

Abstract
V(D)J recombination is initiated by RAG1 and RAG2, which together with HMGB1 bind to a recombination signal sequence (12RSS or 23RSS) to form the signal complex (SC) and then capture a complementary partner RSS, yielding the paired complex (PC). Little is known regarding the structural changes that accompany the SC to PC transition or the structural features that allow RAG to distinguish its two asymmetric substrates. To address these issues, we analyzed the structure of the 12RSS in the SC and PC using fluorescence resonance energy transfer (FRET) and molecular dynamics modeling. The resulting models indicate that the 12RSS adopts a strongly bent V-shaped structure upon RAG/HMGB1 binding and reveal structural differences, particularly near the heptamer, between the 12RSS in the SC and PC. Comparison of models of the 12RSS and 23RSS in the PC reveals broadly similar shapes but a distinct number and location of DNA bends as well as a smaller central cavity for the 12RSS. These findings provide the most detailed view yet of the 12RSS in RAG–DNA complexes and highlight structural features of the RSS that might underlie activation of RAG-mediated cleavage and substrate asymmetry important for the 12/23 rule of V(D)J recombination.
INTRODUCTION
The genes encoding the variable domains of the T and B cell immune receptors are assembled from germline gene segments by the recombination activating gene proteins RAG1 and RAG2 (together referred to as RAG) and high mobility group box protein HMGB1 (or 2), in a process called V(D)J recombination. The germline V (variable), D (diversity) and J (joining) coding gene segments are bound, paired and then cleaved by RAG, which thus orchestrates the first phase of recombination (reviewed in (1)). Processing and joining of the cleaved coding ends into a potentially functional variable region exon is completed in the second phase of recombination by ubiquitously expressed factors of the nonhomologous end joining repair pathway, which include the DNA-dependent protein kinase (DNAPKcs, Ku70, Ku80), Artemis, XRCC4 and Ligase IV (reviewed in (2)).
Each germline coding segment is flanked by a recombination signal sequence (RSS) that is bound by RAG and that specifies the site of a sequential pair of RAG-mediated phosphoryl transfer reactions. RSSs contain two well-conserved elements, a palindromic heptamer (consensus 5′-CACAGTG-3′) and an AT-rich nonamer (consensus 5′-ACAAAAACC-3′) separated by a poorly conserved spacer region of 12 or 23 bp, hence their 12RSS or 23RSS designations (Figure 1B). Recombination occurs efficiently only between gene segments in which one is flanked by a 12RSS and the other by a 23RSS, an observation referred to as the 12/23 rule.
Figure 1.
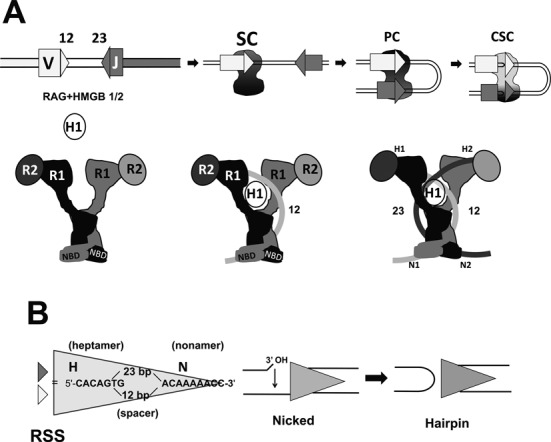
Features of RAG-mediated DNA cleavage during V(D)J recombination. (A) The steps involved in RAG-mediated DNA cleavage are represented schematically (upper panel), indicating V and J gene segments (rectangles), the 12RSS and 23RSS (light and dark gray triangles, respectively) and the RAG1/RAG2/HMGB1 complex (black/dark gray shape). SC: signal complex; PC: paired complex; CSC: cleaved signal complex. The lower panel presents a cartoon representation of the RAG heterotetramer (left), the RAG heterotetramer after binding the 12RSS (curved light gray line) and incorporating HMGB1 (white oval) (12SC; middle) and after incorporation of the 23RSS (curved darkest gray line) (PC; right). This depiction of the RAG heterotetramer and its DNA complexes derives from the shape of the signal end complex described by Grundy et al. (30) and incorporates strong bending of the 12RSS (this paper) and the 23RSS (26), with black and dark gray used to indicate the RAG subunits engaged in cleavage of the 12RSS and 23RSS, respectively. (B) The RSS and the mechanism of RAG-mediated DNA cleavage. The sequences of the consensus heptamer and nonamer elements and the structures of the nicked intermediate (with 3′ hydroxyl (3′ OH) group) and the products of double strand break formation, including the hairpin coding end, are shown.
Due to their higher solubility than full-length RAG proteins, most biochemical studies have used the ‘core’ domains of murine RAG1 (residues 384–1008 of 1040aa) and RAG2 (residues 1–383 of 527aa) (3–5), which are catalytically active and can mediate recombination in vivo. The RAG1 core forms a tight homodimer and is the central player in both DNA binding and catalysis (6–10). It interacts with RAG2, makes direct contacts with the nonamer and the heptamer, and contains three acidic catalytic residues (D600, D708 and E962, DDE motif defined) (11–13) that constitute a DDE motif thought to resemble the catalytic center of certain transposases and retroviral integrases (14–16). The acidic residues coordinate divalent cations essential for catalytic activity. Mg2+ is thought to be physiologically significant because it is the only divalent cation to support DNA binding and cleavage in accord with the 12/23 rule (17).
Much less is known about the role of the RAG2 core protein, which appears to function as an accessory factor. Although the RAG2 core is important for RSS binding specificity and critical for catalysis by RAG1, the mechanism by which it facilitates these processes is not known, although its interaction with RAG1 is thought to be important. A cleavage competent RAG protein complex is thought to contain a dimer of RAG1 and two subunits of RAG2 (6,9,18) (Figure 1A, schematic diagram).
The RAG cleavage mechanism involves two steps (Figure 1B). The recombinase first activates a water nucleophile, which nicks the phosphodiester bridge between the coding flank and the 5′ C of the heptamer, leaving a 3′ hydroxyl at the coding sequence upper strand. In the second step, this 3′ OH group serves as a nucleophile that attacks the phosphodiester of the bottom strand. This is thought to require significant distortion of the DNA and movement of the DNA or protein to place the new target phosphodiester bond in the active site, and hence predicts conformational alterations of the protein–DNA complex. DNA cleavage occurs in an ordered series of protein–DNA complexes. Initially, RAG binds together with HMGB1/2 to a 12RSS or a 23RSS to form the 12 signal complex (12SC) or the 23SC, respectively (Figure 1A). The SC, though capable of nicking, does not support hairpin formation. For this to occur, the SC must bind a 12/23 complementary RSS to form the paired complex (PC) (Figure 1A)(17). Despite its central role in hairpin formation, the mechanism by which the PC is assembled is poorly understood.
RAG1 alone binds to the 12RSS but with limited discriminative power over nonspecific DNA (7,18–20). Although RAG2 does not bind DNA by itself, the RAG1–RAG2 complex has higher affinity and specificity for the RSS than does RAG1 alone. RAG binding to a 12RSS does not require the presence of HMGB1/2, whereas 23SC and PC formation are strongly dependent on these ubiquitous nonspecific DNA binding/bending proteins (21,22). HMGB1 binds with higher affinity to DNA in the presence of RAG1 than in its absence, an effect that suggests binding synergy between the two proteins during RAG–RSS complex assembly (23).
Detailed footprinting and photo-crosslinking studies have been performed on RAG–RSS complexes (reviewed in (24)). In the PC, both the 12 and 23RSS display a DNAse I hypersensitive site near the center of the spacer (bottom strand), in contrast with the SCs in which a hypersensitive site is observed in the upper strand at the nonamer–spacer border (25). Since DNAse I hypersensitivity can reflect sites of DNA bending, these results suggest changes in RSS structure during the SC to PC transition. In the SC and PC, the 12 and 23 RSS display similar RAG–phosphate backbone interactions that localize primarily to one face of the DNA helix. These indicate a similar engagement of the proteins with the two RSSs in these complexes, which in turn suggests differential bending of 23RSS versus 12RSS given the DNA length difference of their spacers. HMGB1 is required for appropriate bending of RSS DNA and its contacts appear to lie near the spacer–nonamer and spacer–heptamer borders of both the 12 and 23RSSs in the SC and PC (7,21,24,26,27).
Little structural information is available for the RAG proteins or RAG–DNA complexes. The structure of the RAG1 nonamer binding domain (NBD; amino acids 389–464) in complex with a short nonamer oligonucleotide revealed a tight protein dimer making contact with two antiparallel, tilted DNA molecules (28). RAG2c was proposed to adopt a 6-bladed β-propellor structure based on sequence analysis (11,29). Elegant biochemical studies demonstrated that the RAG heterotetramer adopts an intricate inter-subunit division of labor in which the NBD and DDE motifs are located in trans on the two RAG1 subunits with respect to one RSS (15), a common theme for transposases.
A low-resolution electron microscopy structure of an RAG heterotetramer plus HMGB1 bound to cleaved 12/23RSSs (the signal end complex, or SEC) revealed a symmetric complex shaped like an anchor (30). The shank of the anchor was proposed to be composed of intertwined RAG1 monomers that diverge at the arms (‘hooks’) of the anchor and make contact with RAG2 monomers, which constitute the tips of the arms (see cartoon representation in Figure 1A). Neither the location of HMGB1 nor the path of the DNA could be determined, although the nonamer (and NBD) was inferred to lie at the bottom end of shank (30). Furthermore, RAG–RSS complexes formed on DNA molecules of various length have been imaged by Atomic Force Microscopy (AFM) in the SC (31) and PC (32). These studies revealed substantial bending (estimated at 60° by (31)) in the region of the DNA occupied by the RAG proteins.
To better understand the organization of substrate DNA in RAG–DNA complexes, we have used fluorescence resonance energy transfer (FRET) to measure distances between different points on one DNA molecule (fluorophores positioned in cis), or in some cases between two different DNA molecules (fluorophores in trans). Our initial study of the PC, using fluorophores in trans, suggested that the two RSSs are bent and cross one another (33). Subsequently, using fluorophores in cis, we demonstrated that the 23RSS in the PC is indeed strongly bent, adopting a U-shaped structure with the center of the bend located in the spacer (26). Little is known regarding the structure of the 12RSS in RAG–DNA complexes.
Here, we have analyzed the 12RSS in both the SC and PC, allowing us to address two interrelated questions: to what extent is the 12RSS bent in the PC, and, how different is the 12RSS configuration in the SC from that in the PC? Our results demonstrate that the 12RSS is strongly bent in both the SC and PC, that there are clear differences in its structure between the two complexes and that within the PC, the 12RSS and 23RSS exhibit structural differences. Our findings provide a starting point for construction of detailed, testable models of the PC and have implications for the putative conformational changes that occur upon synapsis and enable the DNA double strand break step of RAG-mediated DNA cleavage.
