


Abstract
Of the ∼1.3 million Alu elements in the human genome, only a tiny number are estimated to be active in transcription by RNA polymerase (Pol) III. Tracing the individual loci from which Alu transcripts originate is complicated by their highly repetitive nature. By exploiting RNA-Seq data sets and unique Alu DNA sequences, we devised a bioinformatic pipeline allowing us to identify Pol III-dependent transcripts of individual Alu elements. When applied to ENCODE transcriptomes of seven human cell lines, this search strategy identified ∼1300 Alu loci corresponding to detectable transcripts, with ∼120 of them expressed in at least three cell lines. In vitro transcription of selected Alus did not reflect their in vivo expression properties, and required the native 5′-flanking region in addition to internal promoter. We also identified a cluster of expressed AluYa5-derived transcription units, juxtaposed to snaR genes on chromosome 19, formed by a promoter-containing left monomer fused to an Alu-unrelated downstream moiety. Autonomous Pol III transcription was also revealed for Alus nested within Pol II-transcribed genes. The ability to investigate Alu transcriptomes at single-locus resolution will facilitate both the identification of novel biologically relevant Alu RNAs and the assessment of Alu expression alteration under pathological conditions.
INTRODUCTION
Almost half of the human genome sequence is accounted for by mobile DNA elements, of which Alu elements, belonging to the class of retrotransposons called SINEs (short interspersed elements), are one of the most successful, being present in more than 1 million copies (1). The body of a typical Alu element is about 280 bases in length, and is formed from two diverged, 7SL-related monomers separated by a short A-rich region. A longer poly(A) region is located at the 3′ end of the element. An internal, bipartite RNA polymerase (Pol) III promoter element, composed of an A and a B box both located within the left monomer, make Alus potential targets for the Pol III transcription machinery, which can initiate transcription at the beginning of the Alu and terminate at the closest poly(dT) termination sequence encountered downstream of the Alu body (2)3,4 (Figure 1A). In particular, Alu transcription by Pol III requires the recognition, within the Alu left monomer, of the internal promoter by the assembly factor TFIIIC, which in turn recruits the Pol III-interacting initiation factor TFIIIB on a ∼50-bp region upstream of the transcription start site (TSS) (5,6). Even though TFIIIB-DNA association is generally sequence-independent, an influence of the 5′-flanking region on Alu transcription was put in light in early studies and later confirmed in vitro and in transfected cell lines (7–9).
Figure 1.
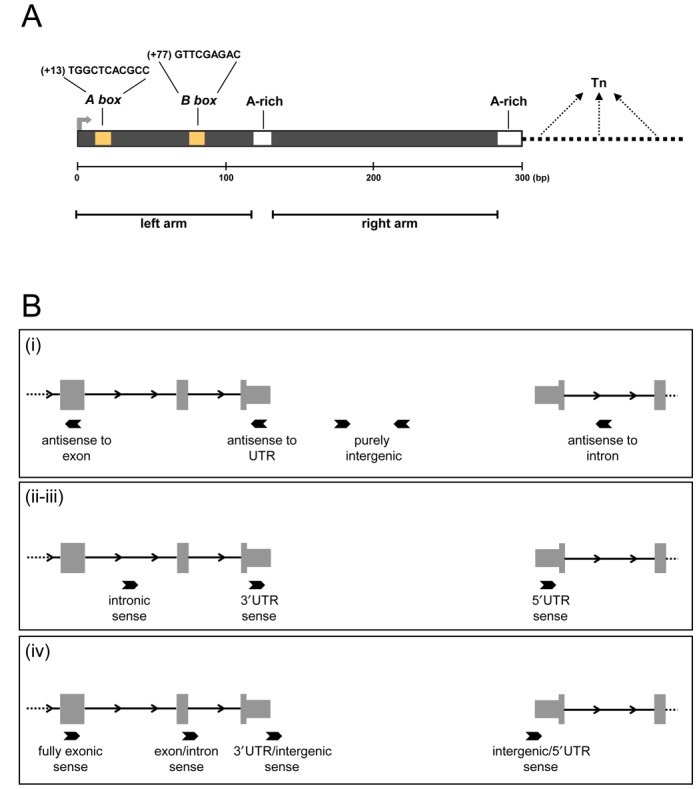
Architecture of Alu elements considered as RNA polymerase III transcription units. (A) Schematic representation of a typical Alu element, ∼300 bp in length (indicated by graduated bar). Alu transcription by RNA polymerase III requires A box and B box internal promoter elements (orange bars) (6), which form together the binding site for TFIIIC. The consensus sequences for Alu A and B boxes are reported above the scheme. While the Alu B box sequence perfectly matches the canonical B box sequence found in tRNA genes, the sequence of Alu A box slightly diverges from canonical A box sequence (TRGYnnAnnnG; (5)). Transcription is thought to start at the first Alu nt (G) (3,4). The A box starts at position +13, the B box 53 bp downstream, at position +77. The left and right arms of the Alu, each being ancestrally derived from 7SL RNA, are separated from each other by an intermediate A-rich region, starting 35 bp downstream of the B box, whose consensus sequence is A5TACA6. Another A-rich tract is located 3′ to the right arm, at the end of the Alu body, starting at ∼150 bp downstream of the middle A-rich region. Transcription termination by RNA polymerase III is expected to mainly occur at the first encountered termination signal (Tn) downstream of the 3′ terminal A-rich tract. Such a signal, either a run of at least four Ts or a T-rich non-canonical terminator (25), may be located at varying distances from the end of the Alu body, thus allowing for the generation of Alu primary transcripts carrying 3′ trailers of different lengths and sequences. (B) Possible localizations of Alu elements with respect to other transcription units: (i) intergenic/antisense, comprising purely intergenic Alus as well as Alus which are not included in longer transcription units on the same strand, but overlap in antisense orientation to transcription units located on the opposite strand; (ii and iii) gene-hosted, comprising Alus fully contained within introns or UTRs of protein-coding or lincRNA genes in a sense orientation; (iv) all other cases, including Alu RNAs fully or partially mapping to exons, or partially mapping to UTRs, in a sense orientation.
Pol III-synthesized Alu RNAs are generally expressed at very low cellular levels. For example, a typical HeLa cell has been estimated to express only 100 molecules of Alu RNA (10). Pol III-dependent Alu expression can increase under cellular stress conditions like heat shock and viral infection (11,12). In any case, however, given the ubiquitous presence of Alu elements throughout the human genome, their preference for gene-rich regions and their high density in introns of Pol II-transcribed genes, most Alu-containing RNA is accounted for by either primary nuclear transcripts (hnRNAs) or mature mRNAs deriving from Pol II transcription activity. Therefore, genuine Pol III-derived Alu transcripts, which may be involved in the retrotransposition process, are difficult to identify and measure with respect to those that, being part of longer host RNA molecules, lack any significant retrotransposition potential (1,13).
Previous attempts to identify individual, Pol III-derived Alu transcripts and the corresponding genomic elements have exploited either various combinations of size fractionation, primer extension and 3′ RACE (rapid amplification of cDNA ends) (10,14–15) or, more recently, genome-wide chromatin immunoprecipitation profiling (through ChIP-Seq) of Alu loci bound by the Pol III machinery (16–18). A careful inspection of such data collections recently led to the confirmation that only a small minority of Alu loci are likely to be expressed, and that the profile of expressed Alu loci tends to vary by cell type, transformation state and even in response to tiny variations in growth conditions (19). This is in agreement with the results of a recent comprehensive analysis of the human transcriptome, showing that the main characteristic of transcripts originating from repeat regions of the human genome, including LINEs (Long Interspersed Elements) and SINEs, is cell-line specificity (20). On the other hand, it has been shown that when the paucity of Alu expression is experimentally overcome by plasmid-directed Alu overexpression, hundreds if not thousands of Alu elements, all belonging to the AluS and AluY lineages, are potentially retrotransposed (21). The issue of Alu transcriptional control is thus highly relevant to human genome stability. Moreover, the reported existence of Alu-related non-coding (nc) RNAs expressed from unique Alu-derived transcription units and playing highly specific regulatory roles (22–24) makes it urgent to adequately explore this hidden part of the human transcriptome.
