

Abstract
Toxin-antitoxin (TA) modules are pairs of genes essential for bacterial regulation upon environmental stresses. The mazEF module encodes the MazF toxin and its cognate MazE antitoxin. The highly dynamic MazE possesses an N-terminal DNA binding domain through which it can negatively regulate its own promoter. Despite being one of the first TA systems studied, transcriptional regulation of Escherichia coli mazEF remains poorly understood. This paper presents the solution structure of C-terminal truncated E. coli MazE and a MazE-DNA model with a DNA palindrome sequence ∼10 bp upstream of the mazEF promoter. The work has led to a transcription regulator-DNA model, which has remained elusive thus far in the E. coli toxin–antitoxin family. Multiple complementary techniques including NMR, SAXS and ITC show that the long intrinsically disordered C-termini in MazE, required for MazF neutralization, does not affect the interactions between the antitoxin and its operator. Rather, the MazE C-terminus plays an important role in the MazF binding, which was found to increase the MazE affinity for the palindromic single site operator.
INTRODUCTION
Toxin–antitoxin (TA) systems are ubiquitous on bacterial chromosomes and bacterial plasmids. Depending on the nature of the antitoxin and the mechanism by which it neutralizes the toxin, TA modules can be categorized into five distinct types (1–8). Type II TA systems, where both toxin and antitoxin are proteins, are the most common. Their expression is regulated at the level of transcription through the antitoxin, which acts as a repressor, the activity of which is modulated by the toxin (9–12). Multiple roles have been suggested for TA modules ranging from plasmid stabilization (13–16) to altruistic suicide (17,18). Recent reports indicate that TA modules are associated with generation of nondividing but viable persister cells (19–21). Modulation of the persister state requires entangled molecular mechanisms that link protein activity to transcription regulation via the intrinsically disordered nature of the antitoxin C-terminal domain (22).
The mazEF operon was the first TA system found on the Escherichia coli chromosome (17). It is related to the kis/kid module on plasmid R1 (1,23), and it is homologous to the E. coli chromosomal TA module chpBIK (24). The mazEF operons encode a long-lived ribonuclease MazF, which cleaves mRNAs at specific sites (25).
In addition, MazF targets the 16S rRNA within the E. coli 30S ribosomal subunit at the decoding center, thereby removing 43 nucleotides from the 3′-terminus. The resulting truncated ribosomes preferentially translate the subset of leaderless mRNAs (26). The activity of the MazF toxin is neutralized by the short-lived antitoxin MazE, which is degraded by the ClpPA serine protease (17).
MazE proteins consist of two domains. The N-terminal domain has a DNA binding function and adopts a swapped-hairpin β-strand motif, typically of the AbrB/MazE/MraZ superfamily (27,28). This fold is common among bacterial transcription regulators and is found e.g. in the transition state regulator Abh (29) and the transcription regulator SpoVT (30,31). X-Ray diffraction studies have shown that the C-terminal domain of MazE is intrinsically disordered and upon binding to MazF adopts a unique and mostly extended conformation (27,32). The MazE binding to MazF stabilizes and protects its own vulnerable C-terminus from specific protease's cleavage, by which the TA system would be induced, leading to antitoxin degradation and toxin activation.
The E. coli mazEF operon contains a 47-bp operator region that contains three binding sites for the MazE dimer (‘cab’) (27,33,34). These sites are termed ‘c’, ‘a’ and ‘b’ when moving downstream toward the mazE start codon. Of these, only site ‘a’ contains the perfect palindrome 5′-ATATAT-3′, a hallmark of TA operator sequences (35). The details of the interaction between E. coli MazE and its operator DNA are not known yet and two distinct models were proposed based on the crystal structures of a EcMazEF complex (32) and a complex between EcMazE and a dromedary heavy chain antibody fragment (27,34). DNA binding by MazE/MazF complexes is thought to be primarily due to the antitoxin, with the toxin serving to enhance binding affinity (33).
In this work we studied in detail the structural and (thermo-)dynamic features of E. coli MazE binding to the ‘a’ site DNA. We show that the N-terminal domain of the E. coli MazE (EcMazE1–50) is solely involved in DNA binding, excluding any participation of the disordered C-terminus in DNA interaction. We confirm that the functional role of the intrinsically disordered region is purely related to toxin neutralization, which is essential for transcription regulation. Moreover, EcMazF cooperatively increases the EcMazE-DNA affinity on the single palindrome suggesting how the toxin/antitoxin molar ratio can control the self-regulation of the TA locus transcription.
MATERIALS AND METHODS
Expression and purification of full-length EcMazE
The pQE30-mazE plasmid containing a 21 residue N-terminal tag, six of which are histidines, was transformed into E. coli MC4100ΔmazEF-lacIq (relA+) and cells were grown in 1 l of M9 minimal medium at 310 K supplemented with 15N-labeled NH4Cl and/or 13C-labeled glucose, respectively, and 100 μg/ml ampicillin. At an OD600 of 0.8 protein expression was induced with 1 mM IPTG and after additional 5 h of incubation the cells were harvested by centrifugation (30 min/4.000 g/277 K) and resuspended in 50 mM K2HPO4, 300 mM NaCl, 10 mM imidazole, pH 8.0. After sonication the suspension was again centrifuged (60 min/15.000 g/277 K) and the supernatant filtered through a 0.45 μm sterile filter prior to loading it onto a Ni-CAM column (Sigma Aldrich, St. Louis, MO, USA), pre-equilibrated in 50 mM K2HPO4, 300 mM NaCl, 10 mM imidazole, pH 8.0. The protein was eluted from the column with 50 mM K2HPO4, 300 mM NaCl, 500 mM imidazole, pH 8.0 with a 10–500 mM imidazole gradient as a single peak at an approximate concentration of 100-mM imidazole. The fractions containing the protein were combined and dialyzed first against 2 l of distilled water followed by 1 l of 20 mM KH2PO4, 100 mM NaCl, pH 6.5 as nuclear magnetic resonance (NMR) buffer. In a final step the concentrated protein was heated up to 358–363 K for 2 min and slowly cooled down to room temperature. This heat treatment makes EcMazE more stable for long-term storage at room temperature, since it denatures contaminating proteins, such as proteases, while EcMazE can be refolded upon heat denaturation like other bacterial antitoxins (36,37).
Expression and purification of truncated EcMazE1–50
In parallel, the pQE30-mazE-truncated plasmid containing EcMazE1–50 with 18 residues N-terminus tag, six of which are histidines, was transformed into E. coli BL21 (DE3) competent cells. Cells were grown in 13C, 15N-enriched minimal medium (SPECTRA 9, purchased from Cambridge Isotope Laboratories). Expression of 13C, 15N-labeled EcMazE1–50 was induced with 1 mM IPTG at an OD600nm of 0.6 and the culture was incubated overnight at 310 K, 120 rpm. The cells were harvested by centrifugation (30 min/4.000 g/277 K) and resuspended in lysis buffer, 20 mM Tris-HCl, 150 mM NaCl, 10 mM imidazole, pH 7.0, 0.1 mg/ml p-aminoethylbenzenesulfonyl fluoride (AEBSF) and 1 μg/ml leupeptin. After breaking the cells, passing them twice though a french press (1000–1200 bar, 12000 psi), the suspension was again centrifuged (20 min/15.000 g/277 K) and the supernatant filtered through a 0.45 μm sterile filter prior to loading it onto a 5 ml Ni-NTA resin (Qiagen) pre-equilibrated with 20 mM Tris-HCl, 150 mM NaCl, 10 mM imidazole, pH 7.0. The proteins were eluted using 20 mM Tris-HCl, 150 mM NaCl, 1 M imidazole, pH 7.0. The EcMazE1–50-containing peak started to elute at 330 mM of imidazole concentration. To obtain highly pure samples, EcMazE1–50 was consecutively loaded on a high-resolution Superdex 75PG 16/60 in 20 mM Tris-HCl, 150 mM NaCl, pH 7.0. The 13C, 15N-labeled EcMazE1–50 was further dialyzed against 50 mM Na phosphate pH 6.5, 50 mM NaCl, as suitable NMR buffer, adding a proteases inhibitor cocktail (10 mM ethylenediaminetetraacetic acid, 50 μg/ml AEBSF, 100 μg/ml leupeptin).
NMR spectroscopy on full-length EcMazE
All full-length EcMazE spectra for the assignment were acquired at 298 K on a Varian Unity INOVA 600-MHz NMR spectrometer. DNA binding experiments and 15N relaxation data were obtained on a Bruker Avance III 700 MHz NMR spectrometer, equipped with a cryogenically cooled 5 mm TCI probe. For the NMR experiments EcMazE was dissolved in 90% aqueous buffer (50 mM KPi pH 6.5, 50 mM NaCl) and 10% D2O, except for the 3D HCCH-TOCSY (100% D2O). Data were processed using NMRPipe (38) and analyzed in NMRView (39). 1H, 15N and 13C resonances were assigned using 2D homonuclear and standard triple resonance experiment (40). {1H}-15N heteronuclear Nuclear Overhauser Enhancements (NOEs) of full-length EcMazE were measured at 700 MHz and 298 K, and determined from the ratio of peak intensities (Ion/Ioff) with and without the saturation of the amide protons for 3 s. Average heteronuclear NOE values and their errors were obtained from a duplicate set of experiments.
NMR spectroscopy on truncated EcMazE1–50
13C, 15N-labeled-truncated EcMazE1–50 was prepared at 1.0 mM in 50 mM Na phosphate pH 6.5, 50 mM NaCl, 10% D2O. All NMR spectra used for the assignment were recorded at 298 K using a Varian 600 MHz NMR Direct-Drive System. A 2D NOESY with a 100 ms mixing time was recorded on a Varian 800 MHz NMR Direct-Drive System, equipped with a salt tolerant triple-resonance PFG-Z cold probe, on the same sample. All NMR data were processed using NMRPipe (38) and analyzed by CCPNMR (41).
Semi-automatic assignment of the protein backbone was performed using CCPNMR software (41). The 1H,15N frequencies of the 15N-HSQC spectrum were used to correlate each peak with its 13Cα and 13Cβ and the ones of the preceding amino acid (by using HNCACB and CBCA(CO)NH spectra) and with the preceding 13CO (via the HNCO spectrum). 1Hα and 1Hβ were assigned using the HBHA(CO)NH spectrum.