MATERIALS AND METHODS
Oligonucleotide RSS substrates
Deoxyoligonucleotides either fluorescently labeled or unlabeled were synthesized and HPLC purified by Integrated DNA Technologies Inc. (Coralville, IA, USA). To assemble double-stranded RSS DNA substrates, 5′ or internally fluorophore labeled, complementary oligonucleotides were mixed in equimolar amounts, heated for 5 min at 95°C, followed by slow cooling to room temperature in binding buffer (BB: 10-mM Tris-HCl (pH 7.5), 50-mM NaCl, 5-mM MgCl2), which was used throughout for FRET and other assays. Oligonucleotide sequences used were (only top strand is shown): 12-RSS (59mer), 5′-GATCTGGCCTGTCTTACACAGTGATACAGACCTTAACAAAAACCTGCACTCGAGCGGAG-3′, 12RSS (81mer) 5′ TCTGAATAGATGATCTGGCCTGTCTTACACAGTGATACAGACCTTAACAAA AACCTGCACTCGAGCGGAGGAGCTGATCTA 3′ and 23-RSS, 5′-GATCTGGCCTGTCTTACACAGTGATGGAAGCTCAATCTGAACTCTGACAAAAACCTCGAGCGGAG-3′. The fluorophores FAM (6-carboxy-fluorescein), Alexa488 and TAMRA-NHS (carboxytetramethylrhodamine ester) were attached to the DNA base via a C6 methylene linker and were incorporated during synthesis using phosphoramidite-labeled nucleotides (dT for FAM and Alexa488, and an amino-modified NHS ester nucleotide in the case of TAMRA).
Protein purification
All experiments used RAG1c (aa 384–1008) fused at its N-terminus to maltose binding protein (MBP) and tagged at its C-terminus with six histidine residues (MBP-RAG1c) purified from bacteria, and RAG2c (aa 1–387) fused at its N-terminus to glutathione S-transferase (GST-RAG2c) purified from HEK293T cells, as previously described (34). Mouse HMGB1 (polyhistidine tagged) was expressed in bacteria and purified as described previously (35).
In solution steady state fluorescence data acquisition and analysis
A PTI C-61 (Photon Technology International) T-format fluorometer equipped with a circulating water bath to control cell temperature was used to record fluorescence emission spectra in 150-μl quartz cuvettes (Starna, Atascadero, CA, USA) at 25°C. Fluorescence emission spectra were recorded with an excitation wavelength of 492 nm using an 8-nm band pass setting for both the excitation and emission monochromators. All fluorescence emission spectra were recorded between 510 and 650 nm (for FAM/TAMRA pairs), using 1-nm steps and two second integration times. Various protein–DNA samples were mixed in ice cold BB buffer followed by 10-min incubation at 25°C. After incubation, each sample mix was moved into the quartz cuvette for recording. No photobleaching was detected between any of two successive recordings. The standard complete reaction contained 15-nM doubly labeled 12RSS DNA, without (SC) or with 45-nM unlabeled 23RSS (PC), 125-nM RAG1 and 250-nM RAG2 and 197-nM HMGB1. Control reactions, in which the labeled DNA contains only the donor (FAM) or the acceptor (TAMRA) fluorophore, were otherwise identical in protein content and unlabeled RSS partner. Spectra were corrected for background created by lamp fluctuations and the wavelength dependence of the lamp and detector. If sample absorption at excitation wavelength exceeded 0.001, inner filter corrections were also applied according to (36).
For each FRET experiment for a particular configuration of donor and acceptor fluorophore probes, emission spectra (λex = 492 nm) were recorded for the following samples that lacked added proteins: (a) substrate labeled with donor RSS only, (b) singly labeled substrates with donor RSS only and acceptor RSS only (in trans), (c) substrate labeled with acceptor RSS, (d) substrate labeled in cis with donor and acceptor RSS; and for the following samples that contained added proteins: (e) substrate labeled with donor RSS only, (f) singly labeled substrates with donor RSS only and acceptor RSS only (in trans), (g) substrate labeled with acceptor RSS and (h) substrate labeled in cis with donor and acceptor RSS. Emission spectra collected for the donor RSS alone (no protein) and for the acceptor RSS alone (no protein) provided data used by the Felix software (Photon Technology International) to calculate correction coefficients. The emission spectra shown (e.g. Figure 2) were obtained by subtracting (c) from (d) and (c) from (b) to yield the ‘(d+a)’ and ‘(d) + (a)’ traces; or (g) from (f) and (g) from (h) to yield the ‘(d) + (a) + Proteins’ and ‘(d+a) + Proteins’ traces. The use of subtracted spectra corrects for the residual emission arising from direct excitation of the acceptor.
Figure 2.
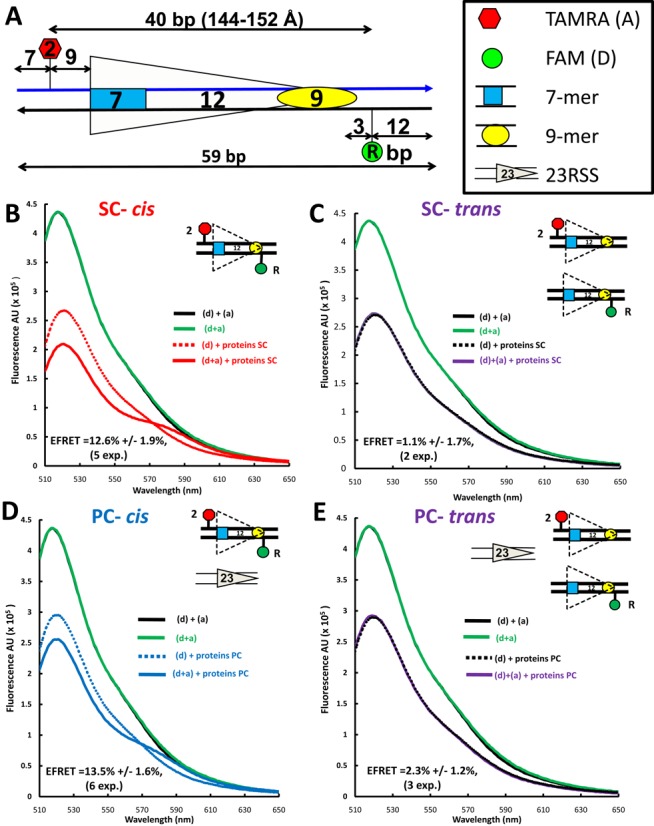
Detection of energy transfer with the 12RSSdR2a substrate. (A) Schematic diagram of the 12RSSdR2a substrate showing the location of the donor and acceptor fluorophores (green and red shapes, respectively), the two DNA strands (blue and black lines) and the heptamer and nonamer (blue rectangle and yellow oval, respectively), separated by a 12-bp spacer. The donor FAM is located at the ‘R’ position in the nonamer flank, while the acceptor TAMRA is located at the ‘2’ position in the coding flank, with distances indicated in bp. (B–E) Representative steady state emission spectra (plotting fluorescence intensity in arbitrary units (AU) against emission wavelength) are shown for the 12RSSdR2a substrate with fluorophores in cis [panels (B) and (D)] or for a mixture of the singly labeled 12RSSdR and 12RSS2a substrates with the fluorophores in trans [panels (C) and (E)] under SC [panels (B) and (C)] or PC [panels (D) and (E)] conditions. The substrates are shown schematically as in Figure 2A. Solid black lines: a mixture of 12RSSdR and 12RSS2a in the absence of protein; green lines: 12RSSdR2a in the absence of protein; dashed lines: 12RSSdR (donor only) in the presence of RAG+HMGB1 without (for SC) or with 23RSS partner DNA (for PC); red line: 12RSSdR2a under SC conditions (RAG+HMGB1); blue line: 12RSSdR2a under PC conditions panel (RAG+HMGB1 and 23RSS partner DNA); purple lines: 12RSSdR + 12RSS2a in the presence of RAG+HMGB1 without (C) or with 23RSS partner (E). Proteins used were MBP-RAG1c, GST-RAG2c and HMGB1.
The energy transfer efficiency (E-FRET) was calculated from emission spectra by the acceptor sensitization method of Fairclough and Cantor (37), as described previously (26,33). Fluorophore to fluorophore (inter-fluorophore) distances for each doubly labeled 12RSS substrate in the PC (rcPC) were calculated based on the average E-FRET for that substrate using R0 = 55 Å for the FAM/TAMRA pair of fluorophores (which assumes a rotational diffusion randomized value of the dipole orientation factor κ2 = 2/3) as described previously (26,33).
Fluorescence correlation data
All fluorescence correlation spectroscopy (FCS) measurements were recorded on a laboratory-built instrument based on an inverted Olympus IX-71 microscope, as described previously (38). Samples were excited using a 488-nm DPSS laser with power adjusted to 5 μW at the microscope incidence. The emitted fluorescence was collected through the objective and separated from laser light by a Z488RDC long-pass dichroic and an HQ500LP long-pass filter (Chroma) before being focused onto the aperture of a 50-μm-diameter optical fiber (OzOptics) directly coupled to an avalanche photo diode (Perkin-Elmer). Autocorrelation curves were generated by a digital correlator (Correlator.com). For each measurement, 10 traces of 10 s each were collected and averaged, then fit to a diffusion equation for a single fluorescent species weighted by the inverse-variance using laboratory-written scripts for MATLAB (MathWorks):
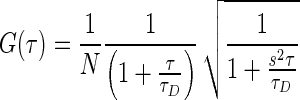 |
in which G(τ) is the autocorrelation as a function of time τ, N is the average number of fluorescent particles in the focal volume and τD is the average diffusion time of those fluorescent particles. The structure factor, s, is the ratio of the radial to axial dimensions of the focal volume, determined experimentally to be 0.2 using solutions of Alexa488 hydrazide dye. While multiple diffusing components were present in most measurements, diffusion times were not separated enough for multi-component fitting of the autocorrelation curves. Therefore, the average diffusion time of all fluorescent species present was used as the output parameter (39).
All samples were placed in eight-well chambered cover glasses (Nunc) passivated by polylysine-conjugated polyethylene glycol to prevent protein adsorption to the surface. For each measurement, Alexa 488-labeled 12RSSdR DNA (5 nM) was incubated with MBP-RAG1c, GST-RAG2c, ± HMGB1 ± 23RSS in BB buffer at a fixed molar ratio of 1:2:1.6:3 for 5 min at 20°C prior to measurements.