Several factors might contribute to our ability to detect transcriptionally active Alus. Most Pol III-generated Alu RNAs display highly distinctive sequence features, due to accumulated mutations in the encoding Alu element, to length and sequence heterogeneity in the terminal A-rich tail, and to the unique 3′ trailer sequence corresponding to the DNA region comprised between the 3′ end of Alu conserved body and the first encountered Pol III terminator [either canonical or non-canonical (25)] in the downstream region (1). The unique 3′ extension of each Alu Pol III transcript is also responsible for length heterogeneity of such transcripts, that are reported to vary from ∼300 to more that 600 nucleotides (nt) in length (26). We reasoned that the combination of such unique DNA sequence features might allow to distinguish the large majority of individual Alus from each other (most human Alu repeats have indeed been previously proposed to be unique; (27)), and thus to define the individual locus from which each expressed Alu RNA originates, provided that sequence information on the transcripts is available. This type of information should be extractable from RNA-seq data sets, especially if transcript sequences are collected and available in the form of long (≥75 nt), paired end RNA-seq reads. This is the case for some of the RNA-seq data sets within the ENCODE project (20), which therefore might represent a largely unexploited resource for Alu expression studies.
MATERIALS AND METHODS
Bioinformatic pipeline for individually expressed Alu identification
An outline of the pipeline is provided in this section. A more detailed description of computational methods can be found in Supplementary Methods.
For Alu RNA identification, we used the Cold Spring Harbor Lab (CSHL) long RNA-seq data within ENCODE (whole-cell polyA+ and polyA- RNAs, two replicates for each sample) relative to the following cell lines: Gm12878, H1-hESC, HeLa-S3, HepG2, HUVEC, K562, NHEK, for a total of 28 data sets (see http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCshlLongRnaSeq). These data sets contain paired-end reads (2 × 76 nt). Reads from each data set were aligned to the reference genome (hg19) using TopHat aligner version 2.0.11 (28) for both ‘unique’ and ‘best match’ alignment strategies (29). Only uniquely mapped paired-end reads (characterized by ‘NH:i:1’ in the aligner-generated bam file) were considered for most of the analyses reported in this study. To this end, the TopHat aligner was used with default settings (allowing to retain reads with up to 20 equally scoring hits in the genome), and uniquely aligned paired-end reads (identified by NH:i:1 in the alignment file) were recognized and counted through the HTSeq Python package (30). Only Alu ids with more than 10 mapped reads were retained. To check for the performance of the aligner and its reliability in unique alignment, we replaced TopHat by the independently developed STAR aligner (31) for the analysis of two data sets (NHEK polyA+, replicates 1 and 2), and found largely (∼96%) overlapping sets of Alus with more than 10 uniquely mapped paired-end reads.
The coordinates of retained Alus were supplied to sitepro script of the Cis-regulatory Element Annotation System (http://liulab.dfci.harvard.edu/CEAS/) along with the corresponding RNA-seq stranded signal profiles. We used sitepro (developed mainly for ChIP-seq data) because it allowed us to calculate the signal profile in a range of ±500 nt from the center of the Alu body with a resolution of 50 nt. In this way we could address the problem of ‘passenger’ Alu RNAs by devising a filter aimed at excluding false positives on the basis of the level of upstream and downstream spurious RNA signals (see Supplementary Methods for details). Only Alu transcripts that passed this filter in both ENCODE RNA-seq replicates were considered to represent autonomously expressed Alu loci (as such, they will be often referred to in the text as ‘expression-positive’). Complete lists of these Alus are reported in Supplementary Table S1. The bam files containing the alignments with uniquely mapped (NH:i:1) paired-end reads, generated through TopHat for all the 28 ENCODE data sets, and through STAR for a subset of them (NHEK polyA+ replicates 1 and 2; HeLa-S3 polyA+ replicate 1; K562 polyA- replicate 1), are deposited at the following link: http://bioinfo.cce.unipr.it/NAR-02564-Z-2014/. Also available at the same link is the above described pipeline in the form of a collection of shell scripts designed to automate the execution of the different publicly available software (such as TopHat and htseq-count, as detailed in the Supplementary Methods along with their specific options).
As a number of Alu transcripts were found both in the polyA+ and polyA− data sets, Supplementary Table S1 also contains a non-redundant list of all expressed Alus obtained by merging expression-positive Alus found in the polyA+ and polyA− fractions of all cell lines (‘All non-redundant’ sheet in Supplementary Table S1).
All analyses were carried out using hg19 (GRCh37) genome assembly. Even though the contribution of novel sequence in GRCh38, that is absent from hg19, to Alu expression profiles was expected to be limited (the total number of bases in GRCh38 being increased by ∼2% only with respect to GRCh37/hg19), we nevertheless screened a pair of ENCODE RNA seq data set replicates (NHEK polyA+, r1 and r2) with our pipeline using GRCh38 assembly as a reference for read mapping, and compared the results with those obtained with hg19 genome assembly. We found that the vast majority (92–95%) of Alus detected as expression-positive in either genome assembly was shared with the other one.
To further support the identification of unique Alu transcripts found in Hela-S3 and K562 cells, we intersected the ChIP-seq peaks of the Pol III machinery components TFIIIC-110, RPC155, BRF1, BRF2, BDP1, derived from ENCODE/Stanford/Yale/USC/Harvard ChIP-seq data (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs/) with the expression-positive Alu coordinates, extended to 200 bp upstream, of these two cell lines. P-values for the association of each Pol III component to expression-positive intergenic Alus were calculated using the Fisher's exact test against total (intergenic) Alus. The lists of Pol III-associated, expression-positive Alus are reported in Supplementary Table S2.
To identify other transcription factors (TFs) associated to expression-positive Alu elements, we intersected, for each cell line, the 500 bp upstream of the Alus with the coordinates of the TF binding sites from ENCODE ChIP-seq (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegTfbsClustered/wgEncodeRegTfbsClusteredWithCellsV3.bed.gz). P-values for the association of TFs to expression-positive Alus were calculated using the Fisher's exact test against total Alus. Lists of TF-Alu interactions are reported in Supplementary Table S3.
Plasmid construction
Using oligonucleotides listed in Supplementary Table S4, nine human Alu loci (whose chromosome coordinates are reported in Table 3), together with 5′- and 3′-flanking regions, were polymerase chain reaction (PCR)-amplified from buccal cell genomic DNA with GoTaq® DNA polymerase (Promega) and cloned into pGEM®-T Easy vector (Promega). Constructs containing targeted mutation of the B box internal control element were obtained by recombinant PCR through the fusion of sub-fragments overlapping in the mutated region, as previously described (32), followed by cloning into pGEM®-T Easy. Upstream deletion constructs employed forward PCR primers generating amplicons truncated to position −12 (or −15, in the case of AluSx_chr10) with respect to Alu 5′ end. Truncated amplicons were inserted into pGEM®-T Easy; the constructs selected for in vitro transcription contained the 5′-truncated insert with the same orientation as its wild-type Alu counterpart, to minimize the influence of vector sequence on transcription efficiency.
Table 3. Alus subjected to in vitro transcription analysis.
| Alu | Expression in cell linesa | Predicted length of primary transcript(s)b |
|---|---|---|
| AluSq2_chr1 (chr1:61523296–61523586) | H1-hESC, HeLa-S3, Hep G2, K562, NHEK | 355 (T4); 361 (T10). |
| AluSx_chr1 (chr1:235531222–235531520) | none | 328 (TAT3); 338 (TAT3); 431 (T4) |
| AluSx1_chr3 (chr3:139109300–139109588) | H1-hESC, GM12878 (sporadical) | 304 (T3GT); 311 (TCT3); 437 (TAT3); 443 (T17) |
| AluY_chr7 (chr7:73761603–73761897) | K562 (sporadical) | 322 (T5) |
| AluY_chr10-a (chr10:103929441–103929803) | H1-hESC (sporadical) | 370 (TCT3); 376 (T4); 397 (T6); 406 (T3GT2) |
| AluY_chr10-b (chr10:69524852–69525156) | NHEK | 397 (T5) |
| AluSx_chr10 (chr10:12236879–12237173) | none | 320 (T4); 456 (T6) |
| AluSp_chr17 (chr17:4295121–4295437) | K562 | 387 (T3CT); 424 (TAT3); 430 (T6) |
| AluY_chr22 (chr22:41932115–41932411) | none | 378 (TGT3); 409 (T4); 590 (T3CT) |
The second column lists, for each Alu element, the cell lines in which it was found to be expressed by RNA-seq data analysis. The transcript lengths (in nts) reported in the third column were calculated by assuming as TSS the G at the first Alu position, located 12 bp upstream of the T with which the A box starts (TRGY…). This assumption is based on early in vitro transcription analyses showing that most Alu transcripts initiate in close proximity to the 5′ end of the consensus Alu sequence (3,6). To estimate the 3′ end of the transcript, both canonical (Tn with n ≥ 4) and non-canonical T-rich (25) Pol III terminators were considered downstream of Alu body sequence (indicated in parentheses after the transcript length); for canonical terminators, the 4 Us corresponding to the first 4 Ts of the termination signal were considered as part of the transcripts; for non-canonical terminators, all the nts of the terminator were considered as incorporated into the RNA. The underlined values are those for which a closely corresponding transcript was detected in transcription gels.
aThis column lists, for each Alu element, the cell lines in which it was found to be expressed by RNA-seq data analysis.
bThe reported transcript lengths were calculated by assuming as TSS the G at the first Alu position, located 12 bp upstream of the T with which the A box starts (TRGY…).