Assignments were extended to the side chain signals using correlations within the C(CO)NH and HCCH-TOCSY for the aliphatic side-chains. Aromatic 1H and 13C frequencies of the single Trp residue were assigned from the 13C-HSQC and 13C-NOESY-HSQC spectra. Side-chain 15N1H2 frequencies of glutamines and asparagines and 15Nϵ1Hϵ of arginines were assigned from HNCACB, CBCA(CO)NH and 3D 15N-NOESY-HSQC spectra. All 1H, 13C and 15N resonances were verified from 3D 15N- and 13C-NOESY-HSQC spectra (with 100 ms mixing times).
{1H}-15N heteronuclear NOEs of EcMazE1–50 were measured at 600 MHz and 298 K, and determined from the ratio of peak intensities (Ion/Ioff) with and without the saturation of the amide protons for 3 s. Average heteronuclear NOE values and their errors were obtained from a duplicate set of experiments.
EcMazE1–50 NMR structure calculations
Truncated EcMazE1–50 NMR solution structure calculations were performed using CYANA version 2.1 (42,43). Sixty-one inter-monomeric NOEs were identified based on the EcMazE X-ray structure (PDB entry 1MVF). These manually assigned NOEs were used together with non-assigned NOEs and dihedral restraints from Talos+ (44) as input for CYANA (42,43)). Non-assigned NOEs were assigned using the automated NOE assignment procedure of CYANA. A standard protocol was used with seven cycles of combined automated NOE assignment and structure calculation of 100 conformers in each cycle. From the three NOESY data sets, 946 NOEs were unambiguously assigned, including 166 inter-monomeric NOEs (Table 1). These unambiguously assigned restraints were used for a final structure refinement in explicit solvent using the RECOORD protocol (45), which runs under CNS (46). The twenty lowest-energy structures were used for final analysis.
Table 1. Structural statistics over the 20 lowest-energy water-refined NMR structures of EcMazE1–50.
| EcMazE1–50 | |
|---|---|
| Distance restraints totala | 1892 (946 A, 946 B) |
| Short range (i − j = 0) | 492 (246 A, 246 B) |
| Medium range (1≤|i − j|≤4) | 920 (460 A, 460 B) |
| Long range (|i − j|≥5) | 480 (240 A, 240 B) |
| Inter-monomer | 332 (166 A->B, 166 B->A) |
| Dihedral restraints | 166 (83 A, 83 B) |
| Phi angles | 80 (40 A, 40 B) |
| Psi angles | 86 (43 A, 43 B) |
| CNS energies (kcal/mol) | |
| Etotal | −4967.6 ± 103.2 |
| Evdw | −526.2 ± 30.9 |
| Eelec | −5554.1 ± 127.8 |
| Restraint statistics | |
| NOE violations >0.5 Å | 0 |
| Dihedral violations >5o | 0 |
| RMSD from averageb (Å) | Residues 3–47 |
| Backbone N, CA, C′ | 0.50 ± 0.16 |
| Heavy atoms | 0.85 ± 0.15 |
| Ramachandran plot | Residues 2–49 |
| Most favored regions (%) | 84.1 |
| Additional allowed regions (%) | 15.2 |
| Generously allowed regions (%) | 0.7 |
| Disallowed regions (%) | 0.0 |
Values are reported for the EcMazE1–50 homodimer consisting of monomers with chain ID: A and B.
aStatistics for residues from −6 to 50. Flexible N-terminal His-tag and the C-terminal residue were omitted from the RMSD analysis and Ramachandran statistics obtained from PROCHECK analysis.
bValues with their corresponding standard deviations are reported for the EcMazE1–50 homodimer.
Isothermal titration calorimetry to study MazE-DNA binding
Isothermal Titration Calorimetry (ITC) experiments were performed on a MicroCal iTC200 system (GE Healthcare). Investigation of the EcMazE binding to its own palindrome promoter sequence was carried out using the ‘a’ site proposed previously by Marianovky et al. (33); (forward: 5′-TTGATATATACTGT-3′; reverse: 3′-ACAGTATATATCAA-5′). Besides the DNA ‘a’ site as main target in this work, we performed ITC experiments on other biologically relevant sites of the mazEF operon, the full three sites ‘cab’ (forward 5′- CTCGTATCTACAATGTAGATTGATATATACTGTATCTACATATGATAGCGT-3′), and the two other single sites ‘c’ (forward 5′-GTATCTACAATGTAGATTG-3′) and ‘b’ (forward 5′-ATATACTGTATCTACATAT-3′), all purchased from Sigma Aldrich. A control experiment was done with DNA fragment ‘X’ (forward: 5′-GATTTTTGATTTT-3′; reverse: 3′-AAAATCAAAAAC-5′), purchased from VBC Biotech (Vienna, Austria), and treated as all the other samples. The double-stranded DNA fragment solutions were generated by dissolving equimolar amounts of single-strands oligonucleotide in water, heated up the solution to 368 K at 275 K/min and then slowly cooled down to 298 K to allow annealing. To exactly match buffer composition, the double strand DNA fragments and both EcMazEs, full-length and truncated, were dialyzed overnight against 2 l of 50 mM phosphate buffer at pH 6.5 and 50 mM NaCl. Prior to titration, the samples were filtrated with 0.22 μm filters and degassed for 10 min at temperature corresponding to the titration temperature. A 14 μM solution of DNA fragments was titrated with full-length EcMazE and EcMazE1–50 (both solutions at 280 μM). The EcMazEs-DNA ‘a’ titrations were measured at three different temperatures: 292, 298, 305 K, while the others only at 305 K. Additionally, heats of dilution, determined by titrating the proteins into solution buffer, were subtracted from the raw titration data before analysis. Data analysis was performed with MicroCal Origin software accompanying the ITC instrument. The binding affinity (KD) and change in enthalpy associated with the binding event (ΔH) were calculated after fitting each data set by least-squares procedures assuming an n identical and independent site-binding model. The change in heat capacity of binding (ΔCp) was determined from the slope of the linear dependence of ΔH with the temperature.
Electrophoretic mobility shift assay
Binding of EcMazE to the DNA ‘a’ was followed by mobility shift electrophoresis (electrophoretic mobility shift assay, EMSA). Prior to hybridization, the DNA fragments were 5′-end labeled with [γ-32P]-ATP by T4 polynucleotide kinase (New England Biolabs). The double strand DNA ‘a’ fragment was purchased from Sigma Aldrich. Labeled probes were incubated with purified proteins (MazE variable concentrations from 7.5–100 μM for following MazE to DNA binding; MazE fixed concentration of 1 μM for following the effect of MazF on the MazE-to-DNA binding using variable concentration of MazF from 0.5–10 μM in 50 mM Tris-HCl pH 7.5, 50 mM NaCl, 50 μg/ml bovine serum albumin (BSA). Reactions were incubated for 20 min at 310 K. DNA bound complexes were separated by native polyacrylamide gel electrophoresis in 6% acrylamide gels with 0.5 X TBE for 3 h at 8 V cm−1. The separation was followed by phosphorimaging.
NMR chemical shift mapping of MazE-DNA binding
Investigation of the EcMazE binding to its own palindrome promoter sequence was carried out by chemical shift mapping using the ‘a’ site. A 1.0 mM DNA stock solution was prepared by dissolving the two single-strands oligonucleotide in 50 mM KH2PO4 pH 6.5, 50 mM NaCl buffer and annealing over night. For the chemical shift mapping a full-length EcMazE reference 1H-15N-HSQC NMR spectrum at a protein concentration of 0.3 mM was recorded prior to a six-step titration series with the corresponding DNA sequence at a concentration range from 0 to 0.3 mM. The final concentration of the protein in the last point of the titration was 0.21 mM. A control experiment was done using DNA fragment ‘X’. The NMR titration experiments were performed under the same conditions and concentrations used for the DNA ‘a’ chemical shift mapping.
In order to investigate the DNA binding to truncated EcMazE1–50, a slightly different palindrome sequence was selected, differing from the ‘a’ fragment used for the full-length EcMazE by only 3 bp at the 5′ and 3′ extremities (forward: 5′-CGTGATATATACTGC-3′; reverse: 3′-GCAGTATATATCACG-5′, purchased from Sigma Aldrich). A 1.4 mM DNA stock solution was prepared by dissolving equimolar amounts of single-strands oligonucleotide in water, heated up the solution to 368 K at 275 K/min and then slowly cooled down to 298 K to allow annealing. Prior to titration, the double strand DNA ‘a’ fragment was dialyzed against the same NMR buffer, 50 mM Na phosphate pH 6.5, 50 mM NaCl. For the chemical shift mapping a EcMazE1–50 reference 1H-15N-HSQC NMR spectrum at a protein concentration of 0.4 mM was recorded prior to the titration. A titration series was done in six steps using the DNA stock solution leading to a concentration range between 0 and 0.4 mM DNA. A 1H-15N-HSQC NMR spectrum was recorded at each step in order to follow the chemical shift perturbations upon DNA binding. The final concentration of the protein in the last point of the titration was 0.33 mM.
The magnitude of the chemical shift perturbation (Δδ) was calculated by Δδ = [(ΔδH)2+(ΔδN/6.51)2]1/2 where Δδ is the difference between the bound and free form combined chemical shifts.
Structure calculations of the EcMazE:DNA complex
The structural model of the complex between EcMazE1–50 and DNA was obtained using the HADDOCK software (47). Ambiguous Interaction Restraints (AIRs) for EcMazE1–50 were obtained from the chemical shift perturbation data. All the atoms showing a higher difference than the corresponding mean were investigated in terms of solvent accessibility with NACCESS software (48) and location in the EcMazE1–50 structure. The active residues used in the docking were 7–12, 16, 18–20 in both EcMazE1–50 monomers. Histidine 3 was included in the docking as passive residue. Active and passive nucleotides in both DNA strands were defined as nucleotides 5–11, and 3–4 and 12–13, respectively. Overall, a total of 34 AIRs were defined between EcMazE1–50 and DNA with upper distances fixed at 2.0 Å.
