


Abstract
The balance between exonuclease and polymerase activities promotes DNA synthesis over degradation when nucleotides are correctly added to the new strand by replicative B-family polymerases. Misincorporations shift the balance toward the exonuclease site, and the balance tips back in favor of DNA synthesis when the incorrect nucleotides have been removed. Most B-family DNA polymerases have an extended β-hairpin loop that appears to be important for switching from the exonuclease site to the polymerase site, a process that affects fidelity of the DNA polymerase. Here, we show that DNA polymerase ε can switch between the polymerase site and exonuclease site in a processive manner despite the absence of an extended β-hairpin loop. K967 and R988 are two conserved amino acids in the palm and thumb domain that interact with bases on the primer strand in the minor groove at positions n−2 and n−4/n−5, respectively. DNA polymerase ε depends on both K967 and R988 to stabilize the 3′-terminus of the DNA within the polymerase site and on R988 to processively switch between the exonuclease and polymerase sites. Based on a structural alignment with DNA polymerase δ, we propose that arginines corresponding to R988 might have a similar function in other B-family polymerases.
INTRODUCTION
The fidelity of DNA replication depends on the accuracy with which the DNA polymerase incorporates a nucleotide, the enzyme's built-in exonuclease activity that removes misincorporated nucleotides, and the mismatch repair system that corrects errors that elude the DNA polymerase (1). In eukaryotes, DNA polymerase ε (Pol ε) is considered to be responsible for leading strand synthesis and DNA polymerase δ (Pol δ) for lagging strand synthesis (2,3). Both enzymes have proofreading capacity with a built in 3′–5′ exonuclease activity, and the exonuclease and the polymerase sites are located approximately 40 Å apart (4,5).
The transfer of the 3′-terminus of the primer strand between the polymerase and exonuclease sites occurs either through an intermolecular mechanism that involves dissociation and reassociation events or through an intramolecular mechanism without dissociating from the template DNA. In general, DNA polymerases are non-exclusive between these options (6).
The ability of the replicative DNA polymerases to both synthesize and degrade DNA allows the polymerases to build DNA with very high fidelity. However, the partitioning between the polymerase and exonuclease sites not only influences the fidelity, it also determines whether the net result is synthesis of a new DNA strand. The concept of a ‘balance’ was used as a metaphor by Reha-Krantz to describe the relationship between the two activities (6). Under normal circumstances, the polymerase activity dominates over the exonuclease activity. However, there are substitutions in DNA polymerases that affect this balance by creating mutator polymerases or antimutator polymerases. Structural studies have revealed that a large change occurs in the positioning of the thumb domain so as to accommodate the transfer of the DNA primer terminus between the polymerase and exonuclease active sites (7). The separation of the primer strand from the template DNA is an energetically unfavorable process that requires assistance to occur.
The mechanism by which the 3′-terminus of the growing DNA strand is transferred between the polymerase site and exonuclease site is still unclear. Crystal structures have shown that at least three to four base-pairs are separated, and this allows the single-stranded DNA to interact with a groove within the exonuclease domain that leads to the exonuclease active site (8). Genetic studies of bacteriophage T4 revealed that mutations leading to substitutions in an extended β-hairpin loop influence the fidelity of the T4 DNA polymerase (9,10). Biochemical studies later showed that the extended β-hairpin loop influences the switch between the exonuclease site and polymerase site in the B-family DNA polymerase from bacteriophage RB69 (RB69 gp43) (11). Pol ε consists of four subunits, Pol2 (catalytic subunit), Dpb2 (interacts with the replicative polymerase), Dpb3 and Dpb4 (Dpb3 and Dpb4 interacts with double-stranded DNA). The recently solved crystal structure of the catalytic domain of Pol2 (Pol2core (aa 1–1228)) revealed that Pol ε lacks the extended β-hairpin loop found in most other B-family DNA polymerases that has been proposed to interact with the template DNA and to function as a wedge by breaking hydrogen bonds between the template and nascent strands (5,11,12). The absence of an extended β-hairpin loop in Pol ε was unexpected because Pol ε replicates DNA with very high fidelity (13).
B-family polymerases such as Pol ε, Pol δ and RB69 gp43 contain a highly conserved motif identified as motif A (14). Motif A contains a conserved leucine residue with the exception of Pol ε that has a methionine at this position (M644 in Figure 1B) (14,15). The conserved Leu/Met has been shown to be critical for selection of the correct dNTP during replication. Substitutions at this residue have proven to be valuable for understanding the biological functions of Pol ε and Pol δ (2,3). Two such variants of Pol ε—M644G and M644L—have been used to study ribonucleotide incorporation by Pol ε, and both enzymes have distinct phenotypes. Compared to wild-type Pol ε, M644G causes an increased mutation rate (a mutator phenotype) whereas M644L has a lower mutation rate (an antimutator phenotype) (16). The contrasting properties have been suggested to be the consequence of altered partitioning between the polymerase and exonuclease active sites (16).
Figure 1.
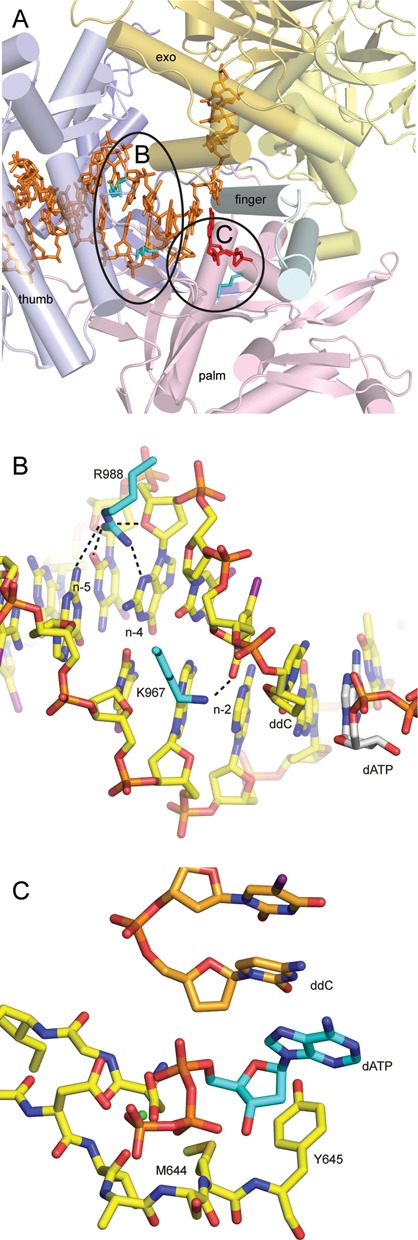
Positions of M644, K967 and R988 in the structure of Pol ε. (A) Overview of the structure of Pol ε (PDB 4M8O(5)). Encircled regions are enlarged in panels B and C. (B) K967 forms a hydrogen bond with the base on the primer strand at position n−2. R988 is able to form hydrogen bonds with both the template and primer strands at positions n−4 and n−5. (C) M644 is positioned under the 3′-primer end and the incoming nucleotide.