Molecular dynamics modeling
The 12RSS-PC/SC models were generated using the calculated interfluorophore FRET distances as the main constraints. The DNA was bent in silico in a two-step procedure similar to that presented in (26). Briefly, in the first step a smooth coarse grained bend was generated by progressively imposing unequal constraints on opposite sides of the linear DNA structure, which minimally affect its local parameters. To minimize departures from the structure of linear B-form DNA, this bend was created by gradually imposing a series of small local changes in tilt and roll parameters of the DNA while blocking base pair openings, extreme changes in propeller twist or buckle parameters as described in (40). The bend created in the first step was consistent with constraints that allowed these parameters to be larger by ∼10% on one side of the helix (residues n, n + 10, etc.) and smaller by ∼10% on the other side of the helix (residues n + 5, n + 15, etc.). The fluorophore-linker moiety was modeled using xLEaP (X Windows based Unit graphical molecule, Atom Properties and a Parmset Editor) Amber software module and Insight II from Accelrys, and was attached to each corresponding base. In the second step, we added the FRET constraints one by one starting with that of the largest linear separation, and continuing the refinement by sequentially imposing the shorter constraints. From each MD (molecular dynamics) step we selected the best frame along the trajectory and used it as an input for the next simulation where we applied an additional FRET-based constraint. The selection involved identifying the frames with least deviation from the imposed constraints and was performed using in-house Tcl/AWK (Tool command language) scripts for the calculation of the RMSD (radius mean square distance) between experimental FRET and MD resulting distances. MD simulations were performed at room temperature in implicit solvent with explicit Mg2+ generated in xLEaP. The linear form of 12RSS was built, based on the nucleotide sequence, using the NAB (Nucleic Acid Builder) module from Amber (41). The trajectories were analyzed using the VMD 1.8.7 software suite (42). The minimization and MD preparation of the input files were performed with xLEaP program from Amber (41). Simulations were performed on a Bull NovaScale R422/R423 high performance computing cluster.
RESULTS
Investigating 12RSS DNA configuration using FRET
In a previous study using an in-solution FRET assay, we investigated the 23RSS configuration in the PC (26). Here we adopted the same methodology to examine and compare the 12RSS structure in the SC and the PC. We designed a series of consensus 12RSS oligonucleotide substrates labeled with a donor (6-carboxyfluoresceine (FAM)) and an acceptor (carboxytetramethylrhodamine (TAMRA)) attached to a DNA base via a C6 methylene linker. The fluorophores were located at various positions relative to one another (shown in the diagram of Supplementary Figure S1). The efficiency of energy transfer (E-FRET) was measured in the absence and presence of proteins and partner 23RSS (to allow either SC or PC formation) and this information was used to calculate the change in interfluorophore distance that occurred as a result of complex formation. In all experiments we used individually expressed MBP-RAG1c (aa 384–1008), GST-RAG2c (aa 1–387) and full-length HMGB1. Fluorophore-labeled 12RSS (15 nM) in the absence (for SC) or presence of a 3-fold molar excess of unlabeled consensus 23RSS partner (for PC) was incubated with MBP-RAG1c (125 nM), GST-RAG2c (250 nM) and HMGB1 (197 nM) (hereafter referred to as the full complement of proteins) in a buffer containing 5-mM Mg2+ (hereafter referred to as the complete reaction). The protein concentrations used were based on the results of optimized conventional RSS mobility shift and cleavage assays (33). Labeled 12RSS DNA substrates were named according to the positions of the fluorophores. The donor position is denoted with a capital letter preceded by ‘d’ and the acceptor by a number followed by ‘a’. The rule is exemplified for the 12RSSdR2a substrate depicted in Figure 2A, which is labeled with the donor at position R and the acceptor at position 2. All fluorophore locations and names for the 12RSS substrates used in this study are shown in Supplementary Figure S1.
Significant RAG-induced DNA bending in 12SC and in the PC
By analogy with a particularly informative 23RSS substrate (23RSSdR2a) used in our previous study (26), we focused initially on a similarly designed substrate (12RSSdR2a) in which the donor lies 3 bp 3′ of the nonamer and the acceptor lies 9 bp 5′ of the heptamer (Figure 2A). In this substrate, the fluorophores are separated by 40 bp (144–152Å in B form DNA, calculated using data from (43)), a distance that exceeds the roughly 90Å limit at which energy transfer is detectable for this pair of fluorophores (26). Indeed, the emission singular spectrum for doubly labeled (fluorophores ‘in cis’) 12RSSdR2a in the absence of protein (Figure 2B-E, green line; indicated as ‘(d+a)’) was almost indistinguishable from those of control reactions containing equimolar amounts of 12RSSdR and 12RSS2a in which the donor and acceptor are on different substrates (fluorophores ‘in trans’; solid black line; indicated as ‘(d) + (a)’). These spectra show the expected emission peak at ∼ 520-nm characteristic of the FAM donor, with no evidence of donor emission quenching or acceptor emission, confirming that the fluorophores on 12RSSdR2a are too widely separated to support energy transfer (background acceptor fluorescence has been subtracted from all spectra as described in the Materials and Methods section).
To determine how the proteins affect donor emission in the absence of the acceptor, the full complement of proteins was added to substrates lacking the acceptor fluorophore (Figure 2B–E, dashed lines; ‘(d) + proteins’). In all cases, protein addition substantially quenched donor fluorescence (compare dashed lines to solid green or black lines). Similar quenching was also observed previously with the 23RSSdR2a substrate and is likely due to direct interaction of DNA bound proteins with the donor (which lies close to the nonamer) (26).
To determine whether energy transfer occurs in the SC or PC, the full complement of proteins was added to doubly labeled 12RSSdR2a in the absence (Figure 2B, solid red line) or presence (Figure 2D, solid blue line) of partner 23RSS. In both cases, donor emission is further decreased and acceptor sensitization (emission peak between 570 and 590 nm) is observed (compare to dashed lines, which represent identical reactions lacking the acceptor). Such spectral changes are directly attributable to the sensitization of the acceptor and indicate that energy transfer is occurring between the fluorophores in both the SC and PC. These changes were observed consistently in independent experiments (n = 5 for SC, n = 6 for PC), and when quantified (see the Materials and Methods section) yielded an average FRET efficiency (E-FRET) of 12.6 ± 1.9% for the SC and of 13.5 ± 1.6% for the PC. Using these E-FRET values and the same approach as in our previous study (26), we calculated the interfluorophore distance of 12RSSdR2a to be 75.9 ± 10 Å in the SC and 74.9 ± 10 Å in the PC.
To confirm that the observed energy transfer was occurring between the donor and acceptor on the same 12RSSdR2a substrate molecule (in cis), rather than by synapsis/aggregation of two (or more) 12RSSdR2a molecules, which would allow energy transfer in trans, the full complement of proteins was added to a mixture (15 nM each) of singly labeled 12RSSdR and 12RSS2a substrates, again in the absence or presence of 23RSS (‘(d)+(a) + proteins’; Figure 2C and E, respectively). The resulting spectra (solid purple lines) were essentially identical to the control reactions lacking the acceptor (dashed lines), demonstrating that there was no energy transfer when the donor and acceptor were on different DNA molecules. Hence, under these conditions, 12/12RSS synapsis/aggregation does not occur to an extent or with a geometry that would allow energy transfer to be detected in trans, and therefore the energy transfer detected with the 12RSSdR2a substrate occurs in cis. We conclude that formation of the 12SC and the PC is accompanied by substantial protein-induced 12RSS bending/distortion that causes a dramatic reduction in the distance between the fluorophores (from ∼145 to 75 Å), much as we observed previously for the 23RSS in the PC (26).
Protein requirements for 12RSS bending in the SC and PC
We next investigated the contribution of the three proteins to the levels of FRET measured with 12RSSdR2a under SC or PC binding conditions. HMGB1 is not essential for the formation of a 12SC that supports nicking, but strongly contributes to 23SC and PC formation and hence to hairpin formation (24). Omission of HMGB1 substantially reduced energy transfer under both SC (Figure 3A) and PC (Supplementary Figure S2A) reaction conditions (compare to Figure 2B and D). In both cases, while no acceptor sensitization was detected, residual donor quenching remained. While other possibilities exist, we interpret this to indicate that the acceptor is sufficiently close to the donor to absorb energy but that some feature of the configuration (presumably related to the nearby protein) prevents excitation of the acceptor dipole to the singlet state, which is required for acceptor emission (36). The level of donor quenching was used to calculate an E-FRET of 6.3 ± 0.5% and 4 ± 0.8% for SC and PC reaction conditions, respectively, with the latter value approaching the background of the FRET assay (2.7 ± 0.2%, the average energy transfer observed in 12 independent experiments on 12RSSdR2a DNA alone). The somewhat lower E-FRET observed under PC conditions most likely reflects reduced levels of 12SC formation due to competition by the 3-fold molar excess of unlabeled 23RSS present in the PC reaction. Overall, these experiments suggest that HMGB1 considerably increases acceptor–donor proximity in 12RSSdR2a SC and is particularly important for allowing acceptor sensitization.
Figure 3.
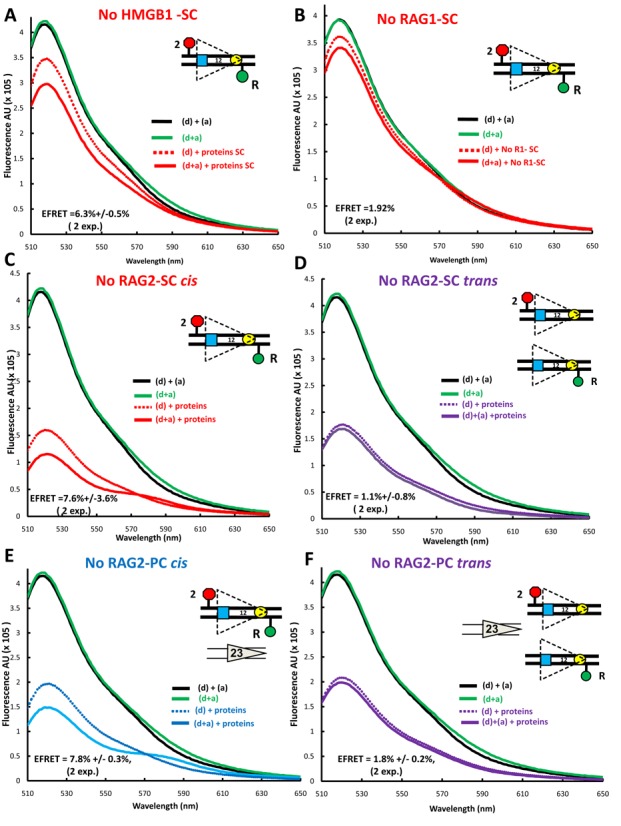
The protein requirements for FRET with the 12RSSdR2a substrate. Steady state emission spectra displayed as in Figure 2 were gathered from reactions similar to those in Figure 2 except that: (A) HMGB1 was omitted from the reaction performed under SC conditions (RAG+HMGB1); (B) RAG1 was omitted from the reaction performed under SC conditions (RAG+HMGB1); (C–F) RAG2 was omitted from reactions with the fluorophores in cis [panels (C) and (E)] or in trans [panels (D) and (F)] performed under SC conditions (RAG+HMGB1) [panels (C) and (D)] or under PC conditions (RAG+HMGB1 and 23RSS partner DNA) [panels (E) and (F)].