In vitro transcription
All plasmids for in vitro transcription reactions were purified with the Qiagen Plasmid Mini kit (Qiagen). Reaction mixtures (25 μl) contained 500 ng of template DNA, 70 mM KCl, 5 mM MgCl2, 1 mM DTT, 2.5% glycerol, 20 mM Tris–HCl pH 7.9, 5 mM phosphocreatine, 2 μg/ml alpha-amanitin, 0.4 U/ml SUPERase-In (Ambion), 40 μg of HeLa cell nuclear extract (33), 0.5 mM ATP, CTP and GTP, 0.025 mM UTP and 10 μCi of [α-32P]UTP (Perkin-Elmer). Reactions were allowed to proceed for 60 min at 30°C before being stopped by addition of 75 μl of nuclease-free water and 100 μl of phenol:chloroform (1:1). Purified labeled RNA products were resolved on a 6% polyacrylamide, 7 M urea gel and visualized and quantified with a Cyclone Phosphor Imager (PerkinElmer) and the Quantity One software (Bio-Rad).
RESULTS
A bioinformatic pipeline for the identification of transcriptionally active Alu loci from RNA-Seq data sets
The availability of RNA-Seq data sets for several human cell lines and tissues offers an unprecedented opportunity to identify individual, transcriptionally active Alu loci from the analysis of raw sequence reads. To this end, it is important to take into account the computational challenges posed by transcripts arising from repetitive elements, in particular the possible occurrence of multireads (i.e. reads aligning to multiple positions on the reference genome) (29). The RNA-Seq data sets we selected for our search are part of those established for the most recent ENCODE project attempt to define the landscape of transcription in human cells, and are all comprised of 76-nt-long paired end RNA-seq reads (20). In particular, we analyzed whole cell long RNA-seq data (polyA+ and polyA−) from ENCODE/CSHL (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCshlLongRnaSeq) for the following cell lines: GM12878 (lymphoblastoid cells), H1-hESC (human embryonic stem cells), K562 (chronic myelogenous leukemia cells), HeLa-S3 (cervical carcinoma), HepG2 (hepatocellular carcinoma), HUVEC (umbilical vein endothelial cells), NHEK (epidermal keratinocytes). We considered in our analysis Alu elements differing in their location and possible mode of expression, in particular: (i) intergenic/antisense Alus, comprising both Alus that are not hosted in any annotated protein-coding or lincRNA gene and Alus that map to introns or exons of annotated genes, but do so in an antisense orientation; intergenic and antisense Alus were grouped together as they are both expected to be transcribed by Pol III as independent transcription units; (ii) Alus fully contained within introns of protein-coding or lincRNA genes in a sense orientation; (iii) Alus located within 5′UTR (untranslated region) or 3′UTR of annotated protein-coding genes in a sense orientation; (iv) all other cases, including Alu RNAs fully or partially mapping to exons in a sense orientation. For groups (ii)–(iv), Alu RNA synthesis should in principle occur mostly as part of Pol II-dependent transcription of the host transcription unit, producing primary or mature mRNA/lincRNA transcripts carrying embedded Alu RNA. These different possibilities for Alu location with respect to other transcription units are illustrated in Figure 1B.
Figure 2 provides a schematic representation of the pipeline we devised to map ENCODE sequence reads to human Alu collections. Our search strategy displays two main features introduced to ensure as much as possible the identification of genuine Alu transcripts (i.e. transcripts whose start, end and sequence closely match those expected from Pol III-dependent transcription of a particular annotated Alu element). The first such feature, aimed at avoiding ambiguous mapping due to Alu repetitive nature, is the reconstruction of base-resolution expression profiles of individual Alus based exclusively on sequence reads that do not map to any other genomic location. This task was accomplished through the TopHat aligner, and was facilitated by the paired-end nature of ENCODE RNA-seq data, allowing unique mapping not simply on the basis of the sequence of individual reads, but also of the combination of sequence and colocalization of the two 76-nt mates in the same read pair (see Materials and Methods and Supplementary Methods for details). Such a ‘unique’ alignment strategy (29) might lead to underestimate the number of expressed Alus (as well as general Alu expression levels); in particular, expressed Alus present in multiple identical copies would be overlooked. To take this possible limitation into account, a parallel and more permissive analysis was also conducted in which each individual read mapping to more than one site was not discarded, but randomly attributed to one of the matching genomic sites [‘best match’ alignment strategy in (29)]. Most of the data presented in this study were based on unique alignment, as the unambiguous identification of expression-positive Alu loci was our main task. The less stringent, ‘best match’ alignment was only employed for some analyses, which would have been compromised by the exclusion of reads that were not uniquely mappable (see below).
Figure 2.
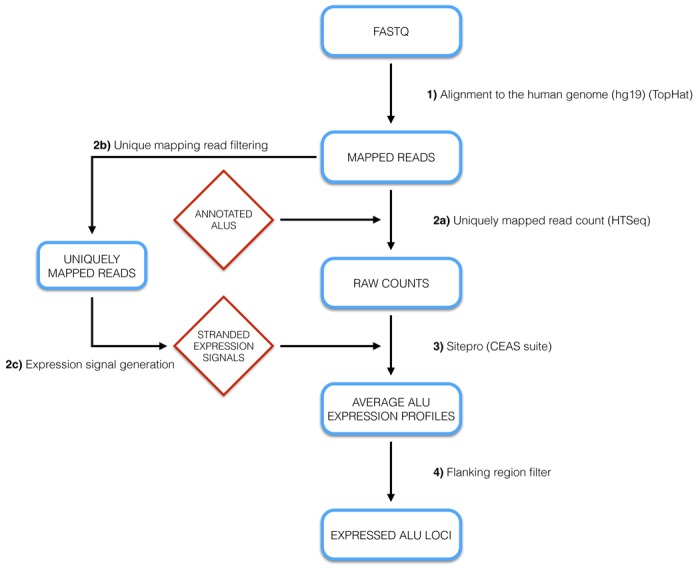
Alu RNA identification pipeline. Shown is a flow-diagram of the bioinformatic pipeline for the identification of autonomously expressed Alu loci from RNA-seq data sets. See Results and Materials and Methods for details.
The second key feature of the search pipeline is a filter step which, by imposing a requirement for significantly lower read densities to the flanking regions immediately upstream and downstream of each Alu element, systematically excludes Alu RNA sequences that are part of longer, Pol II-synthesized transcripts. This filter is also aimed at excluding Alu RNAs that are part of Pol II transcript trailers extending downstream of annotated 3′UTRs. A shortcut for the elimination of embedded Alu RNA could have been the a priori exclusion, from the reference Alu data set, of any Alu mapping in a RefSeq gene in a sense orientation. In this case, however, a number of potentially interesting cases might have been overlooked. Indeed, the Pol III machinery might in principle also act on Alus embedded in Pol II gene introns or UTRs, to produce free (not embedded) Alu RNAs. As a further possibility, intron-located Alu RNAs might be released from the host intron RNA through intron processing, as it occurs for intron-derived microRNAs or snoRNAs (34,35). Both nested Pol III transcription units and Alu RNA maturation from introns would generate Alu RNAs passing the final filter step for independently expressed Alu transcripts in our search pipeline. For each data set our search thus considered, as potential transcript sources, all Alus (either complete or incomplete), while maintaining a distinction among: (i) intergenic/antisense Alus; (ii) intronic sense-oriented Alus; (iii) 5′/3′ UTR-embedded, sense-oriented Alus; (iv) partially or fully exonic sense-oriented Alus. We chose to consider as expressed those Alus with 10 or more uniquely mapped read pairs (see the Materials and Methods section for the rationale for this choice).
General features of Alu transcriptomes emerging from RNA-seq data analysis
The full list of Alus identified as expressed by our search algorithm is provided in Supplementary Table S1. We observed that more Alu RNAs are recovered in the polyA− than in the polyA+ fraction of cellular RNA. In detail, 394 and 968 individual Alu RNAs were collectively identified in polyA+ and polyA− fractions, respectively; among these, Alu RNAs originating from 67 Alu loci were found in both polyA+ and polyA− fractions. Therefore, even though Alu RNAs contain an intermediate A-rich spacer and a 3′-terminal poly(A) or A-rich tract which might facilitate their inclusion into poly(A)-containing cellular RNA fractions (36), the A tracts of the majority of them are not sufficiently long for inclusion in polyA+ RNA.