B-family polymerases also contain a conserved KKRY motif that has been shown to be important for stabilizing the primer terminus in the polymerase active site (17). K967 in Pol ε is part of this KKRY motif and is homologous to K814 in Pol δ, K706 in RB69 gp43 and K498 in the DNA polymerase from bacteriophage ϕ29. It was earlier shown that K706 in RB69 gp43 interacts with the base at position n−2 of the newly formed duplex DNA (17). The K706A substitution almost completely abolishes polymerase activity, and this implies that K706 is required to stabilize the 3′-terminus of the DNA within the polymerase site (18). This also suggests that misincorporations that result in a loss of interaction with K706 will destabilize the 3′-terminus in the polymerase site and shift the balance toward the exonuclease site.
Here, we show that Pol ε is able to transfer the 3′-terminus of the DNA between the polymerase site and exonuclease site in a processive mode via an intramolecular mechanism. We demonstrate that the M644G and M644L mutants, which were previously used in studies demonstrating that Pol ε replicates the leading strand and incorporates ribonucleotides, have a shifted balance between the polymerase and exonuclease sites. Finally, we show that Pol ε depends on R988 and K967 to stabilize the 3′-terminus within the polymerase site and to carry out processive DNA synthesis and that R988 is important for the transfer of DNA from the exonuclease site to the polymerase site.
MATERIALS AND METHODS
DNA substrates
Oligonucleotides were purchased from MWG Operon, Germany. The oligonucleotides were gel purified before primer-templates were prepared by mixing 6 μM primer strand with 7.2 μM template strand in a buffer containing 100 mM Tris–HCl (pH 7.5) and 100 mM NaCl, heating to 85°C for 5 min in a heating block, and slow cooling to room temperature. The oligonucleotides are listed in Table 1.
Table 1. Oligonucleotides used for primer extension assays.
| 50/80-mer | 5′GATCAGACTGTCCTTAGAGGATACTCGCTCGCAGCCGTCCACTCAACTCA3′ |
| 3′CTAGTCTGACAGGAATCTCCTATGAGCGAGCGTCGGCAGGTGAGTTGAGTAGGTCTTGTTGCAGTGACTGATAGTTCGAC5′ | |
| 51T/80-mer | 5′GATCAGACTGTCCTTAGAGGATACTCGCTCGCAGCCGTCCACTCAACTCATT3′ |
| 3′CTAGTCTGACAGGAATCTCCTATGAGCGAGCGTCGGCAGGTGAGTTGAGTAGGTCTTGTTGCAGTGACTGATAGTTCGAC5′ |
Over-expression and purification of proteins
Saccharomyces cerevisiae Pol ε, Pol ε M644G, Pol ε M644L, Pol ε K967A, Pol ε R988A and Pol ε R988K were over-expressed under the control of a galactose-inducible promoter in the S. cerevisiae strain Py116 and were purified by conventional chromatography as previously described (19,20). Briefly, the proteins were purified by conventional chromatography over a phosphocellulose column, a MonoQ column, a MonoS column, and, finally, a Superose 6 column. The proteins were stored in a buffer containing 25 mM Hepes–NaOH (pH 7.6), 10% glycerol, 1 mM dithiothreitol and 400 mM NaAc (pH 7.8). The exonuclease-deficient enzyme (D290A/E292A) was created by site-directed mutagenesis in Pol2. Saccharomyces cerevisiae Pol ε exo−, Pol2core exo− (aa 1–1228), and Pol2core (aa 1–1228) were fused with a glutathione-S-transferase tag (GST-tag) at the N-terminus and purified as previously described (5). Briefly, the proteins were purified over a phosphocellulose column followed by affinity purification on glutathione-sepharose and further purification on a MonoQ column and Superose 6 column. The proteins were stored in a buffer containing 25 mM Hepes–NaOH (pH 7.6), 10% glycerol, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and 800 mM NaAc (pH 7.8).
Primer extension assays
All experiments were performed in RQ reaction buffer containing 20 mM Tris–HCl (pH 7.8), 100 μg/ml bovine serum albumin (BSA), and 1 mM dithiothreitol. DNA and enzyme were preincubated together and each reaction was started by the addition of 16 mM MgAc2 and dNTPs. The reaction was stopped after the indicated time by adding 20 μl of 95% formamide, 20 mM ethylenediaminetetraacetic acid (EDTA) and 0.1% bromophenol blue. A total of 8 μl of the reaction mixture was loaded onto a denaturing 10% polyacrylamide gel containing 8 M urea and 25% formamide in 1× TBE buffer. The gel was scanned with a Typhoon Scanner 9400 (GE Healthcare) at the Alexa 532 nm setting to excite the fluorophore, tetrachlorofluorescein (TET), that was covalently bound to the 5′-end of the primer. The band intensities were quantified with the Image Quant 5.2 software (GE Healthcare).
Polymerase to exonuclease switch
Polymerase to exonuclease switch assays were performed by mixing 10 μl of reaction mixture A with 10 μl of reaction mixture B and incubating for 2.5 min at 30°C. Reaction mixture A contained 10 nM primer-template (50/80-mer) and 22 nM enzyme in RQ buffer, and reaction mixture B contained 2 mM dATP, 2 mM dTTP, 2 mM dCTP, 16 mM MgAc2 and 0.002 mg/ml heparin in RQ buffer (heparin was omitted in reactions that allowed for multiple binding events). Reactions were stopped by adding 20 μl of 95% formamide, 20 mM EDTA and 0.1% bromophenol blue. Band intensities were quantified with Image Quant 5.2, and relative intensities were calculated by dividing the intensity of a specific band by the total intensity of all extension products.
Exonuclease to polymerase switch
Exonuclease to polymerase switch assays were performed by mixing 10 μl of reaction mixture A with 10 μl of reaction mixture B and incubating for 2.5 min at 30°C. Reaction mixture A contained 10 nM mismatched primer-template (51T/80-mer) and 22 nM enzyme in RQ buffer, and reaction mixture B contained 16 mM MgAc2, 2× the estimated in vivo dNTP concentration (dCTP = 78 μM, dTTP = 132 μM, dATP = 44 μM and dGTP = 22 μM (21)) and 0.002 mg/ml heparin in RQ buffer (heparin was omitted in reactions that allowed for multiple binding events). Reactions were stopped by adding 20 μl of 95% formamide, 20 mM EDTA and 0.1% bromophenol blue. Band intensities were quantified with Image Quant 5.2, and relative intensities were calculated by dividing the intensity of the extension products by the sum of the excision and extension products.