Docking was started from the whole ensemble of 20 lowest-energy EcMazE1–50 free structures. For the DNA ‘a’ fragment we used the X-ray structure of the VapBC2–DNA complex (PDB entry 3ZVK) as model (49). We mutated the VapBC2 DNA using UCSF Chimera (50) to obtain the structure of our 15 bp DNA ‘a’ fragment. During docking the DNA was kept rigid, while the protein was kept semi-flexible. The final step of the structure refinement was done in explicit water. The seven structures with the lowest interaction energies and lowest AIR violations were selected for further analysis.
Small angle X-ray scattering
Complexes of full-length EcMazE DNA and EcMazE1–50-DNA from the same pQE30-mazE construct (18 N-terminal His-tag) were analyzed by SAXS; data were collected at Swing, Soleil Synchrotron (Paris, France) at 7–8 mg/ml concentration. The complexes were beforehand dialyzed against 20 mM Tris-HCl pH 7.0, 150 mM NaCl. All samples were subsequently centrifuged at 10 000 rpm for 2 min at 277 K and loaded on Shodex packed HPLC column coupled to the beam capillary. For each data set, 250 frames were collected, averaged and background-subtracted. The initial data process was realized using the program PRIMUS (51,52) for scaling and merging. A Guinier analysis was performed at very low scattering angle and used to estimate the radius of gyration (Rg) of the particle. The indirect Fourier transform package GNOM (51) was used to compute the distance distribution p(r) function from the scattering curve and calculate the maximum dimension of the particle (Dmax). To accurately determine the molecular mass, assess model-data agreement and verify that we did not over-fitted our data, metrics like QR, χ2 free and Rsas were calculated based on Rambo and Tainer (53). To define the minimal set of MazE1–50-DNA NMR structures that can explain the SAXS data, the minimal ensemble algorithm (Minimal Ensemble Search, MES) was used (54). This algorithm searches for the minimal ensemble set of conformations from the pool of all given conformations, systematically evaluating combinations of five models or less. The full-length EcMazE in complex with DNA was built using our best NMR EcMazE1–50 structure in complex with DNA and modeled the missing C-terminal disordered tails using MODELLER (AllosMod FOXS) (55,56). A minimal ensemble was defined for this protein complex as well. The comparison between the theoretical scattering curves of both protein–DNA complexes with the experimental data, which was expressed in χ2 goodness of fit, was done using FoXS (54).
Paramagnetic relaxation enhancement
In order to validate the structural model of the EcMazE1–50–DNA complex we monitored intensity changes in 1H-15N-HSQC spectra of full-length EcMazE upon the binding of double stranded, paramagnetically tagged DNA corresponding to the ‘a’ region in the mazEF operon. A paramagnetic iodoacetamido-proxyl tag was attached to DNA oligos containing a PTO modification on either the 5′- or 3′-end. 0.5 mM double-stranded DNA stock solutions were prepared by dissolving the modified and unmodified oligonucleotide strands, respectively, in 200 mM trishydroxymethyl-aminomethane, pH 8.0 buffer and left the DNA annealed over-night. Subsequently, the spin-label 3-(2-iodoacetamido)-proxyl (Sigma-Aldrich, St. Louis, MO, USA) was added to a final concentration of 20 mM to the modified dsDNA and the solution was stirred for 48 h at room temperature in the dark. Subsequently, the free spin-label was extracted from the solution by phase separation after addition of 500 μl CH2Cl2. This solvent extraction step was repeated twice prior to dialyzing the sample twice against 1 l of distilled water for a complete removal of the organic solvent. Finally, the dialyzed sample was lyophilized. For each titration, with spin-labeled DNA, a EcMazE reference 1H-15N-HSQC spectrum at 0.2 mM final protein concentration was recorded prior to dissolving the lyophilized DNA in the 300 μl 15N-labeled EcMazE sample. After recording the EcMazE spectrum with paramagnetically labeled DNA and adding 10 mM sodium dithionite to reduce the proxyl group to its diamagnetic form, another 1H-15N-HSQC was acquired. Comparison of peak intensities in the diamagnetic and paramagnetic form yields signal reductions due to paramagnetic relaxation enhancements (PREs). High mobility of the tag due to its length and position at the flexible DNA ends prevented a quantitative analysis of the PRE data.
RESULTS
NMR solution structure and dynamics of EcMazE
In order to reveal insights on the EcMazE–DNA interaction, we firstly conducted a structural and dynamic comparison between the ‘wild-type’ full-length EcMazE antitoxin and its truncated version EcMazE1–50. The EcMazE C-terminal intrinsically disordered tail was strategically truncated in order to distinctly characterize the N-terminal DNA binding domain and its direct interaction with the TA promoter fragment. In contrast to the strong overlap in the crowded full-length EcMazE HSQC spectrum (Figure 1), EcMazE1–50 shows a nicely dispersed HSQC, which guaranteed a straightforward peak assignment, and consequently an accurate structural determination. The 1H-15N peaks for 10G and 11N (including those of the side-chain NH2) are not detectable in the full-length EcMazE HSQC, while they show up as weaker signals in the truncated EcMazE1–50 HSQC. Interestingly, overlay of the two monomers in the X-ray structure (PDB entry 1MVF) shows different conformation of the 10G-11N loop, indicating conformational exchange explaining the weakening/disappearing of the NMR signals. Moreover, in one of the monomers, there is no density for the N11 side chain in the X-ray structure. Difference in chemical shifts between EcMazE1–50 and full-length EcMazE is mainly evident for residue E50, which corresponds to the C-terminus in EcMazE1–50 (see Supplementary Figure S1).
Figure 1.
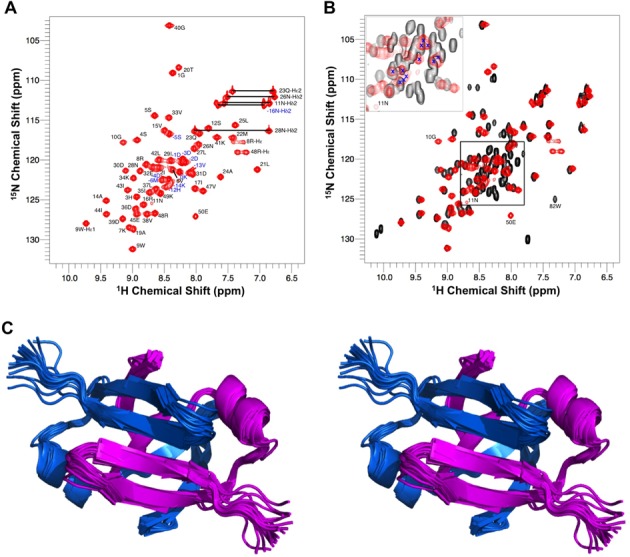
NMR characterization of EcMazE1–50 versus full-length EcMazE. (A) Assigned 600 MHz 1H-15N-HSQC spectrum of 13C-15N-labeled EcMazE1–50 at 298K. EcMazE1–50 peaks of backbone 1H, 15N pairs are numbered with their corresponding position in the amino-acid sequence. Peaks labeled in blue belong to the EcMazE1–50 His-tag. (B) In red the 600 MHz 1H-15N-HSQC spectrum of EcMazE1–50 and in black the 600 MHz 1H-15N-HSQC spectrum of full-length EcMazE. The inset contains peaks of the N-terminal His-tag and all peaks of the C-terminus of full-length EcMazE (aa 50–81), except C-terminal Trp82. The same region contains the peaks of the EcMazE1–50 His-tag, indicated with a blue X. Peaks belonging to G10 and N11 visible in the EcMazE1–50 HSQC and not detectable in the full-length EcMazE HSQC are indicated. (C) Stereo cartoon representation of the 20 lowest energy EcMazE1–50 NMR structures; the two monomers are colored in sky-blue and magenta. The highly flexible N-terminal (His-tag) was removed from the NMR ensemble for clarity. Figure created in PyMol.
The 1H, 15N and 13C assigned resonances of EcMazE1–50 and of full-length EcMazE have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu/) under accession number 25086 and 25093, respectively.
The NMR structure of the truncated version EcMazE1–50 was obtained from the combined use of distance and dihedral restraints. Figure 1C shows the ensemble of the 20 lowest energy conformations. NMR structural statistics are summarized in Table 1. The structural coordinates and experimentally derived restraints have been deposited in the PDB with accession number 2MRN.
The EcMazE1–50 structure possesses a typical swapped hairpin β-strand motif consisting of two N-terminal β-strands, followed by an α-helix and two C-terminal β-strands. The N-terminal and C-terminal strands form two 4-stranded β-sheets in the homodimer. Our EcMazE1–50 solution structure resembles closely the crystal structure of full-length MazE in complex with a nanobody (27), which shows electron density for residues 4 to 47 only, thus missing the disordered C-terminal tail. The backbone rmsd for residues 4–47 of both monomers in the dimer is 0.864 Å between the closest-to-average NMR structure and the X-ray structure, and is 1.07 ± 0.21 Å between all NMR structures and the X-ray structure, using Profit (http://www.bioinf.org.uk/profit/).
Information about the dynamical behavior of both full-length and truncated EcMazEs in their free form was obtained by measuring {1H}-15N steady state NOEs (Supplementary Figure S2). Small and negative {1H}-15N NOEs are indicative of higher flexibility and they are observed mainly for the N-terminal residues (His-tag) and in the disordered C-terminal domain, in agreement with the lack of density in the crystal for the C-terminal tails of full-length EcMazE. Moreover, lack of chemical shift dispersion and high intensity peaks of the C-terminal domain in full-length EcMazE (Figure 1B, inset) confirms the high flexibility of this region.
Additionally, the predicted secondary structure elements from 1Ha, 13Ca, 13C’ and 13Cb chemical shifts, presented as Chemical Shift Index (CSI) patterns in Supplementary Figure S3, are the same for the N-terminal domain of EcMazE in truncated and full-length EcMazE, and correspond well to the secondary structure elements present in the solution structure. The CSI patterns for the C-terminal domain in the full-length EcMazE as well as the low-dispersed and high intensity NMR signals and the small and/or negative {1H}-15N NOEs indicate random structure for this region and point out that the disordered C-terminal tail does not affect the N-terminal domain structure and dynamics.