Energy transfer was not detectable above background in reactions lacking RAG1 (Figure 3B and Supplementary Figure S2B), an observation consistent with the central role played by RAG1 in RSS binding. In contrast, omission of RAG2 reduced but did not eliminate energy transfer under SC (Figure 3C) and PC (Figure 3E) reaction conditions, consistent with our previous findings with 23RSSdR2a (26). FRET in the absence of RAG2 was manifest by both donor quenching and acceptor sensitization, yielding E-FRET values of 7.6 ± 3.6% and 7.8 ± 0.8% under SC and PC conditions, respectively. Using RSSs in trans, we tested if RAG1 and HMGB1, in the absence of RAG2, are able to synapse/bring together two 12RSSs to support FRET. No evidence of energy transfer was detected in the absence of RAG2 under SC (Figure 3D) or PC (Figure 3F) conditions. Our data show that FRET with the 12RSSdR2a substrate occurs only in cis, is strictly dependent on RAG1, is considerably enhanced by HMGB1 and is optimal in the presence of RAG2.
RAG–12RSS complexes investigated by single molecule FCS
To further characterize RAG-HMGB1-12RSS complexes and provide an additional test for possible trans contributions to the FRET signal, the diffusive properties of the complexes were assessed over a wide range of protein and DNA concentrations by FCS. Using confocal optics to record changes in emission fluorescence in a small, well-defined focal volume, FCS assesses the diffusion properties of fluorescent molecules based on a statistical analysis of the fluctuations of their fluorescence emission. The measurements yield an autocorrelation function from which one can derive diffusion time (related to the size of the complex), and the ‘brightness’, or photon counts per molecule, of the fluorescent species (related to the number of fluorophores per complex). Photon emission counts were recorded from samples containing 5-nM Alexa 488-labeled 12RSSdR and increasing concentrations of the other components needed to generate the 12SC (Figure 4A), the PC (Figure 4B) or the RAG1-RAG2-12RSS complex lacking HMGB1 (Figure 4C). For SC analysis, RAG1, RAG2 and HMGB1 were maintained at a fixed molar ratio of 1:2:1.6, while PC analysis was similar but included a 3-fold molar excess of unlabeled 23RSS with respect to RAG1. Calculated diffusion times and brightness per molecule are shown in the left- and right-hand graphs, respectively, of Figure 4 and are plotted with respect to RAG1 concentration.
Figure 4.
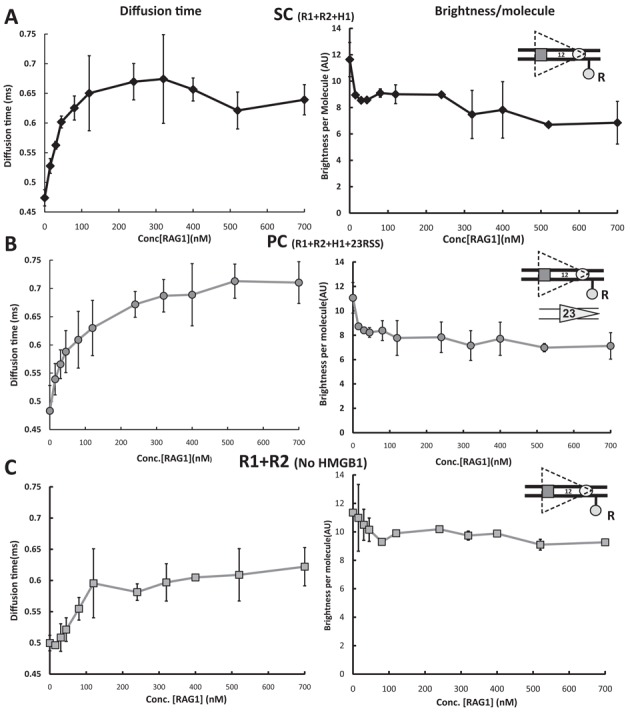
RAG/HMGB1 binding to the 12RSS analyzed by fluorescence correlation spectroscopy (FCS). Photon emission traces were recorded from samples containing 5 nM Alexa 488-labeled 12RSSdR and increasing concentrations of protein mixtures in a fixed molar ratio as follows: (A) 12SC formation, using MBP-RAG1c, GST-RAG2c, HMGB1 in a molar ratio of 1:2:1.6 (black diamonds connected with black lines), (B) PC formation, using MBP-RAG1c, GST-RAG2c, HMGB1 and 23RSS in a molar ratio of 1:2:1.6:3 (gray circles connected with gray lines), (C) 12RSS–RAG complex, using MBP-RAG1c and GST-RAG2c in a molar ratio of 1:2 (gray squares connected with gray lines). Right-hand panels show brightness, or photons per labeled DNA molecule, plotted against RAG1 concentration, whereas left-hand panels show diffusion time versus RAG1 concentration. In (A) and (C), each data point represents the average of three independent measurements, whereas in (B), the average comes from six independent measurements (error bar = standard deviation).
For the 12SC, the diffusion time (and hence average complex size) of the labeled DNA rose rapidly with increasing concentrations of the proteins (RAG1 concentrations less than 100 nM) and reached a plateau of ∼0.65 ms at ∼200 nM RAG1 (Figure 4A, left panel). Diffusion times were stable at RAG1 concentrations from 200–700 nM, arguing against the formation of larger complexes or aggregates even at quite high protein concentrations. With inclusion of the 23RSS (PC conditions), the rise in diffusion time was somewhat more gradual but reached a higher saturation value of ∼0.73 ms (Figure 4B, left panel). The slower rise is likely due to the presence of the 23RSS (which competes with the labeled 12RSS for protein binding), while the higher saturation value presumably reflects the larger size of the PC relative to the SC. In the absence of HMGB1, the increase in diffusion time with increasing RAG concentration was more gradual still (Figure 4C, left panel), suggesting lower affinity binding of RAG to the 12RSS than occurs in the presence of HMGB1, and the saturation value of the diffusion time (approx. 0.6 ms) was lower, reflecting the smaller complex formed.
Importantly, brightness measurements in all cases showed no evidence for synapsis or aggregation of two or more 12RSSdR substrate molecules, which would be manifest as an increase in fluorescence per molecule; instead, brightness declined sharply at lower concentrations of the added components and then remained stable at higher concentrations (Figure 4A–C, right panels). The initial decrease in photon counts per molecule is consistent with the donor quenching observed upon protein addition in the FRET experiments (Figure 2B and D and Figure 3A), and with the results of similar FCS experiments with a labeled 23RSSdR substrate (26). Such quenching could obscure a low level of 12RSSdR substrate synapsis/aggregation, but the absence of any detectable rise in brightness per molecule at high concentrations of the components argues that this is rare if it happens at all. We also note that the concentrations used in the FRET reactions (125/250/197 nM for RAG1, RAG2 and HMGB1, respectively) are below the concentrations required to reach plateau levels of diffusion time, indicating that the FRET assays were performed under non-saturating, dynamic equilibrium conditions. In summary, the FCS experiments provided no evidence for 12RSS synapsis/aggregation and further supported the conclusion that in the FRET assays, most complexes contain only a single labeled 12RSSdR2a substrate and hence that the energy transfer detected occurs in cis.
Mapping distinct 12RSS bending in the paired and signal complex
Our energy transfer data argue that the 12RSS bends similarly in the SC and PC so as to reduce the interfluorophore distance of 12RSSdR2a from ∼150 Å in free DNA to ∼75 Å in these complexes. Such bends might reflect a single large bend or the additive effect of multiple bends/distortions, and while the overall bend appears similar in the SC and PC, there might be subtle differences in the shape/trajectory of the 12RSS in these two complexes. To investigate these issues, we extended the FRET strategy described for 12RSSdR2a to a collection of additional DNA substrates in which the fluorophores were positioned at a variety of positions along the 12RSS substrate (see Supplementary Figure S1 for the location of each fluorophore labeling position). Reactions were performed under the same SC and PC conditions described above for 12RSSdR2a (e.g. Figure 2B and D) and the results obtained are shown for the PC in Figure 5 (16 fluorophore pairs) and for the SC in Figure 6 (11 pairs). In the PC, fluorophores were positioned to report the 12RSS DNA configuration from four regional perspectives: spanning or flanking the nonamer (Figure 5, substrates 1–3), in different portions of the spacer (substrates 4 and 5), spanning or flanking the heptamer (substrates 6–13) and spanning the entire RSS (substrates 14–16). The same four regions were also examined in the SC, although with fewer substrates (Figure 6). In Figures 5 and 6, columns B and D display the E-FRET values measured in the absence or presence of proteins, respectively, while columns C and E show the interfluorophore distances calculated from these measurements. We also calculated the predicted interfluorophore distances in native DNA according to (43,44) (Figures 5 and 6, column C, gray text) (see the Materials and Methods section).
Figure 5.