Processivity assay
Processivity assays were performed by mixing 10 μl of reaction mixture A with 10 μl of reaction mixture B and incubating for 1 or 2 min at 30°C. Reaction mixture A contained 10 nM of perfectly matched primer-template (50/80-mer) and 22 nM enzyme in RQ buffer, and reaction mixture B contained 16 mM MgAc2, 2× the estimated in vivo dNTP concentration (dCTP = 78 μM, dTTP = 132 μM, dATP = 44 μM and dGTP = 22 μM (21)), and 0.002 mg/ml heparin in RQ buffer (heparin was omitted in reactions that allowed for multiple binding events to occur). Reactions were stopped by adding 20 μl of 95% formamide, 20 mM EDTA and 0.1% bromophenol blue.
dNTP concentration-dependent exonuclease to polymerase active site switch
Assays to monitor the effect of dNTP concentration on switching between the exonuclease and polymerase active sites were performed by mixing 10 μl of reaction mixture A with 10 μl of reaction mixture B and incubating for 10 min at 30°C. Reactions with multiple-binding events were performed at low enzyme concentration, high DNA concentration and varying dNTP concentrations. Reaction mixture A contained 10 nM of perfectly matched primer-template (50/80-mer) and 2.3 nM enzyme in RQ buffer, and reaction mixture B contained 16 mM MgAc2 and nucleotide concentrations starting at 2× the estimated in vivo dNTP concentration (21) followed by a 2-fold step dilution series in RQ buffer. Reactions were stopped by adding 20 μl of 95% formamide, 20 mM EDTA and 0.1% bromophenol blue.
Tetrad analysis
A linearized integration plasmid (called p173 (3)) carrying a mutation resulting in a K967A, R988A or R988K substitution was integrated into a diploid E134 yeast strain (ade5-1 lys2::InsEA14 trp1-289 his7-2 leu2-3,112 ura3-52). URA3 was first used for selection of integration, and this was followed by selection on 5-FOA to detect the loss of URA3 that occurred when the URA3 gene was looped out leaving the POL2 gene with the specific mutation. Each diploid strain was sequenced across the POL2 gene to confirm that the selected mutation was correctly integrated in the heterozygote strain. Tetrad dissections were carried out as previously described (22).
RESULTS
Transfer from the polymerase site to the exonuclease site
To determine whether Pol ε is able to transfer the 3′-terminus of the DNA from the polymerase site to the exonuclease site without dissociating from the DNA, we used heparin as a trap to capture any DNA polymerase molecules not bound to DNA. Titration experiments were done to ensure that the concentration of heparin would efficiently trap-free enzyme without competing with the protein bound to the DNA (data not shown). The primer-extension experiment was carried out with 1 mM concentrations of each of the three nucleotides dATP, dCTP and dTTP. The fourth nucleotide, dGTP, was omitted to force a misincorporation opposite the templating C. In the first experiment, Pol2core exo− was added to the reaction mix under conditions allowing multiple binding events (no heparin added) or only a single binding event (heparin added) (Figure 2). Multiple binding events allowed Pol2core exo− to extend mismatches to full-length products after repeated association and dissociation events. The addition of heparin trapped-free Pol2core exo− as soon as the enzyme dissociated from the template, and this resulted in products ending at position m–1, m and m+1 or higher. The mismatch is located at position m. Dissociation prior to the incorporation of a mismatch or the removal of the mismatch results in an oligonucleotide that is shorter by one nucleotide (m–1), and extension by one nucleotide results in the products seen at position m+1 or higher (a small fraction of the mismatches can be extended in the presence of 1 mM dNTP). Next we added Pol2core and monitored the formation of products after multiple binding events or single binding events. If Pol2core is able to transfer the primer terminus from the polymerase site to the exonuclease site without dissociating from the substrate, then less products should be found at positions m and m+1 or higher in the presence of heparin when compared to Pol2core exo−. This was found to be the case in our experiment and the product at position m–1 increased due to the removal of misincorporated nucleotides. To ask whether the accessory subunits and/or the C-terminal domain of Pol2 can influence this switch, we repeated the experiment with Pol ε exo− and Pol ε (Figure 2). To calculate the percent of incorporated mismatch, the band intensity of mismatched and extension products were divided by the total intensity of all the extended bands, including the mismatch. The quantitative analysis showed that 40% of the extended DNA was elongated to or past the mismatch by Pol ε exo−, but only 7% of the DNA was elongated to or past the mismatch by wild-type Pol ε (Table 2). This suggests that 82% of the mismatched primer termini were corrected by wild-type Pol ε by shuttling the 3′-primer terminus to the exonuclease site. Quantification of the replication products from Pol2core showed that Pol2core exo− elongated to or past the mismatch in 36% of the cases, but only 7% of the products formed by Pol2core were elongated to or past the mismatch (Table 2). Thus, 81% of the mismatched primer termini were transferred to the exonuclease site and removed. The results from Pol2core and Pol ε were comparable, and this suggests that Dpb2, Dpb3, Dpb4 and the C-terminus of Pol2 are not required for an intramolecular transfer of DNA from the polymerase site to the exonuclease site.
Figure 2.
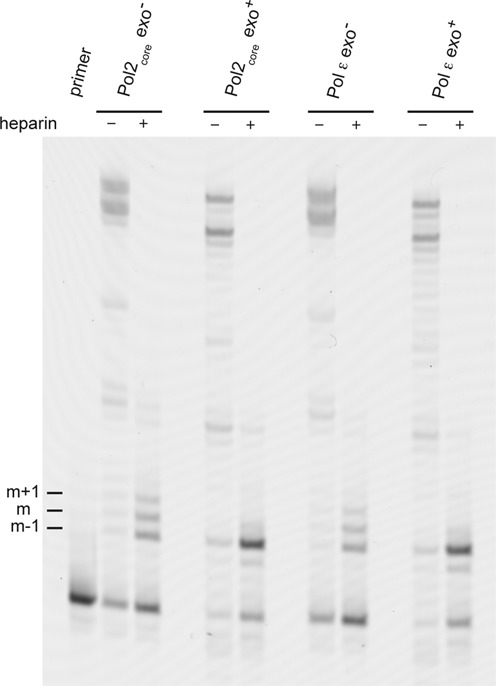
Polymerase site to exonuclease site switch. Preformed enzyme–DNA (50/80-mer) complexes were rapidly mixed with heparin, magnesium acetate and 1 mM dATP, dCTP and dTTP. No dGTP was added, and this forced the polymerase to incorporate mismatches. Reactions allowing multiple binding events in the absence of heparin are indicated in the figure. The reactions were incubated for 2.5 min at 30°C and resolved on a 10% denaturing polyacrylamide gel. The m–1 indicates the nucleotide prior to the mismatch, m indicates the mismatch, and m+1 indicates the extension of the mismatch by one nucleotide. The band intensities of the product were quantified, and the percent mismatch was calculated by dividing the mismatch (m and longer products) by total extension products (Table 2).