Isothermal titration calorimetry on EcMazE binding to its operator DNA fragments
DNA binding is supposed to be limited to the N-terminal domain of EcMazE and specific for the palindrome DNA sequence ‘a’. We performed ITC experiments using both full-length and truncated EcMazE and titrate them with the selected DNA ‘a’ fragment (Figure 2B). These ITC experiments show an equal behavior of these two proteins upon binding the oligonucleotide. The binding of EcMazE1–50 to the other biological relevant operator fragments ‘b’, ‘c’ and ‘cab’ was also measured (Figure 2A and C). Supplementary Figure S4 shows all the ITC data for the systems studied. Table 2 reports the thermodynamic parameters for every ITC measured. The dissociation constants (KD) vary from ∼0.5–5 μM for the different oligonucleotide fragments tested. The enthalpy of DNA ‘a’ binding is rather constant over a temperature range between 292 and 305 K, which leads to a ΔCp close to zero for both EcMazE and EcMazE1–50 (Supplementary Figure S4C and Supplementary Table S1). This indicates very little if any structuring of EcMazE upon DNA binding, neither of its DNA binding domain nor of its disordered tails and agrees with the disordered tails not being involved in the process. This is likely happening as well for all the other fragments, since the thermodynamic values are closely similar. ITC experiments using random DNA segment ‘X’ under the same conditions (Supplementary Figure S4G) show very low affinity binding to EcMazE, indicating that the antitoxin binding is specific to its own operator.
Figure 2.
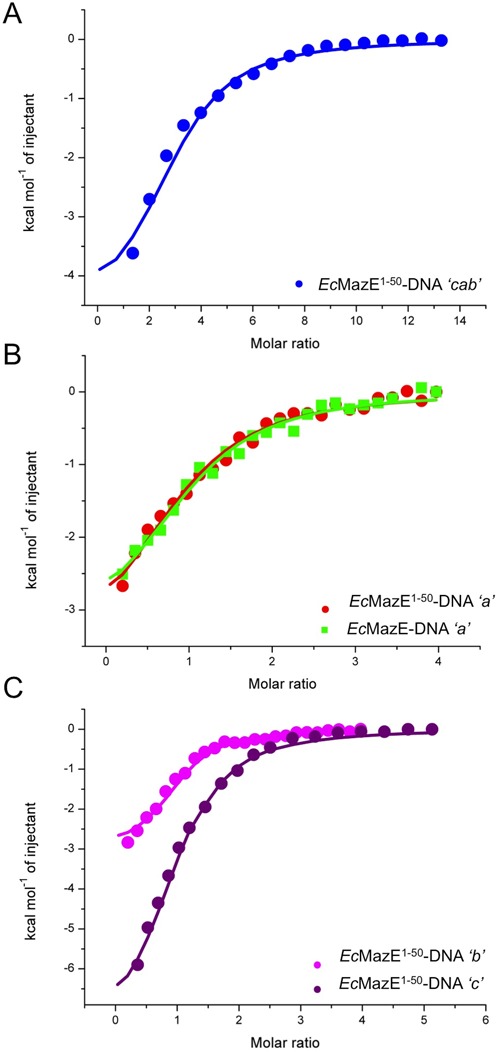
Isothermal titration calorimetry on EcMazE-DNA binding. (A) ITC titration curve for EcMazE1–50 binding to DNA ‘cab’ at 305 K. (B) ITC titration curves for EcMazE1–50 and full-length EcMazE binding to DNA ‘a’ at 305 K in red (circles) and in green (squares), respectively. (C) ITC titration curve for EcMazE1–50-DNA ‘c’ in purple and EcMazE1–50-DNA ‘b’ in magenta measured at 305 K. The solid lines in panels (A), (B) and (C) correspond to the best fit using a n equal to 1 binding site model. The thermodynamic parameters for the EcMazE-DNA binding are reported in Table 2.
Table 2. Thermodynamic parameters for EcMazE-DNA binding from ITC.
| EcMazE1–50- DNA ‘cab’ | EcMazE- DNA ‘a’ | EcMazE1–50- DNA ‘a’ | EcMazE1–50- DNA ‘b’ | EcMazE1–50- DNA ‘c’ | |
|---|---|---|---|---|---|
| Thermodynamic parameters | |||||
| KD (μM) | 0.6 ± 0.5 | 5.1 ± 0.2 | 5.6 ± 0.3 | 2.1 ± 0.6 | 0.6 ± 0.2 |
| ΔH (kcal/mol) | −5.1 ± 0.3 | −3.5 ± 0.3 | −3.8 ± 0.4 | −3.1 ± 0.1 | −9.1 ± 0.5 |
| TΔS (kcal/mol) | 3.4 ± 0.2 | 3.9 ± 0.2 | 3.5 ± 0.1 | 4.3 ± 0.3 | 1.0 ± 0.6 |
| n | 3 | 1 | 1 | 1 | 1 |
The error indicated corresponds to standard deviation. Number of binding site, n, was fixed at the value indicated.
MazE shows higher affinity for the single palindrome operator when titrated with MazF
To elucidate the role of the C-terminus extended domain of antitoxin EcMazE, we performed EMSAs using full-length EcMazE and DNA ‘a’ and consecutively titrating the EcMazE–DNA ‘a’ complex with increasing amounts of toxin EcMazF. First, a gel shift analysis using fixed concentrations of DNA ‘a’ and variable amounts of EcMazE was carried out to probe the antitoxin binding to the palindrome single site. A clear shift corresponding to complex formation is observed, accompanied with diminishing amounts of free DNA (Figure 3A), showing that EcMazE binds specifically to the operator fragment ‘a’.
Figure 3.
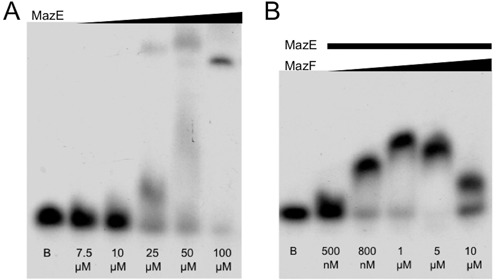
EcMazEF toxin–antitoxin system shows higher affinity binding than the antitoxin alone for its single site operator fragment. (A) Concentration-dependent binding of full-length EcMazE to the single site ‘a’ operator fragment. As the EcMazE concentration increases the DNA shifts from the free state to a EcMazE–DNA complex. Blank sample (DNA without protein) is indicated by a B. (B) Binding of EcMazE to the single site ‘a’ operator fragment enhanced by MazF. All samples include equal concentration of antitoxin EcMazE (1 μM), which is not sufficient to cause a shift of the DNA band. As the EcMazF concentration increases and approaches the 1:1 ratio with the antitoxin, however, a clear mobility shift is observed. At higher ratios, the shift disappears again. B: blank sample (DNA without protein).
However, addition of a variable amount of EcMazF to a lower concentration of EcMazE (1 μM), which is not sufficient to cause a shift of the DNA band by itself, results in an increase in affinity for EcMazE to DNA (Figure 3B). At very high EcMazF:EcMazE ratios, this effect is abolished and coincides with a reduced shift of the band corresponding to the complex. Thus, EcMazF enhances the binding of EcMazE to their DNA operator fragment ‘a’ though its interaction with the C-terminal region of EcMazE.
Structural model of the EcMazE–DNA complex from NMR and SAXS
Since no EcMazE-DNA structure is available, we aimed at obtaining an EcMazE-DNA structure using a combination of techniques. For this, we choose a 15 bp operator fragment containing the ATATAT palindrome sequence labeled as ‘a’ site (33). Using double labeled full-length EcMazE and truncated EcMazE1–50, we carried out NMR chemical shift mapping experiments, titrating both protein solutions with the ‘a’ site DNA (Figure 4). The NMR titration led to close-to-maximum chemical shift changes already at stoichiometric ratios. Substantial chemical shift perturbations and/or signal disappearance in the EcMazE1–50 titration are detected for residues K7, R8, W9, G10, N11, S12, A14, V15, R16, I17, A19, T20, Q23, L27, N28 and I29 (Figure 5A). These chemical shift perturbations induced by the DNA ‘a’ binding on EcMazE1–50 were mapped on the EcMazE structure, as shown in Figure 5C and D.
Figure 4.
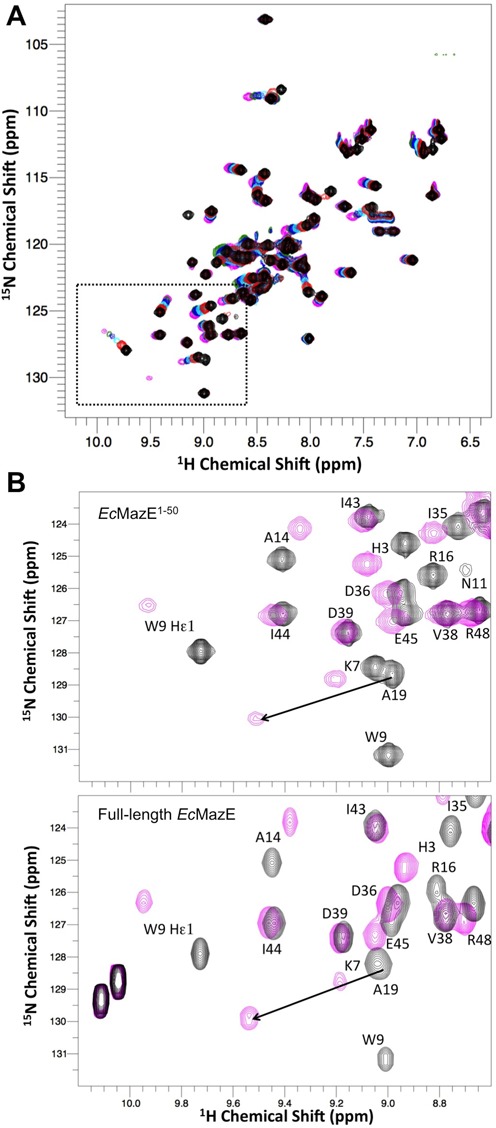
Binding of DNA ‘a’ to EcMazE monitored by NMR. (A) 1H-15N-HSQC spectra recorded during titrations of EcMazE1–50 with DNA ‘a’. All spectra are plotted at the same contour level and are colored from black (free form) to magenta (last titration point). (B) Selected region of 1H-15N-HSQC spectra recorded during titrations of EcMazE1–50 with DNA ‘a’. Only NMR spectra of the EcMazE1–50 free form (black) and of the last titration point (magenta) are shown. For clarification, the black arrow indicates the direction of the chemical shift changes for Ala 19. (C) The same selected region of the 1H-15N-HSQC spectra of free full-length EcMazE (black) and in complex with DNA ‘a’ (magenta).