A preliminary survey of base-resolution expression profiles of individual Alus characterized by different locations suggested that the search pipeline was very effective in identifying intergenic Alus autonomously expressed from their Pol III promoter. Several cases of gene-hosted, sense-oriented Alus, whose transcripts appear to accumulate independently from host gene expression, could also be identified. Among Alu RNAs mapping to gene-hosted elements, however, the final filter step did not appear to be completely effective in excluding spurious Alu RNA sequences that are probably part of longer intronic or messenger RNA molecules. Such ambiguous signals were frequently observed in correspondence of incomplete Alu elements, whose base-resolution profiles, allowing them to pass the filter test, could be explained by the presence of transcribed non-Alu sequences flanking the incomplete Alu upstream and/or downstream, but likely deriving from Pol II transcription of the host gene. For these reasons, we decided to mainly focus on the expression of intergenic/antisense Alus, while a few examples of gene-hosted Alus will be addressed later in the Results section.
As summarized in Table 1, each of the cell lines expressed a limited number of Alu elements (ranging from 149 in the case of Gm12878 cells to 425 in the case of HepG2 cells). Of the whole set of 1295 expression-positive Alu loci, about 30% displayed an intergenic/antisense location (including both purely intergenic Alus and Alus overlapping with annotated genes in an antisense orientation). Among expression-positive intergenic/antisense Alus, a significant percentage (∼22%) actually mapped in antisense orientation to introns of annotated, Pol II-transcribed genes (including 10 lincRNA genes). A consistent fraction (20%) of the expression-positive intergenic/antisense Alus were found to be expressed in more than one cell line. On average, ∼40% of intergenic/antisense Alus expressed in a cell line were also expressed in at least one other cell line. Given the extremely high number of genomic Alus which could in principle be expressed, on the order of hundreds of thousands, such a marked sharing of actually expressed Alus among different cell lines, each of which expresses no more than 0.1% of total Alus, points to the existence of a tiny subset of ‘transcription-prone’ Alu elements, within which cell type-specific differences in Alu expression profiles can be established. Alus have been classified into three main subfamilies, called AluJ, AluS and AluY, and it has been proposed that AluY elements, being the youngest evolutionarily, and thus the less degenerated in sequence, might represent the most transcriptionally active subfamily, in agreement with the observation that the only known Alu elements currently active in retrotransposition in the human genome belong to the AluY subfamily (37). We thus asked whether a higher tendency to be expressed could be put in light for AluY with respect to AluJ and AluS subfamilies. As summarized in Table 2, no significant overrepresentation of any particular Alu subfamily within the set of expressed Alus was observed when intergenic Alus only were considered, while AluY, somehow unexpectedly, appeared to be slightly underrepresented when the full set of expression-positive Alus was considered. Since the above data are based on uniquely mapped reads, the younger AluY subfamily, whose individual members tend to be more homogeneous in sequence, could be underrepresented among expression-positive Alus simply because of a wider exclusion of the corresponding reads as non-uniquely mapped. To avoid such a possible bias, we interrogated for Alu subfamily representation an Alu expression data set generated through a variant of our search pipeline in which the TopHat aligner, through the ‘-g1’ setting, distributes multi-reads randomly across equally good loci (see Supplementary Methods). We reasoned that, in this way, most AluY multi-reads, discarded in the unique alignment, would be attributed to members of the same subfamily. As shown in Table 2, the AluY underrepresentation was less marked in this case, thus leading to conclude that no preferential expression of specific Alu subfamilies is put in evidence by our analysis.
Table 1. Statistics of expression-positive Alu elements in selected cell lines.
| Cell line | Total Alusa | Intergenic/antisense | Intergenic/antisense sharedb | Antisense to intronsc |
|---|---|---|---|---|
| Gm12878 | 149 | 48 | 21 | 13 |
| H1-hESC | 257 | 92 | 28 | 12 |
| HeLa S3 | 276 | 44 | 32 | 7 |
| HepG2 | 425 | 88 | 31 | 19 |
| HUVEC | 326 | 36 | 18 | 4 |
| K562 | 154 | 71 | 33 | 20 |
| NHEK | 231 | 130 | 38 | 34 |
| ALLd | 1295 | 386 | 78 | 87 |
For each cell line, from left to right, the first column reports the number of Alus considered as expression-positive in both ENCODE RNA-seq replicates; the second column reports the number of intergenic/antisense Alus (including purely intergenic Alus and Alus overlapping to other transcription units in an antisense orientation); the third column reports the number of intergenic/antisense Alus that, in addition to the corresponding cell line, were also expression-positive in one or more different cell lines; the fourth column reports the numbers of Alus mapping with an antisense orientation to introns of either protein-coding or lncRNA genes. The numbers in the last (bottom) raw refer to the full set of individual Alus expressed in one or more cell lines.
aFor each cell line, the column reports the number of Alus considered as autonomously expressed in both ENCODE RNA-seq replicates.
bFor each cell line, the column reports the number of intergenic Alus that are also expressed in one or more different cell lines.
cReported in this column are the numbers of intergenic Alus mapping with an antisense orientation to introns of both protein-coding and lncRNA genes.
dThe numbers in this raw refer to individual Alus expressed in one or more cell lines.
Table 2. Subfamily distribution of expression-positive Alus.
| Alu subfamily | Total genomica | Expressed genomica | Total intergenic/antisensea | Expressed intergenic/antisensea (copy number) | Expressed intergenic/antisenseb (read count) |
|---|---|---|---|---|---|
| S | 675 428 (60%) | 735 (57%) | 513 048 (60%) | 219 (57%) | 62% |
| J | 307 612 (27%) | 479 (37%) | 225 907 (27%) | 112 (29%) | 27% |
| Y | 140 707 (13%) | 81 (6%) | 107 922 (13%) | 55 (14%) | 11% |
| TOTAL | 1 123 747 | 1295 | 846 877 | 386 | 100% |
For each Alu subfamily (raws) and subset (columns), the table reports the absolute number of elements and (in parentheses) their percentage. The percentages were calculated by dividing the copy number of each subfamily for the total Alu copy number in the different sets of Alus (from the second to the fourth column: whole genomic Alu set; expression-positive Alus at any genomic location as detected by ‘unique’ alignment; whole intergenic/antisense Alu set; expression-positive intergenic/antisense Alus as detected by ‘unique’ alignment). The rightmost column refers to the data set of expressed intergenic/antisense Alus generated through a variant of the search pipeline in which the TopHat aligner, through the ‘-g1’ setting, distributes multi-reads randomly across equally good loci.
aReported are the absolute copy numbers and (in parentheses) the percentages of Alus of each sub-family considered relative to (from left to right): the total set of genomic Alus (‘Total genomic’); the set of Alus found to be expression-positive in one or more cell line (‘Expressed genomic’); the total set of intergenic/antisense Alus; the set of intergenic/antisense Alus found to be expression-positive in one or more cell lines.
bThe rightmost column refers to the data set of expressed intergenic/antisense Alus generated through a variant of the search pipeline in which the TopHat aligner, through the ‘-g1’ setting, distributes multi-reads randomly across equally good loci.
Survey of expressed intergenic Alus according to location and base-resolution expression profiles
The inspection of individual expression profiles reconstructed through our analysis for intergenic Alus revealed different types of profile that deserve circumstantial examination. In particular, profiles were observed which can be roughly summarized as: whole Alu, left-monomer, right-monomer coverage.