Table 2. Switching between the polymerase and exonuclease sites in Pol2core and Pol ε.
| Polymerase to exonuclease active site switcha | Exonuclease to polymerase active site switchb,c | |
|---|---|---|
| % of primer in polymerase site = (mismatch)/(total extension) | % of primer in polymerase site = (extension)/(extension + excision) | |
| Pol2 exo− | 36.4 | 0 |
| Pol2 | 6.7 | 71 |
| Pol ε exo− | 40.4 | 0 |
| Pol ε | 6.9 | 79 |
To verify that the inactivation of the exonuclease activity did not affect the polymerase activity, we carried out a processivity experiment with Pol ε exo− and Pol ε enzymes and found that they have comparable processivity and positions of pause sites (Figure 3). This indicated that mutations in the exonuclease domain did not affect the function of the polymerase domain, and thus the observed difference was due to the inability of Pol ε exo− to remove a mismatch.
Figure 3.

Processivity. Primer extension assays were performed by mixing preformed enzyme–DNA (50/80-mer) complexes with magnesium acetate, physiological concentration of dNTP (21), and heparin. The reactions were stopped either after 1 or 2 min as indicated in the figure. Reactions allowing multiple binding events in the absence of heparin are indicated in the figure. The reaction products were resolved on a 10% denaturing acrylamide gel.
Transfer from the exonuclease site to the polymerase site
We next asked whether Pol ε could remove a mismatched primer terminus and then transfer the new 3′-terminus from the exonuclease site to the polymerase site for extension without dissociating from the DNA. Pol ε was given a primer-template substrate with a mismatch at the 3′-terminus of the primer. Under these conditions, Pol ε must first remove the mismatch in the exonuclease site prior to transferring the 3′-terminus to the polymerase site for extension of the primer. In other words, the polymerase reaction on this substrate will only be observed in the presence of a heparin trap if exonucleolytic removal of the mismatch and transfer of the edited primer terminus to the polymerase site occur processively.
To verify that the mismatched primer was not extended prior to the exonucleolytic removal of the mismatch, we performed a primer extension assay with Pol ε exo− on the mismatched template, allowing multiple or single binding events. We observed that Pol ε exo− extended the mismatched primer terminus given multiple binding events but was unable to efficiently extend the mismatch when only allowed a single binding event (in the presence of heparin) (Figure 4). Therefore, the exonuclease reaction is a prerequisite to observe polymerization products longer than the starting primer (a 52-mer) when restricted to a single binding event. We next performed the experiment with Pol ε, and, because it was exonuclease proficient, it was able to remove the mismatch. We observed replication products after allowing both multiple and single binding events (Figure 4), and the ability to extend the primer under conditions with only a single binding event suggests that Pol ε is able to transfer the 3′-end from the exonuclease site to the polymerase site via an intramolecular mechanism.
Figure 4.
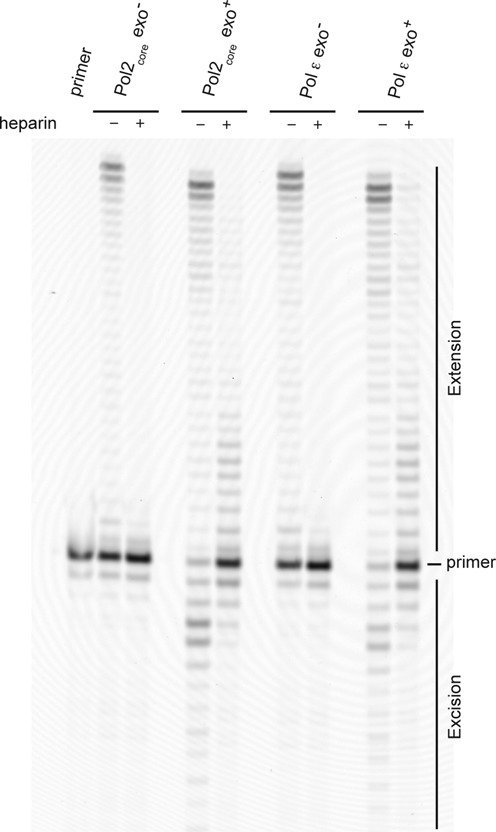
Exonuclease site to polymerase site switch. Primer extension assays were performed by mixing preformed enzyme–DNA (51T/80-mer) complexes with magnesium acetate, physiological concentrations of dNTPs (21) and heparin. Reactions allowing multiple binding events in the absence of heparin are indicated in the figure. The reactions were incubated for 2.5 min at 30°C and resolved on a 10% denaturing acrylamide gel. The band intensities of the products were quantified and the percentage of corrected mismatches was calculated by dividing the extension products by the sum of the excision and extension products (Table 2).
To ask whether the accessory subunits influence the exonucleolytic removal of the mismatched primer terminus and the transfer to the polymerase site, we repeated the experiments with both Pol2core exo− and Pol2core (Figure 4). The results were comparable to when Pol ε was used, and this suggests that there is no pronounced effect of the accessory subunits and C-terminus of Pol2 on the transfer of the primer terminus from the exonuclease site to the polymerase site. To further confirm our observations, we quantified the intensity of the bands and calculated how much of the primer was in the exonuclease site and how much of the primer had been transferred to the polymerase site before the polymerase dissociated. The percentage of primer transferred to the polymerase site for extension after exonucleolytic removal of the mismatch was calculated as the sum of the intensities of the extension products divided by sum of the extension products and the sum of the excision products. We found that Pol2core and Pol ε were able to transfer 71 and 79% of the primer, respectively (Table 2).
The effect of the M644G and M644L substitutions on switching between the polymerase and exonuclease sites
The transfer of the 3′-terminus between the polymerase and exonuclease site involves melting of the DNA and changes in the relative positions of domains within the enzyme. The M644G and M644L enzymes have been used to determine which strand is synthesized by Pol ε at the replication fork and to study ribonucleotide incorporation. M644G has lower fidelity than M644L both in vivo and in vitro. M644G incorporates ribonucleotides with a higher frequency than wild-type Pol ε, but M644L incorporates fewer ribonucleotides than wild-type Pol ε. Because the two mutants are commonly used in genetic experiments, it is of interest to explore the underlying mechanism behind the divergent defects in the two polymerases.
To determine if the partitioning between the exonuclease site and polymerase site was affected in the M644G and M644L polymerases, we repeated the experiments described above by first measuring the transfer from the polymerase site to the exonuclease site when restricted to a single binding event (Figure 5). Pol ε M644G was more efficient than wild-type Pol ε in extending a mismatch (9% versus 4.5%, respectively), whereas Pol ε M644L was less efficient than wild-type Pol ε (0% versus 4.5%, respectively) (Table 3). Thus, Pol ε M644G is less efficient in transferring the 3′-terminus from the polymerase to exonuclease site, but the ability is enhanced in M644L.