Figure 5.
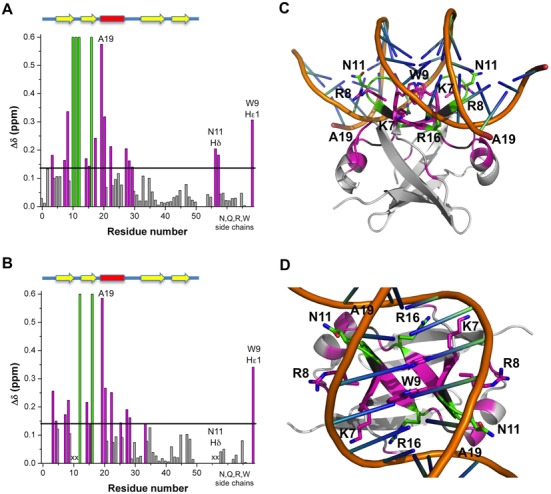
Chemical shift mapping of the DNA ‘a’ binding sites on EcMazE1–50 and full-length EcMazE. (A) Residue-specific DNA ‘a’-induced chemical shift changes EcMazE1–50. The chemical shift perturbations Δδ < Δδav + 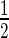 SD are colored gray, Δδ > Δδav +
SD are colored gray, Δδ > Δδav + 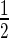 SD colored magenta, in green the residues of which their peak disappear upon addition of the DNA. The black line represents the chemical shift perturbations Δδav +
SD colored magenta, in green the residues of which their peak disappear upon addition of the DNA. The black line represents the chemical shift perturbations Δδav + 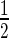 SD. Secondary structure elements within the EcMazE1–50 structure are indicated by yellow arrows (β-strands) and red bars (α-helices). (B) Residue-specific DNA ‘a’-induced chemical shift changes full-length EcMazE. Color coding as in (A). Residues not visible in the HSQC spectra of free full-length EcMazE are labeled as x. (C, D) Chemical shift mapping on the representative free NMR structure of EcMazE1–50 superimposed on VapB within the VapBC2–DNA complex (showing only the DNA within the complex, PDB entry 3ZVK) as in Supplementary Figure S6. Color coding as in (A). Figures prepared using PyMol.
SD. Secondary structure elements within the EcMazE1–50 structure are indicated by yellow arrows (β-strands) and red bars (α-helices). (B) Residue-specific DNA ‘a’-induced chemical shift changes full-length EcMazE. Color coding as in (A). Residues not visible in the HSQC spectra of free full-length EcMazE are labeled as x. (C, D) Chemical shift mapping on the representative free NMR structure of EcMazE1–50 superimposed on VapB within the VapBC2–DNA complex (showing only the DNA within the complex, PDB entry 3ZVK) as in Supplementary Figure S6. Color coding as in (A). Figures prepared using PyMol.
We performed the NMR titrations also on full-length EcMazE. The chemical shift perturbations upon DNA binding are very similar between the EcMazE1–50 and full-length EcMazE (Figure 5A and B, respectively), underlining that both proteins bind specifically to the oligonucleotide fragment used and that the C-terminal region in EcMazE is not involved in direct binding to DNA. Additionally, an NMR titration with the random DNA sequence ‘X’ was performed as negative control (Supplementary Figure S5). The DNA-binding-induced EcMazE shifts in this NMR titration experiment are much smaller and for some residues different than the ones upon binding DNA ‘a’, indicating that EcMazE indeed binds DNA ‘a’ specifically with much higher affinity.
The 1H and 15N assigned resonances of EcMazE1–50 and of full-length EcMazE in complex with DNA ‘a’ have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu/) under accession numbers 25092 and 25094, respectively.
Based on the chemical shift perturbations shown in Figure 5, we used this information to drive the docking of the DNA ‘a’ fragment onto the EcMazE1–50. The chemical shift perturbations are in agreement with the X-ray structure of the Rickettsia felis VapBC2–DNA complex (PDB entry 3ZVK), which adopts the same fold as the EcMazE N-terminal domain (Supplementary Figure S6). Besides the similar fold of the homologous proteins, the central region of the DNA involved in binding VapB2 (ATATATACT) is identical to that in the DNA ‘a’ fragment we used, which we have demonstrated to bind specifically to the antitoxin EcMazE.
Figure 6 shows the structural models of the EcMazE1–50–DNA complex resulting from the structure calculation procedure using HADDOCK. The structural statistics are summarized in Table 3. The structural coordinates and experimentally derived restraints have been deposited in the PDB with accession number 2MRU.
Figure 6.
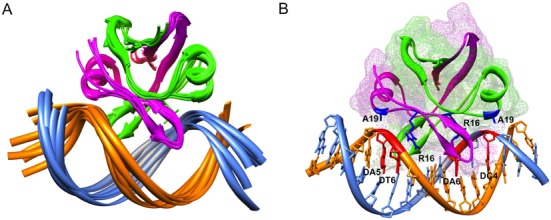
Structural model of the EcMazE1–50–DNA ‘a’ complex. (A) Cartoon representation of the ensemble of the seven HADDOCK structures with the lowest interaction energies and lowest AIR violations. The two EcMazE1–50 monomers are colored green and magenta, the two DNA strands in orange and sky-blue. (B) Details of the EcMazE1–50–DNA complex showing the lowest-interaction energy structure of the ensemble. Color coding as in (A). The EcMazE1–50 dimer is also shown in mesh surface. Residues and nucleotides involved in H-bonding common in the ensemble are shown in blue (EcMazE1–50) and red (DNA) sticks, respectively. Figures prepared using Chimera.
Table 3. Structural statistics over the seven Haddock structures of EcMazE1–50-DNA ‘a’.
| Ambiguous interaction restraints | 34 | |
| CNS interaction energies (kcal/mol) | ||
| Etotal | −208.1 ± 25.8 | |
| Evdw | −70.8 ± 8.6 | |
| Eelec | −501.9 ± 17.5 | |
| Restraint statistics | ||
| AIR violations >0.3 Å | 1.57 ± 0.79 | |
| Buried surface areaa (Å2) | EcMazE1–50 | DNA |
| 953.2 ± 44.8 | 898.5 ± 48.6 | |
| RMSD from averageb (Å) | Residues 2–49 | |
| Backbone N, CA, C′ | 0.52 ± 0.12 | |
| Heavy atoms | 0.87 ± 0.10 | |
| Ramachandran plotb | Residues 2–49 | |
| Most favored regions (%) | 79.6 ± 3.9 | |
| Additional allowed regions (%) | 19.3 ± 4.1 | |
| Generously allowed regions (%) | 1.0 ± 1.3 | |
| Disallowed regions (%) | 0.0 | |
aBSAs with their corresponding standard deviations calculated between the EcMazE1–50 dimer and the double-stranded DNA using PDBePISA (http://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver).
bValues with their corresponding standard deviations are reported for the EcMazE1–50 homodimer. Ramachandran statistics obtained from PROCHECK analysis.
A closer view into this structural model reveals that the homodimer EcMazE1–50 binds into the major groove of double-stranded DNA ‘a’, involving key residues W9, N11, R16 for the main interactions with the oligonucleotide. The complex shows a large concave surface for protein interaction in the center of the oligonucleotide fragment, resulting from widening of the major groove. In this model, the R16 side-chain and the A19 backbone amide make specific hydrogen bonds with the nucleotide bases at positions 6 and 4 or 5, respectively. The formation of an H-bond of the A19 amide proton is in agreement with the downfield shift observed for this proton upon DNA binding (Figure 4B). Additional electrostatic interactions between the positive-charged residues K7 and R8 with the DNA backbone stabilize the EcMazE1–50–DNA complex. Residues W9, N11 and R16 are three key residues in EcMazE1–50–DNA complex which correspond to N9, Q11 and R16 in VapB2 for DNA binding (49). The side-chains of these homologous residues show striking similar structural conformations (Supplementary Figure S6B), indicating the common key-role for these three residues in DNA recognition.
In order to compare the structural and dynamical characteristics between truncated EcMazE1–50-DNA with full-length EcMazE–DNA complexes, we employed SAXS on both EcMazE-DNA ‘a’ complexes. SAXS is particularly suitable for studying less structured systems, and especially complementary with NMR (37,57,58). No other techniques so far have reported the behavior of EcMazE with its long disordered tails in solution. Complementary to validating our NMR EcMazE1–50-DNA structure, we aimed to investigate the structural dynamics of the DNA-binding domain and the extended toxin-neutralizing domain in the full-length protein. The SAXS data collection, structural parameters and model statistics derived from the Guinier analysis of both full-length EcMazE-DNA and truncated EcMazE1–50–DNA complexes are given in Table 4. The estimated molecular masses determined by Guinier I(0) analysis, SAXSMoW (59) and QR (53) agree well with the one predicted from the corresponding sequences. The overall size of both systems was examined by monitoring the Rg and Dmax values. As expected, a comparison of these parameters for EcMazE1–50-DNA and full-length EcMazE-DNA, respectively, indicates that there is a substantial difference in terms of size between both scattering particles (Rg 25 Å versus 30 Å, and Dmax 71 Å versus 91 Å; see also Figure 7C). Furthermore, a comparison between the normalized Kratky plots of both EcMazE–DNA complexes (Figure 7D) reveals that the EcMazE1–50-DNA shows diminished internal flexibility compared to full-length EcMazE-DNA. This is in perfect agreement with our NMR relaxation data (Supplementary Figure S2), and can be explained by the absence of the long disordered C-terminal tail in the truncated EcMazE1–50. Such plots show a maximum value of ∼1.3 at a q.Rg value of around 2.2 for the EcMazE1–50–DNA ‘a’ complex and around 2.6 for the full-length EcMazE–DNA ‘a’ complex. None of the two normalized Kratky return to zero, indicating the presence of highly flexible regions in the scattering particle mainly due to the His-tag in both complexes and increased flexibility is even more present in the full-length EcMazE–DNA ‘a’ complex due to the extended disordered C-terminal tails, missing in the EcMazE1–50–DNA ‘a’ complex. Because of the significant degree of flexibility present in both systems, it is unlike that a single conformer can account for the experimental SAXS data. We therefore determined the minimal ensemble of structures (MES) sufficient to describe the SAXS data and the peculiar dynamics of the complex. In the case of EcMazE1–50-DNA ‘a’, the MES turned out to be as little as two models (χ2 = 0.64), whereas a minimal ensemble of three structures is needed for full-length EcMazE-DNA ‘a’ (χ2 = 0.96) (Figure 7A and B). The major source of variability required for a good agreement with the SAXS data is likely attributed to the flexible C-terminus more than at the N-terminal His-tag.
