105524]; Jonsson Family Fellowship in Bioengineering and an Industry Advisory Board Graduate Fellowship (to R.M.); Academic Excellence Scholarship (to A.S., M.L., S.P.); Welch Foundation (to S.P.). Funding for open access charge US NSF.
Conflict of interest statement. None declared.
REFERENCES
- 1.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei C., Liu J., Yu Z., Zhang B., Gao G., Jiao R. TALEN or Cas9–rapid, efficient and specific choices for genomic modifications. J. Genet. Genomics. 2013;40:281–289. doi: 10.1016/j.jgg.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Kabadi A.M., Ousterout D.G., Hilton I.B., Gersbach C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42:e147. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cong L., Ran F., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang W., Li W., Ma N., Steinhoff G. Non-viral gene delivery methods. Curr. Pharm. Biotechnol. 2013;14:46–60. [PubMed] [Google Scholar]
- 10.Giacca M., Zacchigna S. Virus-mediated gene delivery for human gene therapy. J. Controlled Release. 2012;161:377–388. doi: 10.1016/j.jconrel.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson D.G., Young L., Chuang R., Venter J.C., Hutchison C.A., Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 14.Chang C., Lai Y., Pawlik K.M., Liu K., Sun C., Li C., Schoeb T.R., Townes T.M. Polycistronic lentiviral vector for ‘hit and run’ reprogramming of adult skin fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27:1042–1049. doi: 10.1002/stem.39. [DOI] [PubMed] [Google Scholar]
- 15.Kelmanson I. Enhanced red and far-red fluorescent proteins for in vivo imaging. Nat. Methods. 2009;6 [Google Scholar]
- 16.Li X., Zhao X., Fang Y., Jiang X., Duong T., Fan C., Huang C.C., Kain S.R. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 1998;273:34970–34975. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- 17.Sage D., Unser M., Salmon P., Dibner C. A software solution for recording circadian oscillator features in time-lapse live cell microscopy. Cell. Div. 2010;5:17. doi: 10.1186/1747-1028-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimasu H., Ran F., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen B., Zhang W., Zhang J., Zhou J., Wang J., Chen L., Wang L., Hodgkins A., Iyer V., Huang X. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- 20.Bleris L., Xie Z., Glass D., Adadey A., Sontag E., Benenson Y. Synthetic incoherent feedforward circuits show adaptation to the amount of their genetic template. Mol. Syst. Biol. 2011;7:519. doi: 10.1038/msb.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deans T.L., Cantor C.R., Collins J.J. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell. 2007;130:363–372. doi: 10.1016/j.cell.2007.05.045. [DOI] [PubMed] [Google Scholar]
- 22.Kiani S., Beal J., Ebrahimkhani M.R., Huh J., Hall R.N., Xie Z., Li Y., Weiss R. CRISPR transcriptional repression devices and layered circuits in mammalian cells. Nat. Methods. 2014;11:723–726. doi: 10.1038/nmeth.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nissim L., Perli S.D., Fridkin A., Perez-Pinera P., Lu T.K. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell. 2014;54:698–710. doi: 10.1016/j.molcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lienert F., Lohmueller J.J., Garg A., Silver P.A. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol. 2014;15:95–107. doi: 10.1038/nrm3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber W., Fussenegger M. Emerging biomedical applications of synthetic biology. Nat. Rev. Genet. 2012;13:21–35. doi: 10.1038/nrg3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purnick P.E., Weiss R. The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 2009;10:410–422. doi: 10.1038/nrm2698. [DOI] [PubMed] [Google Scholar]
- 28.Calado S.M., Oliveira A.V., Machado S.C., Haase R., Silva G.A. Sustained gene expression in the retina by improved episomal vectors. Tissue Eng. 2014;20:2692–2698. doi: 10.1089/ten.tea.2013.0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeFrancesco L. Clinical gene therapies progress. Nat. Biotechnol. 2012;30:60. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.