Figure 5.
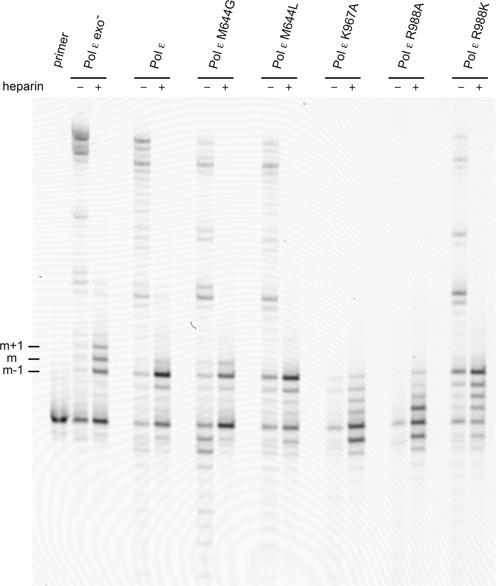
Polymerase site to exonuclease site switch in mutants. Preformed enzyme–DNA (50/80-mer) complexes were rapidly mixed with heparin, magnesium acetate and 1 mM dATP, dCTP and dTTP. No dGTP was added, and this forced the polymerase to incorporate mismatches. Reactions allowing multiple binding events in the absence of heparin are indicated in the figure. The reaction products were resolved on a 10% denaturing acrylamide gel. The m–1 sign indicates the nucleotide prior to the mismatch, m indicates the mismatch and m+1 indicates the extension of the mismatch by one nucleotide. The band intensities were quantified, and the percent mismatch was calculated by dividing the mismatch (m and longer products) by the total extension products (Table 3).
Table 3. Switching between the polymerase and exonuclease sites in wild-type and mutant Pol ε.
| Polymerase to exonuclease active site switcha | Exonuclease to polymerase active site switchb,c | |
|---|---|---|
| % of primer in polymerase site = (mismatch)/(total extension) | % of primer in polymerase site = (extension)/(extension + excision)) | |
| Pol ε exo− | 41 | 0 |
| Pol ε | 4.5 | 79 |
| Pol ε M644G | 9 | 68 |
| Pol ε M644L | 0 | 53 |
| Pol ε K967A | 0 | 0 |
| Pol ε R988A | 0 | 0 |
| Pol ε R988K | 1 | 8 |
Next we measured the ability of the M644G and M644L mutants to transfer the 3′-terminus from the exonuclease site to the polymerase site on mismatched primer termini when restricted to a single binding event (Figure 6 and Table 3). Under conditions allowing multiple binding events, the polymerization of DNA by both mutants is similar to the wild-type polymerase. When restricted to a single binding event, the M644G mutant was less efficient in transferring the 3′-terminus to the polymerase site compared to wild-type Pol ε (68 and 79%, respectively). The M644L mutant was even less efficient than M644G when switching from the exonuclease site to the polymerase site. M644G was able to transfer 68% of the primer from the exonuclease site to the polymerase site, but the M644L mutant only transferred 53% (Table 3). Thus, 21% of the primer was retained in the exonuclease site in wild-type Pol ε and 32 and 47% were retained in M644G and M644L, respectively.
Figure 6.
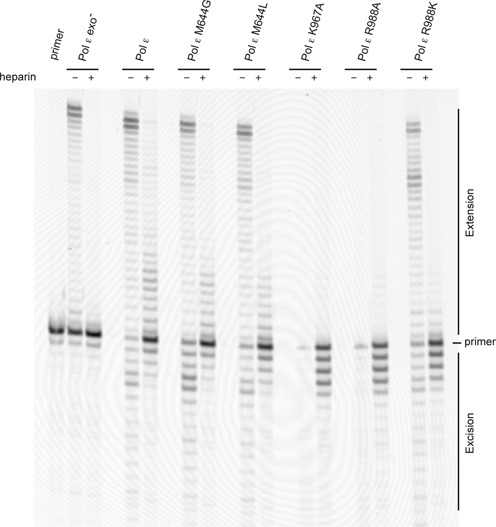
Exonuclease site to polymerase site switch in mutants. Primer extension assays were performed by mixing preformed enzyme–DNA (51T/80-mer) complexes with magnesium acetate, physiological concentrations of dNTPs (21) and heparin. Reactions allowing multiple binding events in the absence of heparin are indicated in the figure. The reactions were incubated for 2.5 min at 30°C and resolved on a 10% denaturing acrylamide gel. The band intensities of products were quantified and the percentage of corrected mismatches was calculated by dividing the extension products by the sum of the excision and extension products (Table 3).
We observed that the processivity of the M644G and M644L mutants might be affected during the switch from the exonuclease site to the polymerase site (Figure 6). To clarify if this was the case, we compared the processivity of wild-type Pol ε with the M644G and M644L mutants when given a matched 3′-terminus (Figure 3). Both the M644G and M644L mutants have reduced processivity, but they are still able to reach the end of the template used in this assay.
Role of K967 and R988 in stabilizing the 3′-terminus in the polymerase active site
K967 in Pol ε is structurally conserved in Pol δ (K814) and in RB69 gp43 (K706). The crystal structure of RB69 gp43 suggests that the role of K706 is to stabilize the hydrogen bonds between the two last base pairs of the nascent strand and thus stabilize the 3′-terminus in the polymerase active site. The K706A substitution almost completely abolishes the polymerase activity in RB69 gp43 (18). Based on the position of R988 in Pol ε, we hypothesized that the hydrogen bonds between R988 and the nucleotides at position n–4 and n–5 should not affect the positioning of the 3′-OH in the polymerase active site. However, R988 might stabilize the interaction with the double helix and thus influence the partitioning between the exonuclease site and polymerase site as well as the processivity of the polymerase. To explore the role of K967 and R988, we purified the K967A, R988A and R988K mutants. The K967A substitution almost completely abolished the polymerase activity in Pol ε, but when restricted to a single binding event at very high dNTP concentrations K967A was still able to synthesize DNA (Figure 5). Experiments only allowing a single binding event and measuring the transfer from the exonuclease site to the polymerase site and processivity at moderate dNTP concentrations (such as those found during S-phase) showed that K967A did not incorporate nucleotides and instead processively degraded the DNA with its 3′–5′ exonuclease activity (Figures 3 and 6).