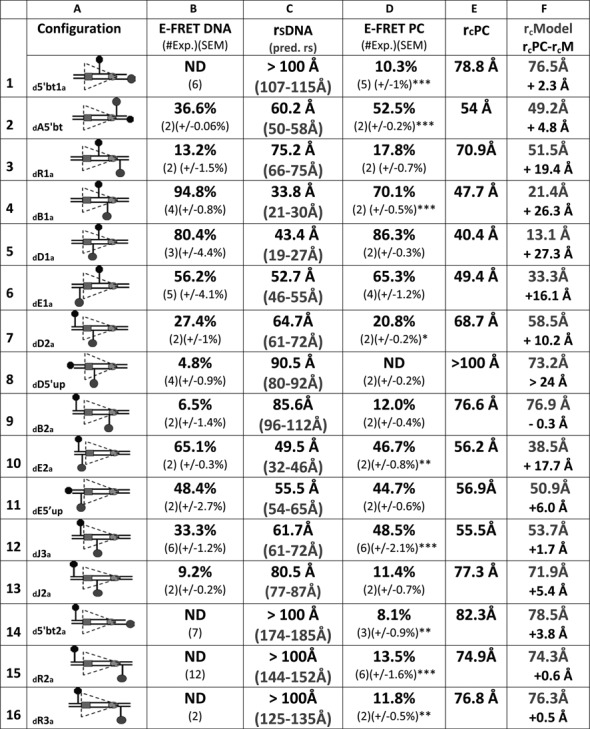
FRET analysis of the 12RSS in the PC. Data for 16 fluorophore-labeled substrates under PC conditions; in each panel, the columns designate the following features: column (A), schematic depiction of the substrates as in Figure 2; column (B), average energy transfer efficiency for substrate in the absence of protein (E-FRET DNA), with the number of independent experiments and SEM in parentheses (ND, no energy transfer above background); column (C), calculated distance between the donor and acceptor fluorophores in the substrate in the absence of protein (rsDNA) based on the E-FRET in column (B), with the predicted interfluorophore distance in the DNA (pred. rs) in gray in parentheses; column (D), average energy transfer efficiency for substrate in the complete reaction E-FRET PC, with the number of independent determinations and SEM in parentheses. Statistical comparison of E-FRET PC versus E-FRET DNA: *P < 0.05; **P < 0.01; ***P < 0.001; column (E), calculated distance between the donor and acceptor fluorophores in the substrate in the PC reaction rcPC, based on the E-FRET in column (D); column (F), interfluorophore distances in the PC models shown in Figure 7 of the 12RSS (rcModel, gray), estimated by measuring the distance between the 5′ carbons of the sugars of the two fluorophore-labeled nucleotides. Below (black), interfluorophore distance derived experimentally minus the interfluorophore distance in the model for the PC (rcPC-rcM).
Figure 6.
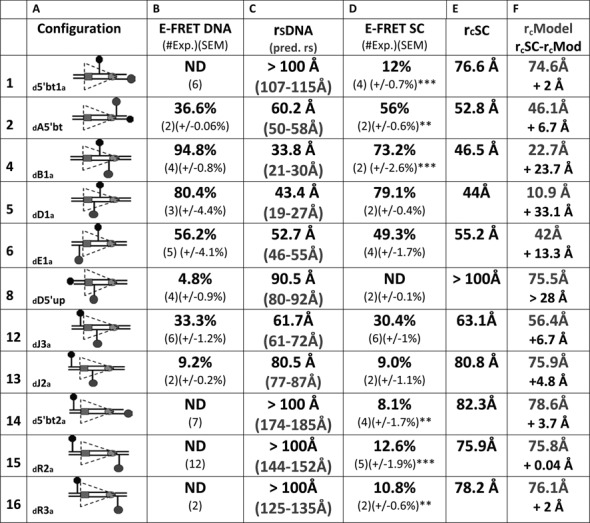
FRET analysis of the 12RSS in the12SC. Data for 11 fluorophore-labeled substrates under SC conditions. Columns (A)–(C) designate the same features described in the Figure 5 legend. Column (D), average energy transfer efficiency for substrate in the complete reaction E-FRET SC, with the number of independent determinations and SEM in parentheses. Statistical comparison of E-FRET SC versus E-FRET DNA: *P < 0.05; **P < 0.01; ***P < 0.001; column (E), calculated distance between the donor and acceptor fluorophores in the substrate in the SC reaction rcSC, based on the E-FRET in column (D). Column (F), interfluorophore distances in the 12SC models shown in Figure 7 of the 12RSS (rcModel, gray), estimated by measuring the distance between the 5′ carbons of the sugars of the two fluorophore-labeled nucleotides. Below (black), interfluorophore distance derived experimentally minus the interfluorophore distance in the model for the 12SC (rcSC- rcM).
For most substrates, formation of the 12SC or PC significantly altered the level of energy transfer detected relative to that obtained with naked DNA (Figures 5 and 6, compare columns B and D; statistical significance indicated in column D with asterisks). Two of the substrates with fluorophores flanking/adjacent to the nonamer (substrates 1 and 2) showed large but similar increases in FRET in the SC and PC relative to naked DNA, and FRET levels with the third substrate in this class (substrate 3, investigated only in the PC) also increased, but not significantly, upon complex formation. These data support bending in the vicinity of the 12RSS nonamer.
Substrates 4 and 5, with fluorophores in the spacer, behaved similarly in the SC and PC, with substrate 4 (but not 5) exhibiting a significant change in energy transfer in the complexes relative to naked DNA (Figures 5 and 6). Because of the short distance between donor and acceptor in these substrates, these data should be interpreted cautiously (see below), but they are consistent with bending/distortion in the spacer.
For substrates 6–13 with fluorophores flanking/adjacent to the heptamer, significant changes in FRET levels compared to naked DNA were seen with three of them (numbers 7, 10 and 12) and only in the PC (although of these three, only number 12 was examined in both the SC and PC). Substrate 12 (12RSSdJ3a) exhibited a significant increase in FRET in the PC but not the SC, resulting in significantly higher levels of energy transfer in the PC versus the SC (paired t test PC versus SC P = 0.0017) (representative energy transfer traces shown in Supplementary Figure S3). A similar phenomenon was observed with substrate 6 (12RSSdE1a), where energy transfer (relative to naked DNA) decreased in the SC but increased in the PC; while neither of these changes was statistically significant, energy transfer in the PC was significantly higher than in the SC (paired t test PC versus SC P = 0.0045). In substrates 6 and 12, the fluorophores span the heptamer and are positioned similarly, with one located near the middle of the spacer and the other immediately flanking the heptamer (Supplementary Figure S1). In both cases, the fluorophores are separated by about 1.5 helical turns (14 and 17 bp in substrates 6 and 12, respectively), placing the donor and acceptor on nearly opposite sides of the helix. Notably, the interfluorophore distance in these substrates (∼50–60 Å in naked DNA) is one in which FRET with these fluorophores is quite sensitive. These results suggest a markedly shorter distance between the center of the spacer and the immediate heptamer flank of the 12RSS in the PC as compared to the SC. In substrate 13 (12RSSdJ2a), where the acceptor is moved 5 bp further from the heptamer (Supplementary Figure S1) and to the opposite side of the helix relative to substrate 12 (12RSSdJ3a), no significant difference in energy transfer was observed between the SC, PC and naked DNA (Figures 5 and 6). This, and the results with substrate 8 (no significant FRET in any condition), suggests that as one fluorophore moves further into the coding flank and away from the vicinity of the heptamer, the FRET assay becomes less sensitive to the differences between the SC and PC. Finally, we note that for substrate 7 (12RSSdD2a), in which the fluorophores also span the heptamer, substantial quenching of both donor and acceptor was observed under PC conditions (data not shown), suggesting strong protein interactions with both fluorophores. It is unclear how to interpret the decrease in energy transfer observed in the PC relative to naked DNA in this substrate (Figure 5).
All three substrates with fluorophores flanking the entire RSS (numbers 14–16) exhibited equivalent levels of energy transfer in the SC and PC, which in all cases were significantly above that of naked DNA (Figures 5 and 6). These findings demonstrate that in three different substrates in which the fluorophores are separated by at least 125 Å in linear DNA, formation of the 12SC and PC leads to clearly detectable energy transfer and a calculated interfluorophore separation of less than 80 Å. This provides strong support for the conclusion that the 12RSS undergoes a major bend in the 12SC and PC.
Might the degree of bending be reduced if additional flanking DNA were present in our 12RSS substrates (perhaps due to charge repulsion of the two DNA backbones)? To examine this, we extended the length of the 12RSSdR2a reporter substrate from 59 to 81 bp and repeated the FRET experiments. E-FRET values were not reduced with the longer substrate; if anything, they were slightly increased (Supplementary Figure S4). Hence, addition of an additional helical turn of DNA at each end of the substrate did not interfere with RAG/HMGB1 binding or reduce the apparent extent of bending. Instead, it is possible that the additional DNA allows for additional protein–DNA contacts and somewhat increased bending.
In summary, our detailed FRET analysis of the 12RSS suggests a local reconfiguration of the DNA in the vicinity of the heptamer during the 12SC to PC transition, and indicates a large degree of global DNA bending that brings the flanking DNA (coding flank and nonamer flank) much closer together in the protein–DNA complexes than in the naked DNA substrate.
Molecular dynamics modeling of the 12RSS in the SC and PC
The calculated interfluorophore distances resulting from the energy transfer experiments (Figures 5 and 6, column E) were used as the constraints for Molecular Dynamics modeling of the structures of the 12RSS in the SC and the PC, using an approach similar to that we used previously to derive a structural model of the 23RSS in the PC based on FRET measurements (26). First, a large bend was introduced into the DNA, and this was then subjected to the FRET constraints, a process that was performed independently for the 12RSS in the SC and PC. The models were refined to maximize conformity with the experimental data and to minimize energy penalties and structural deviations from B form DNA in the final structures (see the Materials and Methods section).
Both of the resulting models show a strongly bent V-shaped DNA molecule with the spacer occupying the center of the bend and the heptamer/coding flank and nonamer/nonamer flank constituting their arms (Figure 7A for 12SC and Figure 7B for PC; Supplementary data files sc12RSS.pdb, pc12RSS.pdb). When viewed from the ‘front’ (Figure 7A and B), the shape of the DNA in the two models is generally similar, with the overall large bend arising from a series of smaller, distinct bends, the two most prominent of which are located near the heptamer/spacer border and the spacer/nonamer border. Interestingly, protein–DNA backbone interactions in the 12SC (9) map to the inner (concave) surface and one face of the spacer in the 12SC model (Figure 7A, yellow), whereas a DNase I hypersensitive site maps to the outer (convex) surface of the DNA at the spacer/nonamer border, precisely at the site of the sharpest bend in the model (pink arrow). The front view also reveals two differences between the models. First, the opening between the heptamer and nonamer arms is slightly wider in the 12SC than in the PC model. Second, the spacer is rotated almost one half helical turn in the 12SC compared to the PC model, visible as distinct succession of major and minor grooves that face the viewer in each structure.
Figure 7.