Figure 3 shows examples of the occurrence of these expression profiles for both purely intergenic and intron-antisense Alus. For the AluSg reported in Figure 3A, found to be expressed in three different cell lines (H1hESC, K562 and NHEK), a complete and precise coverage of the Alu by uniquely mapping sequence reads was observed. An inspection of its sequence revealed that this Alu possesses canonical A- and B-boxes, as well as a Pol III termination signal located ∼20 bp downstream of the 3′ poly(dA) tail, thus suggesting that Pol III transcription of this intergenic transcription unit generates a specific, ∼300-nt-long Alu RNA. Figure 3B and C show typical examples of expression profiles corresponding to truncated Alu transcripts. Frequently, sequence reads tended to cover either the left or the right monomer of a complete Alu element. For the AluY of Figure 3B, sequence reads precisely covered the left monomer sequence, up to the short A-rich region (A5TACA6) separating the two Alu monomers. Given the absence of Pol III termination signals within the body of this Alu, the short transcript likely belongs to the previously reported family of small cytoplasmic (sc) Alu RNAs (38), being generated by processing of a full-length primary Alu transcript (14). Truncated Alu transcripts like the one reported for the AluSc in Figure 3C are more difficult to interpret based on our current understanding. In this case, the transcript appears to start just downstream of the internal A-rich region, suggesting that right monomer Alu RNA fragments might also be generated through processing of full-length precursors. Incomplete coverage of some Alus might in principle be due to the fact that these Alus possess sequence tracts (corresponding to the uncovered regions) that are identically repeated at other genomic locations, such that mapping reads would be non-unique and thus discarded. To explore this possibility, we looked at the coverage profiles obtained for some of these Alus (those in Figure 3B and C) using the TopHat bam file generated with default settings, and thus reporting up to 20 alignments for multi-mapped reads. We still observed the same incomplete coverage for all of these Alus (Supplementary Figure S1). Incomplete expression profiles are thus unlikely to be due to multi-mapping issues, as they are not appreciably changed by multi-read permissive alignment. Furthermore, the fact that the same partial coverage profiles were also observed with STAR alignment (also shown in Supplementary Figure S1) argues against partial coverage being an aligner artifact. In Figure 3D, the whole AluY element (mapping with antisense orientation to the second intron of the COL4A1 gene) is covered by sequence reads all along its extension, but with a double-humped profile in which two peaks are approximately centered on the left and right monomer of the Alu element. This type of profile was much more frequently observed in our analyses than the more continuous type of profile, such as the one shown in Figure 3A. As a tentative explanation, we reasoned that, since a full-length Alu transcript is on average 300-nt long, the post-fragmentation selection of 200 bp fragments during RNA-seq library preparation (20) should produce a relative enrichment in fragments containing either the 5′ or the 3′ end of the Alu cDNA. Sequencing of the 3′ and 5′ ends of such cDNA fragments would lead to an underrepresentation of the central part of the transcript, and thus to the generation of two-humped base-resolution profiles.
Figure 3.
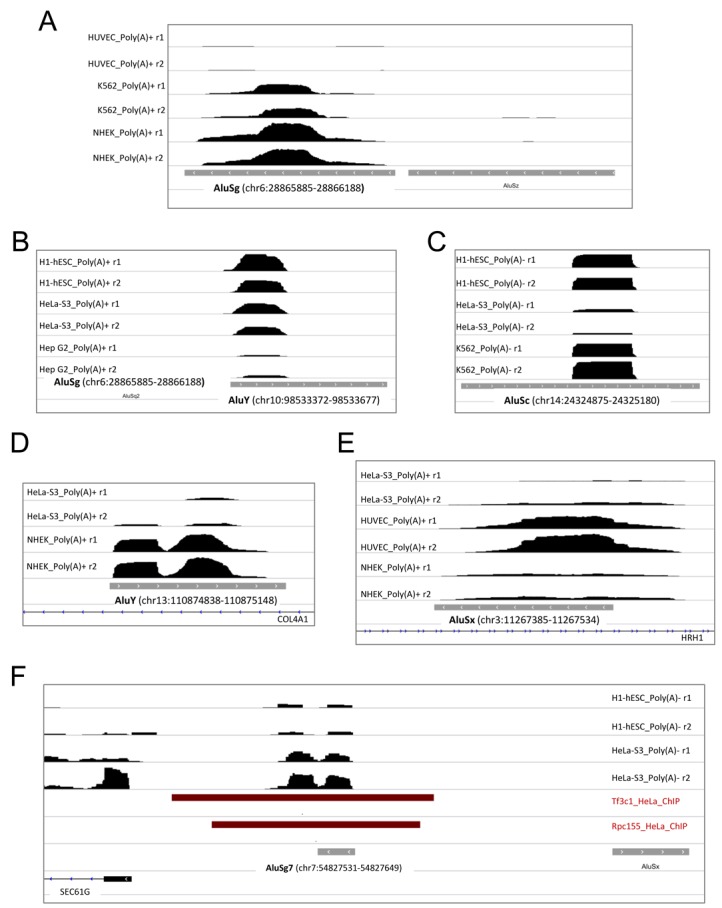
Base-resolution expression profiles for six representative Alus of the intergenic/antisense type. Panels A–C and F refer to purely intergenic Alus, panels D and E to two antisense Alus. Shown are the Integrative Genomics Viewer (IGV; http://www.broadinstitute.org/igv/home) visualizations of RNA-seq stranded expression profiles (in bigwig format) around Alu loci in the cell lines indicated either on the left (A–E) or on the right (F) of each panel. r1 and r2 indicate the two independent replicates found in ENCODE data. The orientation and chromosomal coordinates of each Alu, as well as the overlapping (antisense) or nearby RefSeq genes, are indicated in each panel. The dark red bars in panel F indicate regions associated to either TFIIIC (Tf3c1 track) or Pol III (Rpc155 track) in HeLa cells as derived from ENCODE ChIP-seq data.
We also identified cases of incomplete Alu elements (Alu monomers) whose corresponding RNAs extend upstream or downstream of the annotated Alu monomer. Figure 3E shows the case of a 150-bp long, right AluSx monomer (mapping with an antisense orientation to the first intron of HRH1, just upstream of the next-to-last exon), whose sequence read coverage extend ∼60 bp upstream, delineating a transcription unit starting upstream of the Alu monomer, within an Alu-unrelated region, and including the Alu right monomer as the downstream moiety of the transcript. A complementary example of an Alu left monomer being part of a longer transcription unit extending downstream is reported in Figure 3F, showing the expression profile of a purely intergenic AluSg7. Here a ∼120-bp left monomer containing A- and B-boxes appears to direct the synthesis of a transcript ending ∼180 bp downstream, at a position which is only ∼400 bp upstream of the TSS of the SEC61G gene. Through parallel analysis of ENCODE ChIP-seq data of Pol III components, we noted the existence of Pol III and TFIIIC association peaks precisely mapping to this Alu, an observation supporting the conclusion that it constitutes a bona fide Pol III transcription unit. (A more exhaustive account of parallel analysis of ENCODE ChIP-seq data will be provided below.) Through the ‘-g1’ variant of our search algorithm, attributing multi-reads randomly to one of the hits, we observed another interesting case of an Alu left monomer directing the transcription of a longer transcription unit (see below ‘Identification of a novel AluYa5-derived Pol III transcript’). Expression of Alu monomers is thus likely to be more frequent than commonly thought, in agreement with the observation of recent Alu monomer insertions, some of which generated through retroposition (39). Interestingly we observed, as a general trend for expression-positive Alu monomers, that transcripts mapping to left and right Alu monomers extend downstream and upstream of the monomer, respectively, in agreement with the fact that Alu left monomers generally contain a functional Pol III promoter, able to direct transcription of the monomer itself followed by downstream sequences until a Pol III terminator is encountered, while Alu right monomers do not contain a Pol III promoter and thus their expression requires incorporation into an upstream initiated transcript.
Evidence for independent expression of gene-hosted, sense-oriented Alus
Even though Alus located within intron or exons (including UTRs) of Pol II-transcribed genes are expected to be mostly transcribed as part of longer Pol II transcripts, we addressed the possibility that a few of them might be transcribed as autonomous Pol III transcription units or, more generally, that the corresponding Alu RNAs might accumulate to a detectable extent independently from host gene expression. The final filter step of our search algorithm is devised to produce an enrichment of such Alu RNA species, as it imposes a strong reduction in the number of sequence reads mapping to regions flanking the gene-hosted Alus, thus favoring isolated expression signals centered on Alu elements. By inspecting the profiles of many gene-hosted (especially intron-hosted) Alus that had been identified as expression-positive in our search, we confirmed the presence of Alu-centered expression signals as expected on the basis of our filter step; the Alu peaks, however, were frequently preceded and/or followed by expression peaks mapping to Alu-less surrounding regions, thus suggesting the possibility that Alu signals, as well as the surrounding signals, might represent fragments of longer intron RNAs. In a limited number of cases, however, Alu expression profiles were suggestive of the presence of autonomous Alu transcription units. One such case is illustrated in Figure 4A, showing the base-resolution expression profiles of an AluSx1 located, in a sense orientation, within the first intron of SRGAP2, a gene involved in human brain development and evolution (40). The AluSx1 is followed immediately downstream by an AluSp with the same orientation, to which a few sequence reads also map. The left monomer of the AluSx1 has canonical A- and B-boxes, but the first potential Pol III terminator is located downstream of the AluSp, thus suggesting that these two Alus might be transcribed into a dimeric Alu primary transcript. A similar situation is illustrated by the example in Figure 4B, showing the base-resolution profile of an intronic sense-oriented AluY located between exons 9 and 10 of ZC3H3 gene. This Alu Y is endowed with A- and B-boxes, and even if there is no recognizable Pol III termination signal separating the AluY from the AluSq located immediately downstream with the same orientation, transcription appears to terminate just downstream of the first Alu, given the absence of sequence read coverage of the second Alu. However, through parallel analysis of ENCODE ChIP-seq data of Pol III factors, we noted that Pol III (and TFIIIC) appear to be associated with a region encompassing both AluY and the downstream AluSq, as if both were part of the same transcription unit. An intriguing example of independent accumulation of intronic Alu RNA is provided by the AluJb located within the intron separating exons 35 and 36 of USP34 (Figure 4C). The expression levels of this left Alu monomer (whose transcripts extend downstream by ∼70 bases) appear to be inversely correlated with the levels of exon 37 expression in the different cell lines, suggesting mutual expression interference. A few cases of independently expressed Alus located within lincRNA gene introns were also observed. One of them is illustrated in Figure 4D, showing the base-resolution profiles of an AluY hosted in a sense orientation between exons 4 and 5 of lincRNA gene TCONS_I2_00015350 on chromosome 2. ChIP-detected association of Pol III and TFIIIC with this Alu locus further argues that it is a genuine Pol III transcription unit. Finally, as exemplified in Figure 4E, 3′ UTRs can also host sense-oriented Alus whose transcripts accumulate independently from the corresponding mRNA.