Table 4. SAXS data collection and scattering-derived parameters for EcMazE–DNA complexes.
| EcMazE1–50-‘a’ | EcMazE-‘a’ | |
|---|---|---|
| Data collection parameters | ||
| Beam line | SWING | SWING |
| HPLC column | KW402.5–4F | KW402.5–4F |
| Wavelength (Å) | 1.03 | 1.03 |
| q range | 0.012–0.299 | 0.012–0.331 |
| Injected concentrations | 90 μl at 7 mg/ml | 90 μl at 8 mg/ml |
| Temperature (K) | 283 | 283 |
| Structural parameters | ||
| I(0) (from Guinier) | 0.52 ± 0.30.10−2 | 0.24.10−3 ± 0.38.10−6 |
| Rg (Å) (from Guinier) | 25.51 ± 0.19 | 30.26 ± 0.07 |
| I(0) (from p(r)) | 0.51 ± 0.10.10−2 | 0.24.10−3 ± 0.21.10−6 |
| Rg (Å) (from p(r)) | 24.78 ± 0.06 | 30.96 ± 0.03 |
| Dmax (Å) | 71 | 91 |
| Molecular mass determination | ||
| SAXSMoW (kDa)a | 29.0 | 32.3 |
| QR (kDa)a | 28.1 | 30.2 |
| Theoretical MM from sequence (kDa) | 24.3 | 31.7 |
| Model statistics | ||
| χ2 | 0.64 | 0.96 |
| χ2free | 0.92 | 1.15 |
| RSAS (%) | 0.9927 | 0.0126 |
aFor the q range reported above in the table.
Figure 7.
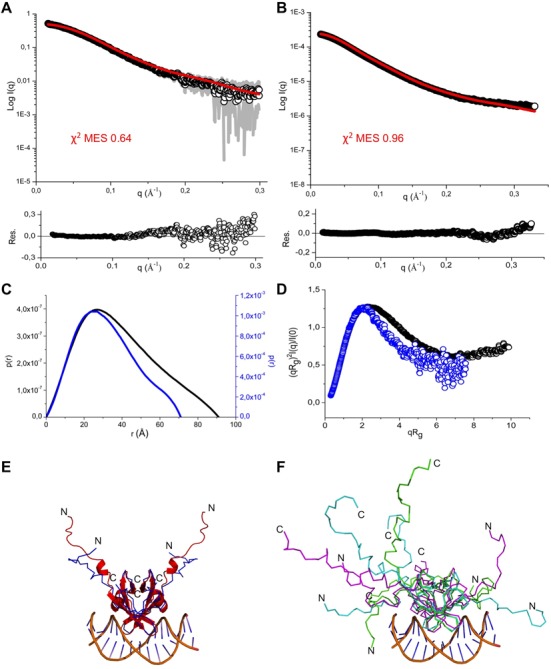
Small angle X-ray scattering of EcMazE-DNA ‘a’. (A, B) The experimental SAXS curve for EcMazE1–50–DNA ‘a’ and full-length EcMazE–DNA ‘a’ complexes are shown in open dots, while the error margins are shown in gray. The fit of the minimal search ensemble (MES) of two structures for EcMazE1–50-DNA ‘a’ is reported in red (A), while for full-length EcMazE-DNA ‘a’, the fit of the MES is with three structures (red line in (B)). The residual fitting is reported below for both systems. (C) Overlay of two p(r) function of EcMazE1–50-DNA ‘a’ (blue) and full-length EcMazE-DNA ‘a’ (black). (D) Overlay of two normalized Kratky plots corresponding to the EcMazE1–50-DNA ‘a’ (blue) and full-length EcMazE-DNA ‘a’ (black) shown as open dots. (E, F) A cartoon representation of the minimal set of two NMR structures of the EcMazE1–50–DNA ‘a’ complex and three structures of the full-length EcMazE–DNA ‘a’ complex. The N-terminal His-tag and the extended flexible C-terminal tails are indicated by N and C. Panels (E) and (F) were created using PyMol.
To verify that our model-data were accurately determined, we calculated the χ2 free and Rsas for both complexes (53). From the high quality of these values (see Table 4), we can conclude that our analysis was not overfitted and the models are in good agreement with the experimental SAXS data.
To confirm the correctness of our EcMazE-DNA structural models, paramagnetic spin labels were introduced into the ‘a’ oligomer. The PRE was measured by monitoring peak intensities in 2D 1H-15N-HSQC spectra upon the reduction of the paramagnetic iodo-acetamido-proxyl labels to its diamagnetic form. Paramagnetic probes like this nitroxide spin-label influence the relaxation behavior of nearby signals (distance <∼10 Å). Transverse relaxation (T2) enhancement leads to broader signals, which in turn lowers their intensity. Due to the relatively short-range effect only NH signals close to the paramagnetic DNA tag are expected to show an effect. While most signals of EcMazE are only slightly or not affected by the paramagnetic probe, the signals of H3, but especially of A19, T20 and M22, change their intensities significantly, in agreement with the position and orientation of the DNA relative to the EcMazE1–50 dimer in the complex (Supplementary Figure S7).
DISCUSSION
The E. coli mazEF operon was the first TA module that was identified on a chromosome (33) and remains one of the best characterized TA modules in terms of biochemical and physiological functions of the toxin. E. coli mazEF is autoregulated with MazE being the primary transcription factor and MazF modulating its activity. The mazEF operator consists of three consecutive independent MazE binding sites that differ in affinity up to one order of magnitude. Our binding data thus improve the model of three non-interacting, quasi-equivalent binding sites published earlier (34). Three EcMazE dimers bind at the promoter ‘cab’ sequence with apparent binding constants in the micromolar range and favorable enthalpic components dominating the Gibbs free energy. The interaction is specific and the presence of the intrinsically disordered EcMazF-neutralizing tail does not significantly influences the affinity of the protein for either a single site or the complete operator (34).
We determined an accurate structural model of the EcMazE–DNA complex using a combination of NMR and SAXS. This structure is in agreement with previous mutagenesis data and confirms that the C-terminal tail of EcMazE remains disordered and is not directly involved in DNA-binding upon interaction between the N-terminal EcMazE domain with DNA. EcMazE binds into the major groove of double-stranded DNA ‘a’, involving side-chains of residues W9, N11, R16 for the main interactions with the oligonucleotide. Indeed, the R16A mutant is essentially inactive (27). In addition we could identify further electrostatic interactions that likely participate in stabilizing the EcMazE–DNA complex, in particular between the positive-charged residues K7 and R8 and the DNA backbone. K7 and R8 were previously pointed as the primary DNA anchors for the MazE/MazF heterocomplex (32) while the R8A mutant shows reduced binding to the operator (27). Moreover, superposition of our EcMazE–DNA complex on the complex between EcMazE and a dromedary heavy chain antibody fragment indicates no structural clash (Supplementary Figure S8B) and thus confirms the correctness of our structural model as the presence of the heavy chain antibody fragment was shown to have no effect on the DNA-binding properties of MazE (27).
Our structural model resembles strongly the structure of Rickettsia felis VapB2 (RfVapB2) in complex with its operator. While cataloged as a ‘VapB’ due to its association with a VapC toxin, this antitoxin contains an AbrB-type DNA binding domain similar to EcMazE. Interestingly, RfVapB2 recognizes the same palindrome as EcMazE (5′-ATATAT-3′) using identical interactions with the N-terminal β-strand and hairpin. Interactions differ nevertheless at the periphery of the combining site, where the structures of both proteins diverge. There alternative contacts are seen, such as the backbone NH of A19 in EcMazE mimicking the interaction of the side chain of K19 from R. felis VapB with a DNA backbone phosphate.
The presence of EcMazF influences operator recognition by EcMazE (33). MazF proteins structurally resemble CcdB proteins and have a similar binding site for their cognate antitoxin. The antitoxins and toxins from both ccdAB and mazEF modules form chains of alternating toxin and antitoxin dimers (60,61). The F-plasmid ccdAB operon also contains an operator with multiple sites for the antitoxin, and the enhanced affinity of CcdA for its operator in the presence of CcdB is believed to stem from an avidity effect (10). Native mass spectrometry data on the Kis/Kid module (36), a homolog of E. coli MazEF, support a similar model of regulation within the mazEF modules. We observed, however, that MazF can increase the affinity of MazE even for a single operator site where no avidity effects are present. This is unexpected, but can be explained by either direct interactions between EcMazF and DNA or by thermodynamic stabilization of the N-terminal domain of EcMazE through interaction with MazF. Such stabilization-induced affinity enhancement was previously observed in the phd/doc module (9). Superposition of our EcMazE–DNA complex on the crystal structure of the EcMazE–EcMazF complex (32) (Supplementary Figure S8C) indicates additional protein–DNA interaction via the flanking basic regions of the EcMazF homodimer. This favors a model where the enhancement in DNA binding by EcMazF is caused by co-operative binding of the antitoxin and toxin to the DNA instead of an allosteric effect.
At very high EcMazF to EcMazE ratios, the affinity of MazE for the ‘a’ operator site diminishes again. This resembles the conditional co-operativity phenomenon previously observed for the ccdAB, phd/doc and relBE modules (9–12). The phenomenon however occurs only at EcMazF to EcMazE ratios that are never attained in vivo, and its physiological relevance is thus uncertain. We are currently also unable to provide a satisfactory mechanistic explanation.