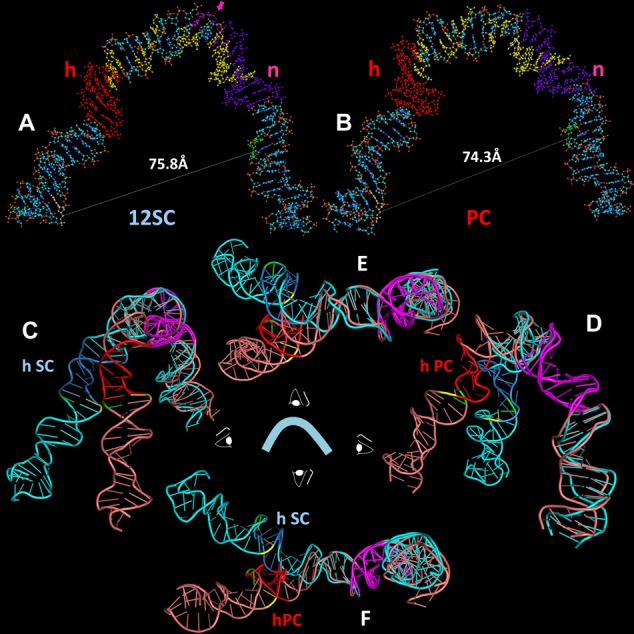
Models of the structure of the 12RSS in the (A) SC and (B) PC, derived from molecular dynamics modeling of the data of Figures 5 and 6. DNA is depicted as a ball and stick model with N in blue, O in red, C in gray, P in orange and H in white. Indicated are the heptamer (h), shaded orange, nonamer (n), shaded purple, the bases that are donor (green) and acceptor (yellow) fluorophore labeled in the 12RSSdR2a substrate, the summative sites of ethylation interference in the RAG1-RAG2-HMGB1–12RSS (12SC and PC)complexes (yellow) and the site of DNAse I hypersensitivity (pink, with arrow) in the 12SC, as determined by Swanson (24). (C–F) Ribbon diagrams showing a superposition of the models of the 12RSS in the SC and the PC from four orthogonal perspectives of the ‘front’ view in (A) and (B), with the SC in blue (heptamer in dark blue) and PC in pink (heptamer in red). The nonamers in both models are in purple and were aligned to create the superposition. Also indicated are the scissile phosphates for nicking (green) and hairpin formation (yellow). (C) View from the left (heptamer/coding flank arm), (D) view from the right (nonamer arm), (E) view from above and (F) view from below.
Additional differences are visible when the models are viewed from other angles. Because the FRET data suggested structural differences in the vicinity of the heptamer, we chose to illustrate the differences between the two models by aligning their nonamers (maximizing backbone and base overlap). The result of this superposition is shown in four orthogonal views in Figure 7C–F (blue, 12SC model; pink, PC model; heptamers in dark blue and red, respectively; nonamers in purple). The most striking feature to emerge is a >400 angle of divergence of the heptamer arms of the two models, most clearly seen in the top and bottom views (Figure 7E and F, respectively). This results in the scissile phosphates (yellow, nicking; green, hairpin formation) being located about 25 Å from one another. In addition, while the 12RSS DNA in the PC is largely coplanar (similar to our model of the 23RSS in the PC; (26)), in the 12SC, the heptamer and nonamer arms deviate from a single plane. These differences arise largely from structural alterations in the vicinity of the heptamer and differential DNA unwinding in the spacer, as reported by the substrates with fluorophore pairs flanking the heptamer, particularly 12RSSdE1a and 12RSSdJ3a.
How well do the models fit the FRET data? To assess this, it was necessary to determine the interfluorophore distances predicted by the model, a task complicated by the flexible six carbon methylene linker that connects each fluorophore to the DNA. This linker is predicted from molecular dynamics simulations to result in a 13–14 Å separation between the fluorophore and its attachment point on a DNA base (see (26)), presumably with the linker projecting outward from the DNA axis. As in our previous study (26), we calculated the interfluorophore distances in the models using the 5′ carbons of the sugars of the two fluorophore-labeled nucleotides to approximate the fluorophore positions. This almost certainly underestimates the distance of fluorophores from the DNA axis because the 5′ carbon of the sugar is only 5.5 Å from the linker attachment point on the base, compared to the 13–14 Å predicted for the linker. This needs to be taken into account when comparing the estimated interfluorophore distances in the models (Figures 5 and 6, column F, rcModel, gray text) to the distances calculated from the FRET data (column E, rcPC and rcSC).
Several trends are clear in this comparison. First, the models almost invariably yield smaller distances than do the data (the calculated difference, in black text in column F, is almost always positive). This is likely due to our use of the 5′ carbon sugar to approximate the fluorophore position, as discussed above. Second, the agreement between model and data is excellent (less than 4 Å) for substrates 14–16 with fluorophores flanking the entire 12RSS. This is perhaps not surprising given the importance of these measurements in determining the large bend that was introduced into the DNA during the first step of the modeling. Third, the models match the data fairly well (within 7 Å) for substrates 1 and 2 with fluorophores flanking or adjacent to the nonamer (although less well for substrate 3, which is also in this category), while agreement is more heterogeneous for substrates 6–13 with fluorophores flanking/adjacent to the heptamer. And fourth, the distances calculated from the data greatly exceed (by >20 Å) those calculated from the models for the two substrates (numbers 4 and 5) where the fluorophores are located very close to one another; in these cases, it is likely that the C6 linkers make a large contribution to the separation between the fluorophores that is not taken into account by the models.
Contributions of partner heptamer and nonamer to 12RSS bending in the PC
Our data, particularly from substrates 6 and 12, and the models suggest a structural change in the 12RSS in the vicinity of the heptamer upon synapsis with a partner 23RSS. What are the relative contributions of the 23RSS heptamer and nonamer to this? To address this, we carried out a new set of FRET measurements with substrates 6 and 12 (12RSSdE1a and 12RSSdJ3a, respectively) either alone, in the SC, in the PC (intact 23RSS partner), or with a 23RSS partner with a scrambled heptamer (5′-CACAGTG changed to 5′-TGAATAC) or a crippled nonamer (5′-ACAAAAACC changed to 5′-ACAAGTCCC). The mutant nonamer partner had little or no effect on the 12RSS, leaving E-FRET similar to that observed in the SC (Figure 8A and B), as expected (26,28,45) given the importance of the nonamer for stable DNA binding. In contrast, the scrambled heptamer partner supported a substantial increase in energy transfer over that observed in the SC for both substrates, although in neither case did E-FRET values reach those obtained with the intact 23RSS partner (Figure 8A and B). Hence, engagement of a partner nonamer, in the absence of a heptamer, appears sufficient to trigger a change in the 12RSS, but additional structural alterations appear to take place when a partner heptamer can also be engaged.
Figure 8.
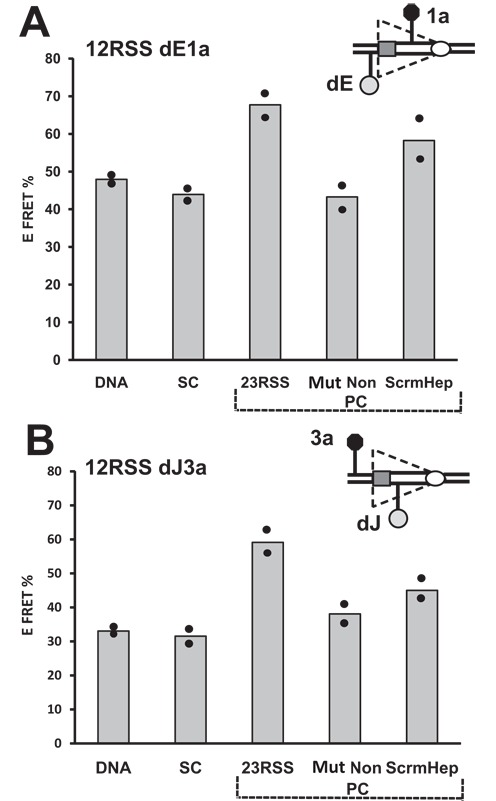
Energy transfer changes in the 12RSS during the SC to PC transition are sensitive to the integrity of the 23RSS heptamer and nonamer. Bars represent the average energy transfer efficiency from two independent measurements, with dots indicating each experimental value and the content of the reaction indicated below each bar. The free DNA, SC and 23RSS partner (PC) reactions were performed as with the 12RSSdR2a substrate in Figure 2B and D. Reactions were also performed with a partner 23RSS with a mutant nonamer (Mut Non) or a scrambled heptamer (ScrmHep). (A) 12RSSdE1a substrate. (B) 12RSSdJ3a substrate.
Together, our data and the models strongly support the existence of a large overall bend in the 12RSS in both the SC and PC, and suggest significant structural differences in the DNA in these two complexes. However, the results also suggest that the models need to be interpreted with caution, as discussed below.
DISCUSSION
The 12/23 rule illustrates a fundamental preference on the part of the RAG proteins for substrate asymmetry in assembling a cleavage-competent synaptic complex. The underlying basis for this preference is unknown. A central motivation for the current work was to address the question of whether this preference for asymmetry is reflected by significant differences in the structure of the two substrates in the PC. While our previous study showed that the 23RSS is strongly bent in the PC (and likely also the SC), it was possible that the 12RSS was configured differently, perhaps substantially less bent because of its shorter spacer. The results presented here argue that this is not the case, indicating instead that the overall shape of the 12RSS and 23RSS in RAG–DNA complexes is similar (Figure 9). Our data also argue that the shape of the 12RSS in the SC and PC is generally similar, and demonstrate that the large bend in the 12RSS arises through the combined action of RAG and HMGB1, with RAG1 playing a particularly critical role. In the absence of high-resolution structures of RAG-HMGB1-DNA complexes, our data provide the most detailed information yet about RSS structure and provide guidance in addressing some of the longstanding questions in the field about the formation and organization of these complexes and the 12/23 rule.
Figure 9.
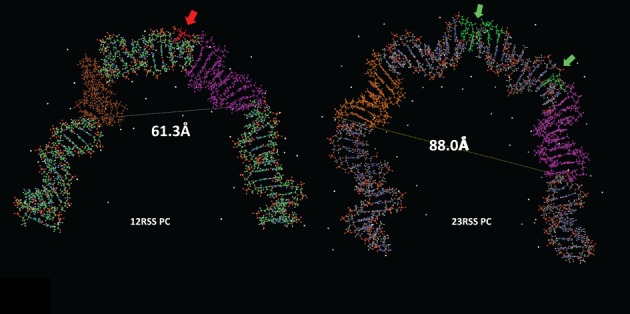
Front view of the models of the (left) 12RSS and (right) 23RSS in the PC. DNA is depicted as a ball and stick modelwith N in blue, O in red, P in orange, and H in white, and C in green for the 12RSS and grey for the 23RSS, the nonamer in purple, and the heptamer in brown (12RSS) or orange (23RSS). Sites of DNAse I hypersensitivity are indicated with arrows and shaded red (12RSS) or green (23RSS) (24,27). The distances between the scissile phosphate for nicking and the distal end of the nonamer (dashed lines) are indicated, 23RSS model from (26).