Figure 4.
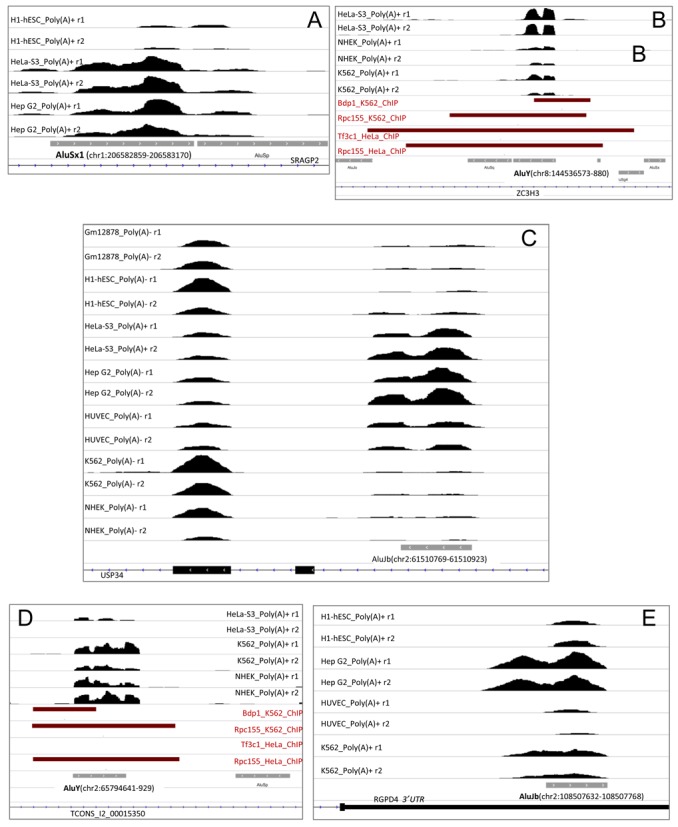
Base-resolution expression profiles for five representative gene-hosted, sense-oriented Alus. Panels A–C refer to Alus hosted within introns of RefSeq genes, panel D to a 3′UTR-hosted Alu, panel E to an Alu hosted within a a lincRNA gene intron. Shown are the IGV visualizations of RNA-seq stranded expression profiles (in bigwig format) around Alu loci in the cell lines indicated either on the left (A–D) or on the right (E) of each panel. r1 and r2 tracks refer to the two independent replicates found in ENCODE data. The orientation and chromosomal coordinates of each Alu, as well as the host RefSeq or lincRNA genes, are indicated in each panel. The dark red bars in panels B and F identify regions associated to the indicated Pol III transcription component (Bdp1, Tf3c1 or Rpc155) in either K562 or HeLa cells as derived from ENCODE ChIP-seq data.
Association of the Pol III machinery to expression-positive Alus
Several genome-wide association studies based on ChIP-seq approaches have been conducted in the last few years with the aim of producing complete inventories of Pol III-transcribed genes (reviewed in (2)). Each of these studies identified a variable (generally small) number of Alus associated to the Pol III machinery. In a recent study, an integrated, comparative evaluation of Pol III-associated Alus was carried out through a synopsis of several ChIP-seq studies (19). We asked whether there is any significant overlap between the set of Alus identified as expressed in our analysis and the Pol III-associated Alus in ChIP-seq studies. To address this point we took advantage of the availability, within the ENCODE data, of ChIP-seq data sets, relative to both K562 and HeLa-S3 cell lines, for key components of the Pol III transcription machinery: Bdp1 and Brf1 (components of TFIIIB), Rpc155 and TFIIIC110 (subunits of RNA polymerase III and TFIIIC, respectively). Supplementary Table S2 lists the expression-positive Alus that are also associated to Pol III components in HeLa and K562 cells. In HeLa cells, 15 out of 276 expression-positive Alus (∼6%) were found among those associated to one or more components of the Pol III machinery in the ENCODE data sets. When the comparison was restricted to the 44 intergenic Alus detected as expressed in HeLa cells, a much higher fraction of them (29%, 13 Alus) were also associated with the Pol III machinery, with 11 Alus being associated with at least two transcription components and 8 with three components representing the whole machinery (TFIIIB, TFIIIC, Pol III). P-values for association of Bdp1, TFIIIC110 and Rpc155 with intergenic expressed Alus (versus the whole set of intergenic Alus) were all <10−14. Similarly, when K562 cells were considered, a significant percentage of expression-positive Alus was Pol III-associated and most strikingly, of the 71 intergenic expression-positive Alus in these cells, 31 (corresponding to 44%) were found associated with at least one component of the Pol III machinery. P-values for association of Bdp1, TFIIIC110 and Rpc155 with intergenic expressed Alus (versus the whole set of intergenic Alus) in K562 cells were <10−15 (Supplementary Table S2). Specifically, of the intergenic Alus whose expression profiles were shown in Figure 3, three were found to be associated to either Pol III (chr13:110874838–110875148, panel D) or Pol III and TFIIIC (chr6:28865885–28866188, panel A; chr7:54827531–54827649, panel F). Interestingly, a few intronic sense-oriented Alus identified as autonomously expressed were also found to be associated with components of the Pol III machinery; among them were those whose profiles are shown in Figure 4B (chr8:144536573–880) and D (chr2:65794641–929). Altogether these findings confirm the effectiveness of our Alu RNA detection procedure, especially in the case of intergenic Alus but also for intron-hosted elements, and suggest the existence, in each cell type, of a very small and specific subset of individually trackable, transcription-prone Alus. We noted that only four Alu elements were found to be expressed and Pol III-associated in both K562 and HeLa cell lines (chr1:61523296–61523586; chr10:5895538–5895651; chr1:28672563–28672802; chr8:144536572–144536880), suggesting a high plasticity of the Alu transcriptome.
Identification of a novel AluYa5-derived Pol III transcript
In parallel with a stringent search procedure based on a ‘unique alignment’ strategy, we also applied to ENCODE RNA-seq data sets a ‘best match’ alignment strategy (29), in which multi-reads are attributed randomly to one of the hits, with the aim of detecting expressed Alus whose presence in multiple identical copies in the genome would prevent their identification as expression-positive in the unique alignment strategy. In this case, the analysis was restricted to intergenic/antisense Alus. As expected, a significantly higher number of intergenic/antisense Alu elements were identified with respect to ‘unique alignment’ search (705 versus 386). Through systematic inspection of Alus found as expression-positive in at least three cell lines, we discovered multiple (∼20) almost identical copies of an AluYa5 left monomer, encompassed within the recently described snaR A/C and snaR A/B/D clusters on the q-arm of chromosome 19 (41). Base-resolution expression profiles of these AluYa5 elements suggest that transcription initiates at the Alu monomer and continues downstream of it, in a 3′-flanking region whose sequence is Alu-unrelated. In this respect these transcription units, hereafter referred to as Ya5-lm (for left monomer of Alu Ya5), resemble the BC200 RNA gene, which can also be described as a transcriptionally active Alu element consisting of an upstream monomeric Alu repeat followed by a non-repetitive domain (23). The base-resolution expression profile of three clustered Ya5-lm elements is shown in Figure 5A, in which their expression can be directly compared with the Pol III-dependent expression of interposed SNAR-A3 elements. Reported in Figure 5B are the sequence and the general organization of Ya5-lm. The upstream AluYa5 monomer contains typical Alu A- and B-boxes (with the A-box differing from canonical tRNA A-box for a C instead of G at the last position; (5)), and ends with an A-rich motif. Downstream of this motif the sequence of the Ya5-lm transcription unit diverges from consensus Alu sequence (Figure 5C). The first potential Pol III termination signal (TTTT) starts at position +260, almost exactly corresponding to the end of sequence read coverage. The snaR genes on chromosome 19 are arrayed in two large inverted regions of tandem repeats, with the two clusters (A/C and A/B/D) separated by a 2-Mb region (41,42). We found that these two clusters contain 11 and 10 copies of Ya5-lm, respectively. In both clusters, all Ya5-lm, separated from each other by ∼5300 bp, have the same orientation as snaR genes, and each of them is separated by ∼1800 and ∼3300 bp from the upstream and downstream snaR gene, respectively. As the Ya5-lm copies on chromosome 19 are almost identical, it is difficult to specifically attribute to one or more of them the mapping sequence reads. Nevertheless, since the non-repetitive sequence domain downstream of AluYa5 monomer is not found at any other locus in the genome, there is no doubt that one or more of these genes are transcribed to produce a novel type of Alu-derived Pol III transcript. In support to this conclusion are ChIP-seq data from Pol III genome-wide location studies available at ENCODE. In one of them, Pol III-associated loci in K562 cells were identified through ChIP-seq using an antiserum against the Pol III largest subunit Rpc155 (18). Analysis of Rpc155 ChIP signals revealed a peak precisely overlapping with the AluYa5 identified by the coordinates chr19:50640453–50640584, and by transcript coverage, in both HeLa and K562 cells (Figure 5D). It is thus likely that only one (or a small subset) of the Ya5-lm elements on chromosome 19 are transcriptionally active. The attribution of multi-reads randomly to one of the hits in the ‘best match’ alignment strategy explains why all Ya5-lm copies are covered by sequence reads (as exemplified in Figure 5A).