The Pol ε R988A mutant showed similar activity as the K967A mutant. Only at very high dNTP concentrations (1 mM dNTP) was any polymerase activity detected, and at dNTP concentrations similar to those found during S-phase only very weak DNA polymerase activity was observed (Figures 3, 5 and 6). The R988A polymerase also exhibited processive exonuclease activity. The processive polymerase activity of Pol ε R988K was affected when compared to wild-type Pol ε, but the mutant was still able to synthesize DNA (Figure 3). Next we asked if R988K is able to transfer a mismatch from the polymerase site to the exonuclease site without dissociating from the DNA (Figure 5). We found that 1% of the mismatch was left uncorrected with the R988K mutant compared to 4.5% for wild-type Pol ε (Table 3). In contrast, R988K was not able to efficiently transfer the primer terminus from the exonuclease site to the polymerase site (8% of the mismatches were corrected and extended with R988K as a result of a single binding event compared to 79% for wild-type Pol ε) (Figure 6 and Table 3). Thus R988 appears to play an important role in guiding the DNA into the polymerase active site when returning from the exonuclease site as well as in stabilizing the DNA within the polymerase active site.
The results obtained in primer-extension assays prompted us to further test the effect of these substitutions in vivo. We established heterozygote diploid strains of S. cerevisiae E134 with a K967A, R988A or R988K substitution in Pol ε. Tetrad analysis revealed that spores carrying K967A were inviable, and R988A spores gave colonies with a severe growth defect (Figure 7). However, R988K spores were viable and gave colonies comparable to wild-type. The genetic results are in agreement with the biochemical properties of the mutants. The strong shift from polymerase to exonuclease activity in K967A and R988A in processivity assays on perfectly primed templates does not allow leading strand synthesis to be completed, and this leads to severe growth defects in these mutants. R988K was only deficient in the transfer of the 3′-terminus from the exonuclease to polymerase site suggesting that the polymerase activity of this enzyme should support cell growth.
Figure 7.
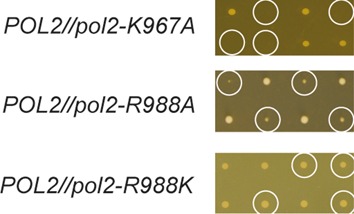
Tetrad analysis of POL2/pol2-K967A, POL2/pol2-R988A, and POL2/pol2-R988K. Tetrads carrying the indicated mutation are circled.
dNTP concentration-dependent exonuclease to polymerase active site switch
It is not only the interactions between the polymerase and DNA that influence the partitioning between the polymerase and exonuclease sites, and the balance between the exonuclease and the polymerase sites is also affected by the dNTP concentration. DNA polymerases with a 3′–5′ exonuclease activity degrade DNA in the absence of dNTPs and synthesize DNA in the presence of high dNTP concentrations. For that reason, we examined the effect of dNTP concentration on the transition of the primer terminus between the exonuclease and polymerase active sites given multiple binding events. A primer extension assay allowing multiple binding events was carried out either in the absence of dNTPs or with increasing amounts of physiologically balanced dNTPs ranging from 0 to 11 μM with respect to dGTP (Figure 8). Thus, the highest concentrations of dNTPs were 11 μM dGTP, 39 μM dCTP, 66 μM dTTP and 22 μM dATP. The shift from exonuclease activity to DNA synthesis for wild-type Pol ε and M644G occurred at about the same dNTP concentration (0.34–0.68 μM dGTP) (Figure 8). The M644G polymerase is less efficient than wild-type Pol ε when degrading double-stranded DNA, and this reveals a defect in separating the primer strand from the template strand. The M644L polymerase is sensitive to low dNTP concentrations and requires >1.4 μM dGTP to shift from exonuclease activity to DNA synthesis (Figure 8). Thus, the M644L mutant requires four times higher dNTP concentrations than wild-type Pol ε and M644G to efficiently maintain the 3′-terminus of the DNA in the polymerase site. In addition, the pattern of degradation reveals that the M644L mutant has the most potent exonuclease activity compared to wild-type Pol ε and M644G. This fits well with our observation that the balance for M644L is shifted toward the exonuclease site even at high dNTP concentrations.
Figure 8.

Nucleotide concentration effect on the balance between the exonuclease and polymerase activities under steady-state conditions. Preformed enzyme–DNA (50/80-mer) complexes were mixed with magnesium acetate and physiologically balanced dNTPs (21). The dGTP concentration is indicated above each lane. The maximum concentrations of added dNTPs were those previously reported to be the in vivo concentrations. The reactions were stopped after 10 min, and the reaction products were resolved on a 10% denaturing acrylamide gel.
DISCUSSION
It was previously shown that an extended β-hairpin loop is important for the transfer of DNA between the polymerase site and exonuclease site in B-family polymerases (11). We demonstrate here that Pol ε can edit a mismatch in the exonuclease site and processively transfer the corrected 3′-terminus of the DNA to the polymerase site for resumed DNA synthesis despite the absence of such as β-hairpin loop. This implies that Pol ε employs a different mechanism compared to other B-family polymerases for the transfer of DNA between the two active sites. We also conclude that the C-terminus and the accessory subunits Dpb2, Dpb3 and Dpb4 do not influence this process. This is in agreement with earlier in vitro fidelity experiments showing that the fidelity of Pol2core and a Pol2–Dpb2 complex are similar to four-subunit Pol ε (13,15).
There is a balance between the forward synthesis, dissociation and exonuclease activity at each position of the template as the new strand is synthesized. Multiple interactions between the DNA and the DNA polymerase influence the outcome each time a nucleotide is added to the growing DNA strand. Here, we have explored three different amino acids and their impact on the process by which Pol ε synthesizes DNA. The methionine at residue 644 is of general interest because it plays an important role in determining where Pol ε replicates DNA and the impact of ribonucleotide incorporation on the fidelity of DNA replication. Earlier studies have shown that M644G is less accurate than wild-type Pol ε, and a signature of M644G is the formation and extension of T–T mismatches and the incorporation of ribonucleotides with higher efficiency than wild-type Pol ε (3,16). The M644L polymerase was previously shown to incorporate ribonucleotides with less efficiency than wild-type Pol ε (16). Thus M644G, Pol ε and M644L form a gradient with regard to the number of ribonucleotides that are incorporated each time the genome is duplicated (16). The insertion and extension efficiencies across a single rNMP in the template suggested that the partitioning of the primer terminus between the exonuclease and polymerase site is affected in the M644L mutant (16). Here, we show that both M644G and M644L are able to transfer the 3′-terminus between the exonuclease and polymerase sites without dissociating from the DNA. The efficiency of transferring the 3′-terminus from the polymerase site to the exonuclease site was enhanced in M644L and reduced in M644G compared to wild-type Pol ε. Going in the opposite direction, both M644G and M644L were less efficient in transferring the 3′-terminus from the exonuclease site to the polymerase site. Thus, substitutions of M644 might not just affect the nucleotide binding pocket and the selectivity for dNTPs, and they might also affect the stability of the primer terminus within the polymerase active site. Both our experiments allowing a single binding event and the nucleotide titration experiment under steady-state conditions agree well with previously published data showing that the partitioning between the exonuclease site and polymerase site varies in M644G and M644L (16). In M644G, the balance is shifted toward the polymerase site, but in M644L the balance is shifted toward the exonuclease site.