The antitoxin EcMazE belongs to a large family of transcription regulators called the AbrB family, for which structural data on DNA recognition are relatively under-represented compared to other major families of DNA binding domains. In order to gain more insight into the DNA recognition and specificity in binding by AbrB-like domains, we performed a comparative study using all structurally homologous proteins in the PDB. AbrB-like transcriptional factors show a swapped hairpin β-strand motif. Conservation of the βαβ core as the main structural unit supports a common evolutionary origin between this AbrB-like fold and the double-psi β barrels (62). A combined sequence and secondary structure alignment of representative proteins of this superfamily show two main families: one formed by AbrB itself and its closest relatives, and the other by the bacterial EcMazE-like domain (Figure 8). The two families mainly differ in the positions of residues crucial for DNA binding: while family I is characterized by the presence of key residues in turn LP1 at positions 9 (W/N/S), 11 (Q/N/R) and the additional arginine at position 16, family II lacks this loop extension and DNA binding is driven by four key arginines at positions 8, 15, 23 and 24 (61). Figure 8B highlights the similar binding characteristics within family I, represented by EcMazE and RfVapB2, and the different binding surface within family II, represented by BsAbrB. Despite their similarities, EcMazE and RfVapB2 differ in the position of additional positively charged residues important for protein–DNA stabilization, N-terminal of the LP1 turn in the case of EcMazE and C-terminal in the case of RfVapB2.
Figure 8.
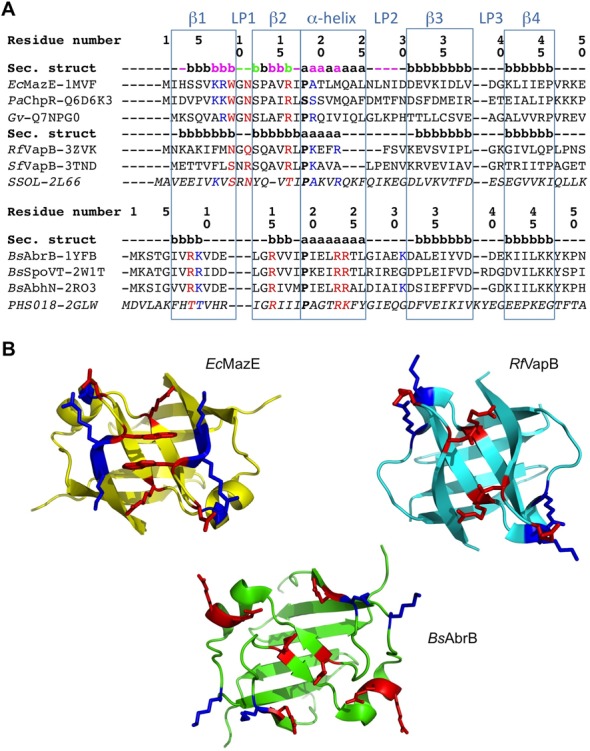
Structure-based sequence alignment of AbrB-like domain superfamily members. (A) Sequence alignment of the superfamily divided into two main families; the first one contains two subgroups. The consensus secondary structure within the superfamily is highlighted in light blue squares. Secondary structure elements within each family and sub-group are also given, representing the first one in each. The one belonging to EcMazE is color-coded by the CSP given in Figure 5 (Δδ > Δδav +  SD colored magenta, in green the residues of which their peaks disappear upon addition of the DNA). Residue numbering for the two families corresponds to that of EcMazE and BsAbrB, respectively. Key residues for DNA interaction are colored red, additional residues stabilizing the protein–DNA interaction in blue. The sequences from top to bottom are (corresponding PDB or Uniprot entries are given between parenthesis): EcMazE: Escherichia coli MazE antitoxin (1MVF); PaChpR: Pectobacterium atrosepticum ChpR suppressor of growth inhibitor (Q6D6K3); Gv: Gloeobacter violaceus cell growth regulatory protein (Q7NPG0); RfVapB: Rickettsia felis VapB antitoxin (3ZVK); SfVapB: Shigella flexneri VapB antitoxin (3TND); SSOL: Sulfolobus solfataricus transcription regulator (2L66); BsAbrB: Bacillus subtilis AbrB transition state regulator (1YFB); BsSpoVT: Bacillus subtilis SpoVT stage V sporulation protein T (2W1T); BsAbhN: Bacillus subtilis AbhN putative transition state regulator (2RO3); PHS018: Pyrococcus horikoshii S018 putative uncharacterized protein (2GLW). (B) Representative structures of the two AbrB-like domain families. Structures were superimposed using Pymol and thus are in the same orientation. Residues important for DNA binding are given in sticks and colored as defined in (A).
SD colored magenta, in green the residues of which their peaks disappear upon addition of the DNA). Residue numbering for the two families corresponds to that of EcMazE and BsAbrB, respectively. Key residues for DNA interaction are colored red, additional residues stabilizing the protein–DNA interaction in blue. The sequences from top to bottom are (corresponding PDB or Uniprot entries are given between parenthesis): EcMazE: Escherichia coli MazE antitoxin (1MVF); PaChpR: Pectobacterium atrosepticum ChpR suppressor of growth inhibitor (Q6D6K3); Gv: Gloeobacter violaceus cell growth regulatory protein (Q7NPG0); RfVapB: Rickettsia felis VapB antitoxin (3ZVK); SfVapB: Shigella flexneri VapB antitoxin (3TND); SSOL: Sulfolobus solfataricus transcription regulator (2L66); BsAbrB: Bacillus subtilis AbrB transition state regulator (1YFB); BsSpoVT: Bacillus subtilis SpoVT stage V sporulation protein T (2W1T); BsAbhN: Bacillus subtilis AbhN putative transition state regulator (2RO3); PHS018: Pyrococcus horikoshii S018 putative uncharacterized protein (2GLW). (B) Representative structures of the two AbrB-like domain families. Structures were superimposed using Pymol and thus are in the same orientation. Residues important for DNA binding are given in sticks and colored as defined in (A).
Based on the binding characteristics of both families we included and accordingly categorized some uncharacterized bacterial proteins, Pectobacterium atrosepticum ChpR suppressor of growth inhibitor (PaChpR) and Gloeobacter violaceus cell growth regulatory protein (Gv), and archaea structures, such as the Pyrococcus horikoshii S018 putative uncharacterized protein (PhS018) and thermoacidophilic Sulfolobus solfataricus Sso7c4 (SSOL). Both PaChpR and Gv possess the key and additional residues for DNA binding as in EcMazE and are thus predicted to have the same DNA-recognition site as the other members of this family.
PhS018 represents an archaeal intermediate between a double-psi (six β-strands) and a swapped-hairpin β-barrel consisting of four β-strands with a βαβ core. Despite the fact that PhS018 differs from the AbrB-like fold by the addition of one β-strand, it has been considered to be the dimeric ancestor of swapped-hairpin dimer (62). Alignment predicts this protein to recognize DNA in a similar way as members belonging to family II.
Differently, the archaeal DNA-binding homodimer Sso7c4 possesses residues that are highly conserved in the archaeal homologs (R11, N12, R22). Based on our alignment (Figure 8A), Sso7c4 aligns well with the EcMazE family, including key residues at positions 9, 11 and 16 important for the DNA binding. Interestingly it contains two residues necessary for DNA stabilization, K8 like in the case of EcMazE and R22 like in RfVapB2, thus representing a characteristic of both subgroups in family I.
DATABASE DEPOSITION
The 1H, 15N and 13C assigned resonances of EcMazE1–50 have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu/) under accession number 25086. The structural coordinates and experimentally derived restraints have been deposited in the PDB with accession number 2MRN.
The 1H and 15N assigned resonances of EcMazE1–50 in complex with DNA ‘a’ have been deposited in the BioMagResBank under accession number 25092. The structural coordinates and experimentally derived restraints have been deposited in the PDB with accession number 2MRU.
The 1H, 15N and 13C assigned resonances of full-length EcMazE have been deposited in the BioMagResBank under accession number 25093. The 1H and 15N assigned resonances of full-length EcMazE in complex with DNA ‘a’ have been deposited in the BioMagResBank under accession number 25094.