Figure 5.
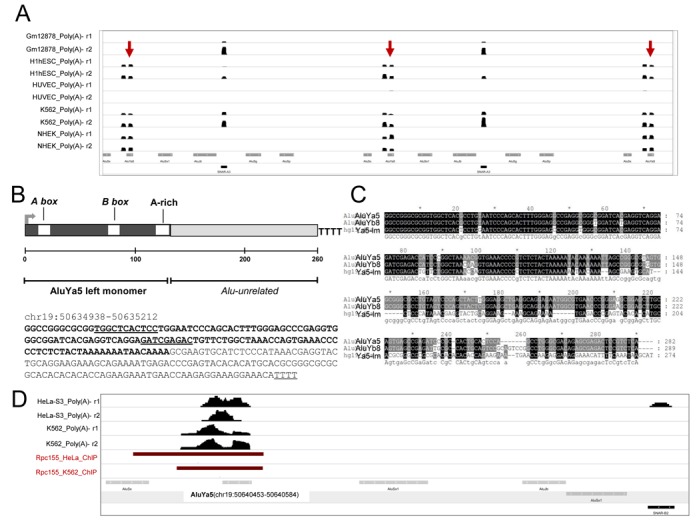
Novel AluYa5-derived transcription units associated to snaR clusters. (A) Genome browser visualization of RNA-seq stranded expression profiles of three AluYa5-derived transcription units (Ya5-lm, indicated by red arrows) within the snaR A/C/D cluster on chromosome 19 (41). (B) Transcription unit architecture and sequence of a Ya5-lm repeat (coordinates in parentheses). (C) Sequence alignment of Ya5-lm with Repbase reference sequences for AluYa5 and AluYb8. (D) Genome browser visualizations of RNA-seq stranded expression profiles around the Ya5-lm element represented in panel B, in the cell lines indicated on the left. The dark red bars identify regions associated to Pol III (Rpc155 subunit) in either K562 or HeLa cells as derived from ENCODE ChIP-seq data.
In vitro transcription analysis of expressed and silent Alu elements
The ability to detect in vivo expression of individual Alu elements prompted us to verify whether Alus with different expression levels can also be differentiated for their in vitro transcription behavior. To this end, we focused on a small subset of Alu loci, representative of different types of expression profiles based on the analyzed RNA-seq data sets. These loci are listed in Table 3. One of them, AluSq2_chr1:61523296–61523586, appears to be expressed in five different cell lines (H1-hESC, HeLa-S3, Hep G2, K562, NHEK), and was also found to be associated to the Pol III machinery in both HeLa and K562 cells (see Supplementary Table S2). This Alu was thus chosen as representative of ubiquitously expressed Alus. Moreover, this Alu is peculiar in sequence as it lacks the internal A-rich motif A5TACA6, which is replaced by A3G. Two other loci, AluY_chr10:69524852–69525156 and AluSp_chr17:4295121–4295437, are expressed above our chosen threshold in only one out of seven cell lines [NHEK and K562 cells, respectively; but lower levels of expression were detectable in other cell types; interestingly, AluY_chr10 is among the few Alus identified as expressed in this study that were also among the candidate source Alus in a recent analysis (19)]. Three loci (AluSx1_chr3:139109300–139109588, Alu Y_chr7:73761603–73761897, AluY_chr10:103929453–103929749) were found to be expressed in a somewhat sporadical manner (i.e. in no more than two cell lines and in one replicate only); however, based on ENCODE ChIP-seq data, each of them is associated to one or more components of the Pol III machinery. The remaining three loci (AluSx_chr1:235531222–235531520, AluSx_chr10:12236879–12237173, AluY_chr22:41932115–41932411) were not found to be detectably expressed by our analysis, even though the AluSx on chromosome 10 was Pol III-associated based on ENCODE ChIP-seq data. Five of these Alus have a purely intergenic location (i.e. they do not overlap with any other transcription unit in either antisense or sense orientation), while four of them are antisense with respect to introns of protein-coding genes. Interestingly one of them, AluY_chr22:41932115–41932411, maps in antisense orientation to intron 2 of POLR3H, coding for the 22.9-KDa subunit of RNA polymerase III (RPC8/RPC22.9), thus suggesting a possible role of this element in POLR3H gene regulation, as already proposed for a MIR elements located on the minus strand within the first intron of both human and mouse genes coding for the RPC5 subunit of Pol III (16,43). The other selected antisense Alus map to: the first intron of TBCE (Tubulin Folding Cofactor E) gene (AluSx_chr1:235531222–235531520); the third intron of CLIP2 gene (AluY_chr7:73761603–73761897); the first intron of NUDT5 gene (AluSx_chr10:12236879–12237173). The nine selected Alu elements were PCR-amplified from human genomic DNA, cloned into pGEM-T-easy vector and tested for their ability to support efficient in vitro transcription using a HeLa cell nuclear extract. To verify that the observed transcripts were produced by the Pol III machinery, reactions were conducted in the presence of α-amanitin at a concentration (2 μg/ml) known to completely inhibit RNA polymerase II activity, and transcription reactions were also programmed in parallel with a mutant version of each Alu element, in which the B box internal promoter element was mutationally inactivated. The results of in vitro transcription analysis are shown in Figure 6. Control transcription reactions were programmed with empty pGEM-T-easy plasmid (lanes 1, 11, 21) and the same vector carrying either (lane 2, 12, 22) a previously characterized, transcriptionally active Alu (AluSx1_chrX:24096144–24096441, producing a 372-nt transcript; A. Orioli and G. Dieci, unpublished data) or (lane 3, 13, 23) a tRNAVal(AAC) gene (TRNAV18, chr6) whose transcription produces three different primary transcripts (of 87, 112 and 142 nt) because of heterogeneous termination at one of three consecutive termination signals (25). Each of the tested Alu elements produced a well-defined pattern of transcription, in which the sizes of the longest and most abundant transcripts matched those predicted on the basis of sequence inspection of the Pol III termination signals, either canonical (a run of at least four Ts) or non-canonical (25), in the 3′-flanking region. The observed transcription efficiencies of all Alus were comparable (with the exception of AluSx_chr1 (lane 7) producing low levels of transcription products heterogeneous in size), indicating that their different tendency to be transcribed in cultured cells is not due to differences in cis-acting elements recognized by the basal Pol III transcription machinery. When the Alu B box was mutationally inactivated (by substituting CG for the invariant TC dinucleotide of the B box consensus sequence GWTCRAnnC), a dramatic reduction in Alu transcription efficiency was observed, thus confirming the essential character of this element for Alu transcription (6).
Figure 6.

In vitro transcription analysis of wild type and B box-mutated Alu loci. In vitro transcription reactions were performed in HeLa nuclear extract using 0.5 μg of the indicated. Alu templates (lanes 5–10, 15–20, 25–30). A previously characterized Alu producing a 372-nt RNA (lanes 2, 12, 22) and a human tRNAVal gene producing a known transcript pattern due to heterogeneous transcription termination (lanes 3, 13, 23) (25) were used as positive controls for in vitro transcription and, at the same time, as a source of RNA size markers. Negative control reactions contained either empty pGEM®-T Easy vector (lanes 1, 11, 21) or no template DNA (no-template control (NTC), lanes 4, 14, 24). For each Alu, both the wild-type and a B box-mutated (Bmut) version were tested.