The high propensity to switch the primer from the polymerase site to the exonuclease site by the M644L mutant contributes to the antimutator properties of this mutant (16) and might decrease the rate of replication in vivo and be one of the causes for the delayed S-phase in strains carrying the M644L mutation (16). The dNTP titration experiment suggests that M644L requires four times higher concentration of dNTP than Pol ε to synthesize DNA. Mutants such as P424L, I417V and A737V have been isolated in T4 DNA polymerase, and these have similar antimutator effects as M644L due to a lower occupancy by the DNA in the polymerase site (23–25). The A737V substitution in T4 DNA polymerase is proposed to affect the closing of the thumb domain and to allow the primer strand to shift toward the exonuclease site.
DNA synthesis requires that the duplex DNA is stabilized in the polymerase active site, and this is achieved by multiple interactions between the DNA and the polymerase active site and residues in the palm domain and the thumb domain. Should a mismatch occur at the primer end, the DNA becomes destabilized in the polymerase active site and is transferred to the exonuclease site. B-family polymerases have a highly conserved lysine (e.g. K706 in RB69 gp43, K498 in ϕ29 DNA polymerase, K814 in Pol δ and K967 in Pol ε) that forms a hydrogen bond with the base at position n–2. This lysine is part of the KKRY motif that has been suggested to stabilize the duplex DNA in the polymerase active site (17). The K706A substitution was shown to almost completely abolish the polymerase activity in RB69 gp43 (18), and the K498T substitution severely affected the capacity to synthesize DNA in ϕ29 DNA polymerase (26). In agreement with these observations, we detected polymerase activity in the Pol ε K967A mutant only at very high non-physiological dNTP concentrations (1 mM dNTP). At physiological dNTP concentrations, the K967A mutant processively degrades the primer.
Less is known about the interaction between the amino acid side chains and the bases at positions n–4 and n–5. Both R988 in Pol ε and R839 in Pol δ form hydrogen bonds with the bases at these positions (4,5), but neither K734 in RB69 gp43 or K555 in ϕ29DNA polymerase are buried in the minor groove and instead engage in water-mediated interactions with these bases (27,28). We found that the R988A substitution in Pol ε gave nearly the same shift in partitioning from the polymerase site to the exonuclease site as the K967A substitution. Under conditions that only allowed a single binding event the 3′-terminus was efficiently transferred to the exonuclease site when the R988A mutant misincorporated a deoxyribonucleotide in the presence of 1 mM dNTP (Figure 5), but the mismatched 3′-terminus was not transferred to the polymerase active site and extended at physiological dNTP concentrations. Instead, the 3′-terminus was processively degraded by the exonuclease active site as previously shown for the K967A mutant. In fact, the biochemical properties of the R988A and K967A mutants were comparable, and this suggests that both residues are essential for stabilizing the 3′-OH of the primer strand within the polymerase active site. Considering that other B-family polymerases have a lysine at the corresponding position, we purified the R988K mutant. The R988K Pol ε mutant was not able to efficiently transfer the 3′-terminus from the exonuclease site to the polymerase site and then extend the primer terminus via an intramolecular mechanism at physiological dNTP concentrations when restricted to a single binding event (Figure 6). Both K967A and R988A resulted in a severe growth defect in yeast, but the R988K mutant appears to grow as wild-type in a tetrad analysis (Figure 7). Interestingly, the effect of losing the interaction with the bases at positions n–4 and n–5 might vary between the B-family polymerases. The K555A substitution in ϕ29 has normal DNA polymerase activity, but it is not as efficient as wild-type ϕ29 when degrading double-stranded DNA (29). Also, K734 in RB69 gp43 and K555 in ϕ29 DNA polymerase are not as deeply buried in the minor groove as the arginines at this position in Pol ε and Pol δ. The ternary structure of ϕ29 DNA polymerase with DNA included two molecules in the unit cell. K555 was oriented toward the minor groove in one molecule and parallel with the DNA in the second molecule, and this suggests that the interaction with the DNA by this lysine in ϕ29 DNA polymerase and RB69 gp43 is more flexible than the arginines in Pol ε and Pol δ.
Earlier studies on the DNA polymerase I fragment from thermophilic Bacillus stearothermophilus (an A-family polymerase) showed that the high fidelity of the polymerase relied on five regions: the template pre-insertion site, the insertion site, the post-insertion site, the catalytic site and the DNA duplex binding region (30). It was shown that mismatches at the 3′-terminus affected the positioning of the 3′-terminus within the polymerase active site and reduced the ability to extend the mismatch. This led to dissociation of the enzyme or transfer of the 3′-terminus to the exonuclease site of the enzyme. Another conclusion from the studies of the Bacillus DNA polymerase I was that there is a short-term memory for mismatches in the duplex DNA up to four nucleotides from the 3′-terminus. A mismatch at position n–2 or n–3 leads to a conformational change that reduces the interactions with the template DNA and the polymerase active site. It was later suggested by Wang and colleagues that this short-term memory was in part mediated by interactions with the five structural waters found in the minor groove between the DNA and the polymerase (31). Here, we propose that Pol ε and Pol δ rely on direct contact with the duplex DNA to sense mismatches four or five nucleotides from the active site. A mismatch at position n–4 or n–5 could affect the hydrogen bonds with the arginine, and this could in turn destabilize the 3′-terminus of the primer within the polymerase site and lead to transfer of the 3′-terminus to the exonuclease active site. We cannot at this time determine the relative contribution of the mechanisms by which mismatches are sensed, but our results correlate well with fidelity measurements showing that Pol ε can efficiently proofread homonucleotide runs up to four or five nucleotides long (13).