ACCESSION NUMBERS
25086, 25092, 25093, 25094, 2MRN and 2MRU.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Fonds Wetenschappelijk Onderzoek-Vlaanderen (FWO); Vlaams Instituut voor Biotechnologie (VIB); Onderzoeksraad of the Vrije Universiteit Brussel (VUB); Hercules Foundation; Biostruct-X; FWO [to V.Z.]; Austrian Science Foundation (FWF) [19902, 25880 to K.Z.]; DOCfFORTE Fellowship, Austrian Academy of Sciences [to E.S.]; ‘European Community-Access to Research Infrastructure Action of the Improving Human Potential Programme’ EU-NMR. Funding for open access charge: Internal VIB grant.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bravo A., de Torrontegui G., Díaz R. Identification of components of a new stability system of plasmid R1, ParD, that is close to the origin of replication of this plasmid. Mol. Gen. Genet. 1987;210:101–110. doi: 10.1007/BF00337764. [DOI] [PubMed] [Google Scholar]
- 2.Brown J.M., Shaw K.J. A novel family of Escherichia coli toxin-antitoxin gene pairs. J. Bacteriol. 2003;185:6600–6608. doi: 10.1128/JB.185.22.6600-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandey D.P., Gerdes K. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerdes K., Wagner E.G.H. RNA antitoxins. Curr. Opin. Microbiol. 2007;10:117–124. doi: 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Buts L., Lah J., Dao-Thi M., Wyns L., Loris R. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 2005;30:672–679. doi: 10.1016/j.tibs.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Fineran P.C., Blower T.R., Foulds I.J., Humphreys D.P., Lilley K.S., Salmond G.P.C. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl. Acad. Sci. U.S.A. 2009;106:894–899. doi: 10.1073/pnas.0808832106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X., Lord D.M., Cheng H.Y., Osbourne D.O., Hong S.H., Sanchez-Torres V., Quiroga C., Zheng K., Herrmann T., Peti W., et al. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012;8:855–861. doi: 10.1038/nchembio.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X., Lord D.M., Hong S.H., Peti W., Benedik M.J., Page R., Wood T.K. Type II toxin/antitoxin MqsR/MqsA controls type V toxin/antitoxin GhoT/GhoS. Environ. Microbiol. 2013;15:1734–1744. doi: 10.1111/1462-2920.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R.D., Charlier D., van Nuland N.A., Loris R. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell. 2010;142:101–111. doi: 10.1016/j.cell.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 10.De Jonge N., Garcia-Pino A., Buts L., Haesaerts S., Charlier D., Zangger K., Wyns L., De Greve H., Loris R. Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain. Mol. Cell. 2009;35:154–163. doi: 10.1016/j.molcel.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 11.Overgaard M., Borch J., Jørgensen M.G., Gerdes K. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008;69:841–857. doi: 10.1111/j.1365-2958.2008.06313.x. [DOI] [PubMed] [Google Scholar]
- 12.Afif H., Allali N., Couturier M., Van Melderen L. The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol. Microbiol. 2001;41:73–82. doi: 10.1046/j.1365-2958.2001.02492.x. [DOI] [PubMed] [Google Scholar]
- 13.Gerdes K., Rasmussen P.B., Molin S. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc. Natl. Acad. Sci. U.S.A. 1986b;83:3116–3120. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts R.C., Ström A.R., Helinski D.R. The parDE operon of the broad-host-range plasmid RK2 specifies growth inhibition associated with plasmid loss. J. Mol. Biol. 1994;237:35–51. doi: 10.1006/jmbi.1994.1207. [DOI] [PubMed] [Google Scholar]
- 15.Christensen S.K., Mikkelsen M., Pedersen K., Gerdes K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14328–14333. doi: 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albrethsen J., Agner J., Piersma S.R., Højrup P., Pham T.V., Weldingh K., Jimenez C.R., Andersen P., Rosenkrands I. Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteomics. 2013;12:1180–1191. doi: 10.1074/mcp.M112.018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aizenman E., Engelberg-Kulka H., Glaser G. An Escherichia coli chromosomal ‘addiction module’ regulated by guanosine 3′, 5′-bispyrophosphate: a model for programmed bacterial cell death. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelberg-Kulka H., Amitai S., Kolodkin-Gal I., Hazan R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006;2:1528–1526. doi: 10.1371/journal.pgen.0020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis K. Persister cells. Annu. Rev. Microbiol. 2010;64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 20.Gerdes K., Maisonneuve E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012;66:103–123. doi: 10.1146/annurev-micro-092611-150159. [DOI] [PubMed] [Google Scholar]
- 21.Tripathi A., Dewan P.C., Siddique S.A., Varadarajan R. MazF-induced growth inhibition and persister generation in Escherichia coli. J. Biol. Chem. 2014;289:4191–205. doi: 10.1074/jbc.M113.510511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loris R., Garcia-Pino A. Disorder- and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chem. Rev. 2014;114:6933–6947. doi: 10.1021/cr400656f. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchimoto S., Ohtsubo H., Ohtsubo E. Two genes, pemK and pemI, responsible for stable maintenance of resistance plasmid R100. J. Bacteriol. 1988;170:1461–1466. doi: 10.1128/jb.170.4.1461-1466.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masuda Y., Miyakawa K., Nishimura Y., Ohtsubo E. chpA and chpB, Escherichia coli chromosomal homologs of the pem locus responsible for stable maintenance of plasmid R100. J. Bacteriol. 1993;175:6850–6856. doi: 10.1128/jb.175.21.6850-6856.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J., Zhang Y., Hoeflich K.P., Ikura M., Qing G., Inouye M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell. 2003;12:913–923. doi: 10.1016/s1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 26.Vesper O., Amitai S., Belitsky M., Byrgazov K., Kaberdina A.C., Engelberg-Kulka H., Moll I. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell. 2011;147:147–157. doi: 10.1016/j.cell.2011.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loris R., Marianovsky I., Lah J., Laeremans T., Engelberg-Kulka H., Glaser G., Muyldermans S., Wyns L. Crystal structure of the intrinsically flexible addiction antidote MazE. J. Biol. Chem. 2003;278:28252–28257. doi: 10.1074/jbc.M302336200. [DOI] [PubMed] [Google Scholar]
- 28.Coles M., Djuranovic S., Söding J., Frickey T., Koretke K., Truffault V., Martin J., Lupas A.N. AbrB-like transcription factors assume a swapped hairpin fold that is evolutionarily related to double-psi beta barrels. Structure. 2005;13:919–928. doi: 10.1016/j.str.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 29.Chumsakul O., Takahashi H., Oshima T., Hishimoto T., Kanaya S., Ogasawara N., Ishikawa S. Genome-wide binding profiles of the Bacillus subtilis transition state regulator AbrB and its homolog Abh reveals their interactive role in transcriptional regulation. Nucleic Acids Res. 2011;39:414–428. doi: 10.1093/nar/gkq780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bagyan I., Hobot J., Cutting S. A compartmentalized regulator of developmental gene expression in Bacillus subtilis. J. Bacteriol. 1996;178:4500–4507. doi: 10.1128/jb.178.15.4500-4507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asen I., Djuranovic S., Lupas A.N., Zeth K. Crystal structure of SpoVT, the final modulator of gene expression during spore development in Bacillus subtilis. J. Mol. Biol. 2009;386:962–975. doi: 10.1016/j.jmb.2008.10.061. [DOI] [PubMed] [Google Scholar]
- 32.Kamada K., Hanaoka F., Burley S.K. Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol. Cell. 2003;11:875–884. doi: 10.1016/s1097-2765(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 33.Marianovsky I., Aizenman E., Engelberg-Kulka H., Glaser G. The regulation of the Escherichia coli mazEF promoter involves an unusual alternating palindrome. J. Biol. Chem. 2001;276:5975–5984. doi: 10.1074/jbc.M008832200. [DOI] [PubMed] [Google Scholar]
- 34.Lah J., Marianovsky I., Glaser G., Engelberg-Kulka H., Kinne J., Wyns L., Loris R. Recognition of the intrinsically flexible addiction antidote MazE by a dromedary single domain antibody fragment. Structure, thermodynamics of binding, stability, and influence on interactions with DNA. J. Biol. Chem. 2003;278:14101–14111. doi: 10.1074/jbc.M209855200. [DOI] [PubMed] [Google Scholar]
- 35.Bailey S.E.S., Hayes F. Influence of operator site geometry on transcriptional control by the YefM-YoeB toxin-antitoxin complex. J. Bacteriol. 2009;191:762–772. doi: 10.1128/JB.01331-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monti M.C., Hernandez-Arriaga A.M., Kamphuis M.B., Lopez-Villarejo J., Heck A.J.R., Boelens R., Diaz-Orejas R., van den Heuvel R.H.H. Interactions of Kid–Kis toxin–antitoxin complexes with the parD operator-promoter region of plasmid R1 are piloted by the Kis antitoxin and tuned by the stoichiometry of Kid–Kis oligomers. Nucleic Acids Res. 2007;35:1737–1749. doi: 10.1093/nar/gkm073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oberer M., Zangger K., Gruber K., Keller W. The solution structure of ParD, the antidote of the ParDE toxin antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci. 2007;16:1676–1688. doi: 10.1110/ps.062680707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 39.Johnson B.A., Blevins R.A. NMR View: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 40.Cavanagh J., Fairbrother W.J., Palmer A.G., Rance M., Skelton N.J. Protein NMR Spectroscopy. 2nd edn. San Diego, CA: Academic Press; 2007. [Google Scholar]
- 41.Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 42.Guntert P., Mumenthaler C., Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 43.Herrmann T., Guntert P., Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- 44.Shen Y., Delaglio F., Cornilescu G., Bax A. TALOS plus: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nederveen A.J., Doreleijers J.F., Vranken W., Miller Z., Spronk C.A., Nabuurs S.B., Guntert P., Livny M., Markley J.L., Nilges M., et al. RECOORD: a recalculated coordinate database of 500 +proteins from the PDB using restraints from the BioMagResBank. Proteins. 2005;59:662–672. doi: 10.1002/prot.20408. [DOI] [PubMed] [Google Scholar]
- 46.Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 47.Dominguez C., Boelens R., Bonvin A.M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 48.Hubbard S., Thornton J. Naccess—Computer Program. 1993 [Google Scholar]
- 49.Maté M.J., Vincentelli R., Foos N., Raoult D., Cambillau C., Ortiz-Lombardía M. Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res. 2011;40:3245–3258. doi: 10.1093/nar/gkr1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 51.Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1282. [Google Scholar]
- 52.Konarev P.V., Petoukhov M.V., Volkov V.V., Svergun D.I. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Cryst. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rambo R.P., Tainer J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pelikan M., Hura G.L., Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneidman-Duhovny D., Hammel M., Sali A. FoXs: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–W544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weinkam P., Pons J., Sali A. Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. U.S.A. 2012;109:4875–4880. doi: 10.1073/pnas.1116274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sterckx Y.G., Volkov A.N., Vranken W.F., Kragelj J., Jensen M.R., Buts L., Garcia-Pino A., Jové T., Van Melderen L., Blackledge M., et al. Small-angle X-Ray scattering- and nuclear magnetic resonance-derived conformational ensemble of the highly flexible antitoxin PaaA2. Structure. 2014;22:854–865. doi: 10.1016/j.str.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 58.Zorzini V., Buts L., Sleutel M., Garcia-Pino A., Talavera A., Haesaerts S., Greve H.D., Cheung A., van Nuland N.A., Loris R. Structural and biophysical characterization of Staphylococcus aureus SaMazF shows conservation of functional dynamics. Nucleic Acids Res. 2014;42:6709–6725. doi: 10.1093/nar/gku266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fischer H., de Oliveira Neto M., Napolitano H.B., Craievich A.F., Polikarpov I. The molecular weight of proteins in solution can be determined from a single SAXS measurement on a relative scale. J. Appl. Cryst. 2010;43:101–109. [Google Scholar]
- 60.Dao-Thi M.H., Charlier D., Loris R., Maes D., Messens J., Wyns L., Backmann J. Intricate interactions within the ccd plasmid addiction system. J. Biol. Chem. 2002;277:3733–3742. doi: 10.1074/jbc.M105505200. [DOI] [PubMed] [Google Scholar]
- 61.Kamphuis M.B., Monti M.C., van den Heuvel R.H., Santos-Sierra S., Folkers G.E., Lemonnier M., Díaz-Orejas R., Heck A.J., Boelens R. Interactions between the toxin Kid of the bacterial parD system and the antitoxins Kis and MazE. Proteins. 2007;67:219–231. doi: 10.1002/prot.21254. [DOI] [PubMed] [Google Scholar]
- 62.Coles M., Hulko M., Djuranovic S., Truffault V., Koretke K., Martin J., Lupas A.N. Common evolutionary origin of swapped-hairpin and double-psi beta barrels. Structure. 2006;14:1489–1498. doi: 10.1016/j.str.2006.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.