Similar large bends in the 12RSS in the SC and PC
Previous footprinting studies revealed strong similarities between the protein–12RSS contacts in the 12SC and PC (24), and our data provide evidence for relatively similar, large bends in the 12RSS in both the SC and the PC. The most important evidence for this comes from the energy transfer efficiencies observed for three independent substrates with fluorophores spanning the entire RSS (Figures 5 and 6, substrates14–16). The data strongly suggest that the fluorophores in these substrates, which are more than 120 Å apart in the free DNA, are separated by less than 80 Å in the complexes. Critical to this conclusion is the assumption that the energy transfer we detect occurs in cis (between two fluorophores on a single 12RSS molecule) and not in trans (between fluorophores on different 12RSS molecules). Numerous controls, both here and even more extensively in our previous study (26), validate this assumption: in no case have we detected energy transfer between fluorophores located on two different 12RSS or two different 23RSS molecules. We conclude that, under our reaction conditions, 12–12 and 23–23 RSS synapsis/aggregation do not take place in such a way as to yield detectable energy transfer, and hence that the energy transfer we do detect occurs between RSSs in cis.
Interestingly, while the 23RSSdR2a substrate studied previously (26) yielded half as much energy transfer in the 23SC as in the PC (8.8% versus 18%), for 12RSSdR2a, the difference between the two complexes was less remarkable (12.6% in 12SC versus 13.5% in the PC). Fluorophore positioning in these two substrates is similar, with donor and acceptor lying 3 and 9 bp from the nonamer and heptamer, respectively. This observation argues for less dramatic structural changes in the 12RSS than in the 23RSS during the SC to PC transition, at least as reported by this pair of fluorophores.
Previous AFM studies indicated considerable DNA bending in both the SC and the PC (31,32), although the magnitude of bending was estimated to be substantially less than what we found here or in our previous study of the 23RSS (26). Lengthening our DNA substrate so as to have coding and nonamer flanks of 27 and 26 bp, respectively, did not reduce E-FRET and hence did not appear to reduce bending. We cannot rule out the possibility that bending is reduced in RAG–RSS complexes formed on much longer DNA substrates. Other issues related to prior studies of DNA bending in RAG–RSS complexes have been discussed previously (26).
Several features of our FRET assay and molecular modeling place limits on the conclusions that can be drawn from our study; these have been discussed in detail previously (26). We note here that interactions with the proteins or DNA can alter fluorophore position in a manner that affects energy transfer. In particular, caution should be used when interpreting data derived from substrates with fluorophores attached to the end of the DNA, which when tethered by flexible linkers have strong tendency to stack onto the adjacent bases (46,47). This effect can influence the orientation term k2 between the transition dipoles of the fluorophores (46,48), resulting in E-FRET values different from those calculated with k2 = 2/3, which assumes freely rotating fluorophores (see the Materials and Methods section). We emphasize that the models that arise from our analysis should be considered working models and should not be interpreted as providing high-resolution molecular information regarding the structure of the 12RSS in RAG–DNA complexes.
Protein requirements for 12RSS bending
Previous studies have demonstrated that RAG is able to bind and nick a 12RSS substrate in the absence of HMGB1 more robustly than a 23RSS substrate (22,24,49), and our FCS data confirmed substantial RAG binding to our 12RSS substrate without added HMGB1 (Figure 4C). Interestingly, the 12RSSdR2a substrate exhibited evidence of RAG-dependent energy transfer in the absence of HMGB1 (Figure 3A), although at a reduced efficiency (6.3% versus 12.6% with the full complement of proteins) and without acceptor sensitization. Such energy transfer implies that the fluorophores are less than 90 Å apart, a large change from the 145–150 Å in the naked DNA substrate. We tentatively conclude that RAG is capable of strongly bending the 12RSS in the absence of HMGB1. Given our inability to detect RAG-dependent energy transfer with the analogous 23RSSdR2a substrate when HMGB1 was omitted (26), we speculate that such 12RSS bending helps establish a DNA configuration compatible with nicking.
RAG1 and HMGB1, in the absence of RAG2, were sufficient for substantial energy transfer with the 12RSSdR2a substrate (Figure 3C and E), a finding that parallels prior results with the 23RSSdR2a substrate (26). Curiously, the magnitude of FRET with RAG1 and HMGB1 was similar for the various substrates/conditions tested: 7.4% for 23RSSdR2a in the PC, 7.6% for 12RSSdR2a in the SC and 7.8% for 12RSSdR2a in the PC. This might reflect a stereotypical bend induced in RSS DNA by RAG1 and HMGB1 in the absence of RAG2. We emphasize however, consistent with our previous findings for the 23RSS in the PC (26), that optimal energy transfer occurs only with the full complement of proteins, conditions compatible with catalysis.
Consequences of 12RSS bending in the SC versus the PC
Given the substantial evidence in favor of DNA distortion near the site of RAG-mediated cleavage (1,24), including base flipping associated with hairpin formation (50), it is intriguing that differences in energy transfer that we observe between the SC and PC occurred when the fluorophores flanked the heptamer (Figures 5 and 6, substrates 6 and 12 and Supplementary Figure S3). If the nonamer remains stationary during the 12SC to PC transition (perhaps because it is anchored to the stable, dimeric NBD (28)), then our models suggest a substantial pivoting of the heptamer and coding flank during synapsis (Figure 7C–F). This is predicted to swing the heptamer arm out of the plane defined by the nonamer and nonamer flank and to move the scissile phosphate bonds (marked in yellow and green) substantially (about 30 Å in the models). While specific details cannot be inferred, it is tempting to think that this pivoting is a reflection of structural changes in the DNA surrounding the site of cleavage that establish a hairpin-competent conformation.
A structural change in the 12RSS that is intimately linked to hairpin formation should require both the nonamer and heptamer of the partner 23RSS to enable sensing of spacer length. We found that a partner containing only a nonamer was able to trigger an increase in E-FRET as reported by 12RSS substrates with fluorophores flanking the heptamer, although this increase was not as large as with an intact 23RSS partner (Figure 8). Perhaps relevant to this is our prior observation that RAG/HMGB1 induce strong bending in a mutant 23RSS substrate containing a nonamer but no heptamer, although again, not to the full extent observed with an intact 23RSS (26). This leads us to speculate that capture and bending of a nonamer-only partner by the 12SC is sufficient to induce a ‘partial’ conformational change in the 12RSS, whereas an intact 23RSS partner, upon capture, undergoes a further degree of bending and induces the full extent of 12RSS structural alteration present in the PC. It is plausible that this is relevant to establishing a hairpin-competent conformation in the 12RSS, although further experiments will be needed to test this. An alternative interpretation is that the smaller increase in E-FRET induced by the nonamer-only partner compared to the 23RSS partner reflects less efficient PC formation in the absence of the heptamer. We think this is unlikely because in an earlier study, we found no significant drop in synapsis (as reported by FRET) upon scrambling of the heptamer (28).
12RSS and 23RSS bending in the PC
Comparison of the models of the 12RSS (Figure 7B) and the 23RSS (26) in the PC reveals broadly similar shapes, but with several distinctive features (Figure 9). Whereas the 23RSS is U shaped, the 12RSS more closely resembles a V, a difference that arises from different numbers and locations of bends. The bend found in the center of the 23RSS spacer (which corresponds closely to a DNAse I hypersensitive site) is absent in the 12RSS spacer, which instead is approximately straight. As with the 23RSS model, a well-documented DNAse I hypersensitive site maps quite close to the largest DNA bend in the 12SC model (Figure 7A). Substantial bends are found at each end of the spacer in both models, but while the 23RSS is bent at the heptamer/coding flank border, the 12RSS is instead bent at the nonamer/nonamer flank border. Despite the bend it contains, the longer spacer of the 23RSS results in a larger central cavity than is found in the 12RSS model, with the heptamer and nonamer modeled to be considerably farther apart in the 23RSS than in the 12RSS (distances from site of cleavage to two positions in the nonamer are indicated in Figure 9). We previously proposed that the RAG proteins occupy the central cavity of 23RSS based on the fact that protein–DNA backbone contacts map largely on the concave (inner) surface of the model (26). In contrast, such contacts map to the inner surface and one side of the 12RSS in the model (Figure 7B). It seems likely that the cavity in the 12RSS structure would not accommodate the proteins in the same manner as the more spacious 23RSS cavity. In particular, the greater distance between the nonamer and heptamer elements predicted in the 23RSS would require a different disposition of heptamer and nonamer binding regions than would be possible with the 12RSS, an asymmetry that we propose is pertinent to the 12/23 rule. Indeed, a similar concept was proposed previously by Jones and Gellert (51), who suggested that the heptamer and NBDs are organized in a spatially distinct manner on the two RAG protomers in the PC: in one, they are relatively close together, suitably spaced for 12RSS recognition (‘12RSS protomer’) while in the other, they are farther apart so as to accommodate the 23RSS (‘23RSS protomer’). Our models are consistent with this notion, as well as with their proposal that while the 12RSS could not be made to accommodate a 23RSS protomer (due to the fixed length of the spacer), the 23RSS could, through additional bending in the spacer, accommodate the 12RSS protomer. It is appealing to think that the distinct shapes of the 12RSS and 23RSS predicted by our models, particularly the unique bend in the center of the 23RSS spacer, reflect mechanistic aspects of the 12/23 rule and help explain situations in which this rule is broken.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank N. Grindley, Scott Strobel and A. Miranker for generously providing access to their fluorometers. They would also like to thank J. Banerjee for helpful discussions and suggestions in writing the manuscript.
FUNDING
National Institutes of Health [AI32524 to D.G.S.; NS079955 to E.R.]; Agentia Nationala pentru Cercetarea Stiintifica (ANCS) [PN-II-ID-PCCE-2012-2-0024 to M.C.]; American Heart Association [to L.A.M.]; Romanian Academy [ANCS Grant ID 3-0342-181/2011 and POSDRU/89/1.5/S/60746 Program to A.J.P., M.D.S.]. Funding for open access charge: National Institutes of Health [AI32524].
Conflict of interest statement. None declared.