FUNDING
Swedish Research Council; Swedish Cancer Society; Kempe Foundations; Knut and Alice Wallenberg Foundation; Insamlingstiftelsen at the Medical faculty of Umeå University. Funding for open access charge: The Swedish Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1.Arana M.E., Kunkel T.A. Mutator phenotypes due to DNA replication infidelity. Semin. Cancer Biol. 2010;20:304–311. doi: 10.1016/j.semcancer.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nick McElhinny S.A., Gordenin D.A., Stith C.M., Burgers P.M., Kunkel T.A. Division of labor at the eukaryotic replication fork. Mol. Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pursell Z.F., Isoz I., Lundstrom E.B., Johansson E., Kunkel T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swan M.K., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat. Struct. Mol. Biol. 2009;16:979–986. doi: 10.1038/nsmb.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogg M., Osterman P., Bylund G.O., Ganai R.A., Lundstrom E.B., Sauer-Eriksson A.E., Johansson E. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat. Struct. Mol. Biol. 2014;21:49–55. doi: 10.1038/nsmb.2712. [DOI] [PubMed] [Google Scholar]
- 6.Reha-Krantz L.J. DNA polymerase proofreading: multiple roles maintain genome stability. Biochim. Biophys. Acta. 2010;1804:1049–1063. doi: 10.1016/j.bbapap.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 7.Shamoo Y., Steitz T.A. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 8.Hogg M., Wallace S.S., Doublie S. Crystallographic snapshots of a replicative DNA polymerase encountering an abasic site. EMBO J. 2004;23:1483–1493. doi: 10.1038/sj.emboj.7600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marquez L.A., Reha-Krantz L.J. Using 2-aminopurine fluorescence and mutational analysis to demonstrate an active role of bacteriophage T4 DNA polymerase in strand separation required for 3′ –> 5′-exonuclease activity. J. Biol. Chem. 1996;271:28 903–28 911. doi: 10.1074/jbc.271.46.28903. [DOI] [PubMed] [Google Scholar]
- 10.Stocki S.A., Nonay R.L., Reha-Krantz L.J. Dynamics of bacteriophage T4 DNA polymerase function: identification of amino acid residues that affect switching between polymerase and 3′ –> 5′ exonuclease activities. J. Mol. Biol. 1995;254:15–28. doi: 10.1006/jmbi.1995.0595. [DOI] [PubMed] [Google Scholar]
- 11.Hogg M., Aller P., Konigsberg W., Wallace S.S., Doublie S. Structural and biochemical investigation of the role in proofreading of a beta hairpin loop found in the exonuclease domain of a replicative DNA polymerase of the B family. J. Biol. Chem. 2007;282:1432–1444. doi: 10.1074/jbc.M605675200. [DOI] [PubMed] [Google Scholar]
- 12.Jain R., Rajashankar K.R., Buku A., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Crystal structure of yeast DNA polymerase epsilon catalytic domain. PLoS One. 2014;9:e94835. doi: 10.1371/journal.pone.0094835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shcherbakova P.V., Pavlov Y.I., Chilkova O., Rogozin I.B., Johansson E., Kunkel T.A. Unique error signature of the four-subunit yeast DNA polymerase epsilon. J. Biol. Chem. 2003;278:43 770–43 780. doi: 10.1074/jbc.M306893200. [DOI] [PubMed] [Google Scholar]
- 14.Zhong X., Pedersen L.C., Kunkel T.A. Characterization of a replicative DNA polymerase mutant with reduced fidelity and increased translesion synthesis capacity. Nucleic Acids Res. 2008;36:3892–3904. doi: 10.1093/nar/gkn312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pursell Z.F., Isoz I., Lundstrom E.B., Johansson E., Kunkel T.A. Regulation of B family DNA polymerase fidelity by a conserved active site residue: characterization of M644W, M644L and M644F mutants of yeast DNA polymerase epsilon. Nucleic Acids Res. 2007;35:3076–3086. doi: 10.1093/nar/gkm132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nick McElhinny S.A., Kumar D., Clark A.B., Watt D.L., Watts B.E., Lundstrom E.B., Johansson E., Chabes A., Kunkel T.A. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 2010;6:774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franklin M.C., Wang J., Steitz T.A. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 18.Zakharova E., Wang J., Konigsberg W. The activity of selected RB69 DNA polymerase mutants can be restored by manganese ions: the existence of alternative metal ion ligands used during the polymerization cycle. Biochemistry. 2004;43:6587–6595. doi: 10.1021/bi049615p. [DOI] [PubMed] [Google Scholar]
- 19.Chilkova O., Jonsson B.H., Johansson E. The quaternary structure of DNA polymerase epsilon from Saccharomyces cerevisiae. J. Biol. Chem. 2003;278:14 082–14 086. doi: 10.1074/jbc.M211818200. [DOI] [PubMed] [Google Scholar]
- 20.Asturias F.J., Cheung I.K., Sabouri N., Chilkova O., Wepplo D., Johansson E. Structure of Saccharomyces cerevisiae DNA polymerase epsilon by cryo-electron microscopy. Nat. Struct. Mol. Biol. 2006;13:35–43. doi: 10.1038/nsmb1040. [DOI] [PubMed] [Google Scholar]
- 21.Sabouri N., Viberg J., Goyal D.K., Johansson E., Chabes A. Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 2008;36:5660–5667. doi: 10.1093/nar/gkn555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isoz I., Persson U., Volkov K., Johansson E. The C-terminus of Dpb2 is required for interaction with Pol2 and for cell viability. Nucleic Acids Res. 2012;40:11 545–11 553. doi: 10.1093/nar/gks880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spacciapoli P., Nossal N.G. A single mutation in bacteriophage T4 DNA polymerase (A737V, tsL141) decreases its processivity as a polymerase and increases its processivity as a 3′–>5′ exonuclease. J. Biol. Chem. 1994;269:438–446. [PubMed] [Google Scholar]
- 24.Reha-Krantz L.J., Nonay R.L. Motif A of bacteriophage T4 DNA polymerase: role in primer extension and DNA replication fidelity. Isolation of new antimutator and mutator DNA polymerases. J. Biol. Chem. 1994;269:5635–5643. [PubMed] [Google Scholar]
- 25.Li V., Hogg M., Reha-Krantz L.J. Identification of a new motif in family B DNA polymerases by mutational analyses of the bacteriophage T4 DNA polymerase. J. Mol. Biol. 2010;400:295–308. doi: 10.1016/j.jmb.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blasco M.A., Mendez J., Lazaro J.M., Blanco L., Salas M. Primer terminus stabilization at the phi 29 DNA polymerase active site. Mutational analysis of conserved motif KXY. J. Biol. Chem. 1995;270:2735–2740. doi: 10.1074/jbc.270.6.2735. [DOI] [PubMed] [Google Scholar]
- 27.Wang J., Sattar A.K., Wang C.C., Karam J.D., Konigsberg W.H., Steitz T.A. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 28.Berman A.J., Kamtekar S., Goodman J.L., Lazaro J.M., de Vega M., Blanco L., Salas M., Steitz T.A. Structures of phi29 DNA polymerase complexed with substrate: the mechanism of translocation in B-family polymerases. EMBO J. 2007;26:3494–3505. doi: 10.1038/sj.emboj.7601780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez-Arnaiz P., Lazaro J.M., Salas M., de Vega M. Involvement of phi29 DNA polymerase thumb subdomain in the proper coordination of synthesis and degradation during DNA replication. Nucleic Acids Res. 2006;34:3107–3115. doi: 10.1093/nar/gkl402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson S.J., Beese L.S. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 31.Wang M., Xia S., Blaha G., Steitz T.A., Konigsberg W.H., Wang J. Insights into base selectivity from the 1.8 A resolution structure of an RB69 DNA polymerase ternary complex. Biochemistry. 2011;50:581–590. doi: 10.1021/bi101192f. [DOI] [PMC free article] [PubMed] [Google Scholar]