REFERENCES
- 1.Schatz D.G., Swanson P.C. V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- 2.Lieber M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuomo C.A., Oettinger M.A. Analysis of regions of RAG-2 important for V(D)J recombination. Nucleic Acids Res. 1994;22:1810–1814. doi: 10.1093/nar/22.10.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadofsky M.J., Hesse J.E., McBlane J.F., Gellert M. Expression and V(D)J recombination activity of mutated RAG-1 proteins. Nucleic Acids Res. 1993;21:5644–5650. doi: 10.1093/nar/21.24.5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadofsky M.J., Hesse J.E., Gellert M. Definition of a core region of RAG-2 that is functional in V(D)J recombination. Nucleic Acids Res. 1994;22:1805–1809. doi: 10.1093/nar/22.10.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailin T., Mo X., Sadofsky M.J. A RAG1 and RAG2 tetramer complex is active in cleavage in V(D)J recombination. Mol. Cell. Biol. 1999;19:4664–4671. doi: 10.1128/mcb.19.7.4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciubotaru M., Ptaszek L.M., Baker G.A., Baker S.N., Bright F.V., Schatz D.G. RAG1-DNA binding in V(D)J recombination. Specificity and DNA-induced conformational changes revealed by fluorescence and CD spectroscopy. J. Biol. Chem. 2003;278:5584–5596. doi: 10.1074/jbc.M209758200. [DOI] [PubMed] [Google Scholar]
- 8.Rodgers K.K., Villey I.J., Ptaszek L., Corbett E., Schatz D.G., Coleman J.E. A dimer of the lymphoid protein RAG1 recognizes the recombination signal sequence and the complex stably incorporates the high mobility group protein HMG2. Nucleic Acids Res. 1999;27:2938–2946. doi: 10.1093/nar/27.14.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swanson P.C. A RAG-1/RAG-2 tetramer supports 12/23-regulated synapsis, cleavage, and transposition of V(D)J recombination signals. Mol. Cell. Biol. 2002;22:7790–7801. doi: 10.1128/MCB.22.22.7790-7801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanson P.C., Desiderio S. RAG-2 promotes heptamer occupancy by RAG-1 in the assembly of a V(D)J initiation complex. Mol. Cell. Biol. 1999;19:3674–3683. doi: 10.1128/mcb.19.5.3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fugmann S.D., Schatz D.G. Identification of basic residues in RAG2 critical for DNA binding by the RAG1-RAG2 complex. Mol. Cell. 2001;8:899–910. doi: 10.1016/s1097-2765(01)00352-5. [DOI] [PubMed] [Google Scholar]
- 12.Kim D.R., Dai Y., Mundy C.L., Yang W., Oettinger M.A. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev. 1999;13:3070–3080. doi: 10.1101/gad.13.23.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landree M.A., Wibbenmeyer J.A., Roth D.B. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes Dev. 1999;13:3059–3069. doi: 10.1101/gad.13.23.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hickman A.B., Chandler M., Dyda F. Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit. Rev. Biochem. Mol. Biol. 2010;45:50–69. doi: 10.3109/10409230903505596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swanson P.C. The DDE motif in RAG-1 is contributed in trans to a single active site that catalyzes the nicking and transesterification steps of V(D)J recombination. Mol. Cell. Biol. 2001;21:449–458. doi: 10.1128/MCB.21.2.449-458.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou L., Mitra R., Atkinson P.W., Hickman A.B., Dyda F., Craig N.L. Transposition of hAT elements links transposable elements and V(D)J recombination. Nature. 2004;432:995–1001. doi: 10.1038/nature03157. [DOI] [PubMed] [Google Scholar]
- 17.Eastman Q.M., Leu T.M., Schatz D.G. Initiation of V(D)J recombination in vitro obeying the 12/23 rule. Nature. 1996;380:85–88. doi: 10.1038/380085a0. [DOI] [PubMed] [Google Scholar]
- 18.Mundy C.L., Patenge N., Matthews A.G., Oettinger M.A. Assembly of the RAG1/RAG2 synaptic complex. Mol. Cell. Biol. 2002;22:69–77. doi: 10.1128/MCB.22.1.69-77.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Difilippantonio M.J., McMahan C.J., Eastman Q.M., Spanopoulou E., Schatz D.G. RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell. 1996;87:253–262. doi: 10.1016/s0092-8674(00)81343-4. [DOI] [PubMed] [Google Scholar]
- 20.Landree M.A., Kale S.B., Roth D.B. Functional organization of single and paired V(D)J cleavage complexes. Mol. Cell. Biol. 2001;21:4256–4264. doi: 10.1128/MCB.21.13.4256-4264.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swanson P.C. Fine structure and activity of discrete RAG-HMG complexes on V(D)J recombination signals. Mol. Cell. Biol. 2002;22:1340–1351. doi: 10.1128/mcb.22.5.1340-1351.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Gent D.C., Hiom K., Paull T.T., Gellert M. Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 1997;16:2665–2670. doi: 10.1093/emboj/16.10.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Little A.J., Corbett E., Ortega F., Schatz D.G. Cooperative recruitment of HMGB1 during V(D)J recombination through interactions with RAG1 and DNA. Nucleic Acids Res. 2013;41:3289–3301. doi: 10.1093/nar/gks1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swanson P.C. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol. Rev. 2004;200:90–114. doi: 10.1111/j.0105-2896.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- 25.Nishihara T., Nagawa F., Imai T., Sakano H. RAG-heptamer interaction in the synaptic complex is a crucial biochemical checkpoint for the 12/23 recombination rule. J. Biol. Chem. 2008;283:4877–4885. doi: 10.1074/jbc.M709890200. [DOI] [PubMed] [Google Scholar]
- 26.Ciubotaru M., Trexler A.J., Spiridon L.N., Surleac M.D., Rhoades E., Petrescu A.J., Schatz D.G. RAG and HMGB1 create a large bend in the 23RSS in the V(D)J recombination synaptic complexes. Nucleic Acids Res. 2013;41:2437–2454. doi: 10.1093/nar/gks1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagawa F., Kodama M., Nishihara T., Ishiguro K., Sakano H. Footprint analysis of recombination signal sequences in the 12/23 synaptic complex of V(D)J recombination. Mol. Cell. Biol. 2002;22:7217–7225. doi: 10.1128/MCB.22.20.7217-7225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin F.F., Bailey S., Innis C.A., Ciubotaru M., Kamtekar S., Steitz T.A., Schatz D.G. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat. Struct. Mol. Biol. 2009;16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Callebaut I., Mornon J.P. The V(D)J recombination activating protein RAG2 consists of a six-bladed propeller and a PHD fingerlike domain, as revealed by sequence analysis. Cell. Mol. Life Sci. 1998;54:880–891. doi: 10.1007/s000180050216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grundy G.J., Ramon-Maiques S., Dimitriadis E.K., Kotova S., Biertumpfel C., Heymann J.B., Steven A.C., Gellert M., Yang W. Initial stages of V(D)J recombination: the organization of RAG1/2 and RSS DNA in the postcleavage complex. Mol. Cell. 2009;35:217–227. doi: 10.1016/j.molcel.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pavlicek J.W., Lyubchenko Y.L., Chang Y. Quantitative analyses of RAG-RSS interactions and conformations revealed by atomic force microscopy. Biochemistry. 2008;47:11204–11211. doi: 10.1021/bi801426x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shlyakhtenko L.S., Gilmore J., Kriatchko A.N., Kumar S., Swanson P.C., Lyubchenko Y.L. Molecular mechanism underlying RAG1/RAG2 synaptic complex formation. J. Biol. Chem. 2009;284:20956–20965. doi: 10.1074/jbc.M109.028977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ciubotaru M., Kriatchko A.N., Swanson P.C., Bright F.V., Schatz D.G. Fluorescence resonance energy transfer analysis of recombination signal sequence configuration in the RAG1/2 synaptic complex. Mol. Cell. Biol. 2007;27:4745–4758. doi: 10.1128/MCB.00177-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fugmann S.D., Villey I.J., Ptaszek L.M., Schatz D.G. Identification of two catalytic residues in RAG1 that define a single active site within the RAG1/RAG2 protein complex. Mol. Cell. 2000;5:97–107. doi: 10.1016/s1097-2765(00)80406-2. [DOI] [PubMed] [Google Scholar]
- 35.Eastman Q.M., Villey I.J., Schatz D.G. Detection of RAG protein-V(D)J recombination signal interactions near the site of DNA cleavage by UV cross-linking. Mol. Cell. Biol. 1999;19:3788–3797. doi: 10.1128/mcb.19.5.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakowicz J.R. Principles of Fluorescence Spectroscopy. 3rd edn. New York: Springer Science Publishers; 2006. [Google Scholar]
- 37.Fairclough R.H., Cantor C.R. The use of singlet-singlet energy transfer to study macromolecular assemblies. Methods Enzymol. 1978;48:347–379. doi: 10.1016/s0076-6879(78)48019-x. [DOI] [PubMed] [Google Scholar]
- 38.Middleton E.R., Rhoades E. Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys. J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hess S.T., Huang S., Heikal A.A., Webb W.W. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 2002;41:697–705. doi: 10.1021/bi0118512. [DOI] [PubMed] [Google Scholar]
- 40.Dickerson R.E. DNA structure from A to Z. Methods Enzymol. 1992;211:67–111. doi: 10.1016/0076-6879(92)11007-6. [DOI] [PubMed] [Google Scholar]
- 41.Case D.A., Cheatham T.E., III, Darden T., Gohlke H., Luo R., Merz K.M. Jr., Onufriev A., Simmerling C., Wang B., Woods R. The Amber biomolecular simulation programs. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33, 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 43.Clegg R.M., Murchie A.I., Zechel A., Lilley D.M. Observing the helical geometry of double-stranded DNA in solution by fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U.S.A. 1993;90:2994–2998. doi: 10.1073/pnas.90.7.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clegg R.M. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 45.Banerjee J.K., Schatz D.G. Synapsis alters RAG-mediated nicking at Tcrb recombination signal sequences: implications for the ‘beyond 12/23’ rule. Mol. Cell. Biol. 2014;34:2566–2580. doi: 10.1128/MCB.00411-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouellet J., Schorr S., Iqbal A., Wilson T.J., Lilley D.M. Orientation of cyanine fluorophores terminally attached to DNA via long, flexible tethers. Biophys. J. 2011;101:1148–1154. doi: 10.1016/j.bpj.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urnavicius L., McPhee S.A., Lilley D.M., Norman D.G. The structure of sulfoindocarbocyanine 3 terminally attached to dsDNA via a long, flexible tether. Biophys. J. 2012;102:561–568. doi: 10.1016/j.bpj.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iqbal A., Arslan S., Okumus B., Wilson T.J., Giraud G., Norman D.G., Ha T., Lilley D.M. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 2008;105:11176–11181. doi: 10.1073/pnas.0801707105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sawchuk D.J., Weis-Garcia F., Malik S., Besmer E., Bustin M., Nussenzweig M.C., Cortes P. V(D)J recombination: modulation of RAG1 and RAG2 cleavage activity on 12/23 substrates by whole cell extract and DNA-bending proteins. J. Exp. Med. 1997;185:2025–2032. doi: 10.1084/jem.185.11.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bischerour J., Lu C., Roth D.B., Chalmers R. Base flipping in V(D)J recombination: insights into the mechanism of hairpin formation, the 12/23 rule, and the coordination of double-strand breaks. Mol. Cell. Biol. 2009;29:5889–5899. doi: 10.1128/MCB.00187-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones J.M., Gellert M. Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J. 2002;21:4162–4171. doi: 10.1093/emboj/cdf394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.