Upstream flanking sequences have previously been shown to influence transcription efficiency of Alu and other SINEs both in vitro and in transfected cells. In particular, upstream deletion mutants of an individual Alu element displayed reduced transcription efficiency, possibly due to the loss of interactions with sequence-specific TF(s) (7). In another study, upstream sequences already known to stimulate transcription of Pol III-transcribed genes (such as vault or U6 RNA genes) were shown to stimulate SINE transcription in chimeric constructs (8). To explore more extensively the role of upstream regions in Alu transcription, we constructed 5′ deletion mutants of the 9 isolated Alus and compared their in vitro transcriptional activity with the one of wild-type constructs. In each case, the natural upstream sequence up to position −12 (or −15 for AluSx_chr10) was replaced by vector sequence, and care was taken to have each wt-deleted Alu pair inserted into plasmid vector with the same orientation, to minimize differences in transcription due do different vector sequence contexts. As shown in Figure 7, as a general trend, upstream sequence deletion negatively affected transcription; however, the extent of transcription inactivation varied markedly among the different Alus. Transcription of upstream deleted Alus was reduced by 4- to 5-fold in the case of AluSx_chr1 and AluSx_chr10 and AluSp_chr17 (cf. lanes 7, 25 and 27 with lanes 8, 26 and 28, respectively), while it was not appreciably affected in AluY_chr10-a (lanes 19 and 20) and only moderately reduced (∼1.5-fold) in the case of AluSq2_chr1 and AluSx1_chr3 (cf. lanes 5 and 9 with 6 and 10, respectively). Overall the data consolidate the notion that the nature of the upstream region may strongly influence Alu transcription; however, they do not reveal any obvious correlation between upstream sequence dependency and in vivo expression profiles.
Figure 7.

In vitro transcription analysis of upstream deleted Alu loci. In vitro transcription reactions were performed in HeLa nuclear extract using 0.5 μg of the indicated Alu templates (lanes 5–10, 15–20, 25–30). A previously characterized Alu producing a 372-nt RNA (lanes 2, 12, 22) and a human tRNAVal gene producing a known transcript pattern due to heterogeneous transcription termination (lanes 3, 13, 23) (25) were used as positive controls for in vitro transcription and, at the same time, as a source of RNA size markers. Negative control reactions contained either empty pGEM®-T Easy vector (lanes 1, 11, 21) or no template DNA (no-template control (NTC), lanes 4, 14, 24). For each Alu, both the wild-type and a mutant version lacking most of the native 5′-flanking region (5′del) were tested. For each of the nine Alus subjected to 5′-flank deletion, the extent of reduction of transcription activity, observed with respect to the corresponding wild-type Alu, is reported below the lanes corresponding to each wt-mutant pair. The values represent the average of two independent transcription experiments that differed by no more than 20% of the mean.
Association with TFs of expression-positive Alus
The influence of upstream region on Alu transcription might be mediated by TFs specifically interacting with this region. The availability of ChIP-seq data sets for several TFs within ENCODE prompted us to assess whether the Alus identified as expressed through RNA-seq data analysis tend to be associated with one or more Pol II TF, in addition to the known components of the Pol III machinery. The results of this analysis are reported in detail in Supplementary Table S3. Since the different cell lines selected for our study have been subjected to ChIP-seq analyses for a highly variable number of TFs (https://genome.ucsc.edu/ENCODE/dataMatrix/encodeChipMatrixHuman.html), a high variability of TF association was observed among them, both in terms of total number of TF-bound Alus (ranging from 17 to 79) and in terms of the number of TFs associated to each Alu. As a general trend, intergenic/antisense Alus tend to be strongly enriched for the presence of TFs associated with their upstream region, with respect to gene-hosted Alus. Apart from the components of the Pol III transcription machinery (including TBP), the transcription proteins most frequently associated to expressed Alus in most cell lines were the transcription regulator and genome organizer CCCTC-binding factor (CTCF), RNA polymerase II (detected through its largest subunit Rpb1) and the Pol II TF JunD. All of these proteins have previously been shown to colocalize with active Pol III-transcribed loci (especially tRNA genes), where CTCF might contribute to the increasingly recognized function of these loci in nuclear organization and insulation (44–47). Their association to expression-positive Alus thus strengthens the notion that the expression-positive Alu loci identified in this study resemble the other Pol III-transcribed genes not only for their transcription properties but also for their extra-transcriptional function in genome organization. Furthermore, our analysis revealed a clear cell line-specific association of expression-positive Alus with CCAAT/Enhancer Binding Protein β (CEBPB), which was one of the two most frequently matching TFs in HeLa, HepG2 and K562 cells (see Supplementary Table S3; P-values for enrichment of CEBPB at expression-positive Alu loci with respect to total Alus were lower than 10−5, 10−10 and 10−6 for HeLa, HepG2 and K562 cells, respectively). This protein was not previously reported to be enriched at other Pol III-transcribed genes; it might thus represent a novel, Alu-specific TF facilitating Alu transcription.
ChIP-seq analyses have provided recently a considerable wealth of information on histone modification marks at Pol III-transcribed genes, revealing a broad similarity between epigenetic marks typical of active Pol II- and Pol III-transcribed genes, together with a few possibly significant differences (44). The search for histone modification profiles typical of expressed Alu loci on the basis of ENCODE ChIP-seq data, that we performed by focusing on purely intergenic expressed Alus, did not produce easily interpretable results, possibly because of the too low number of analyzed loci (data not shown). From the data in Supplementary Table S3, however, we noted that in HepG2, HeLa and K562 cells the P300 acetyltransferase (EP300) is among the top 10 TFs associated to expression-positive Alus (with a P-value < 10−12 for enrichment at expression-positive Alu loci with respect to total Alus in both HeLa and K562 cells) thus suggesting that these Alus might be characterized by high levels of histone acetylation, in agreement with the results of a recent study showing an enrichment of H3K27ac and P300 at enhancer-like Alu elements (48).
DISCUSSION
This work provides the first comprehensive account of transcriptionally active Alu loci in human cells, reveals the existence of novel Pol III-transcribed genes originated from monomeric Alu elements, and supports the notion that Alu expression in human cells occurs rarely, from small, largely cell-specific sets of transcriptionally active Alus regulated by both internal and external cis-acting control elements.
Historically, the tasks of detecting genuine Pol III-transcribed Alu RNAs and of attributing them to individual transcriptionally active Alu loci had to face two challenges: the extremely high copy number and sequence similarity of Alu elements within the human genome, and the frequent location of Alus within introns or untranslated regions of primary or mature Pol II transcripts. Previous studies of Alu expression exploited northern hybridization, producing information on transcript size, as a useful tool in distinguishing genuine (∼300–500 nt) Alu Pol III transcripts from Alu RNA incorporated into longer Pol II transcripts (even though probe cross-hybridization with the closely related, ∼300-nt-long 7SL RNA might frequently represent a problem) (1,49). Distinguishing between the products of individual Alu elements, or even of different Alu subfamilies, however, is unfeasible through northern blot. Alu RNA detection approaches based on reverse transcriptase-PCR are even less effective in distinguishing genuine Pol III Alu transcripts from Alu RNA sequences included into Pol II-synthesized hnRNA or mRNA (1). To date, the only low-throughput approach that has permitted to identify genuine Alu Pol III transcripts, giving the possibility to trace the corresponding Alu loci, was based on a 3′-RACE technique disclosing sequence information on individual Alu RNAs (14). The recent development of unbiased genome-wide location analyses exploiting next-generation sequencing technologies has allowed the identification, through ChIP-seq approaches, of several Alu loci that are bound in vivo by the Pol III transcription machinery, a reasonable indication of a transcriptionally active state (19). Within this context, the original contribution of our work proceeds from the simple remark that appropriate analysis of RNA-seq data, containing full sequence information even on rare transcripts, should allow to successfully face difficulties in both sequence and length determination of Alu transcripts. Indeed, by applying to ENCODE RNA-seq data sets an ad hoc devised computational search strategy, mainly relying on unique alignment and size-selection of RNA-seq signal mapping, we were able to unveil to an unprecedented detail the Alu transcriptomes of several human cell lines. Our search algorithm appeared to work well especially for the identification of expressed intergenic/antisense Alu transcription units whose RNA products, in contrast to the ones of Alus located within Pol II genes in a sense orientation, tend to be less obscured by flanking unrelated RNA-seq signals. In strong support to the genuine nature of expression-positive intergenic/antisense Alus as independent Pol III transcription units is the observation that, in HeLa and K562 cell lines, a remarkable percentage of them (29% and 44%, respectively) was independently found to be bound by one or more components of the Pol III transcription machinery in independent ChIP-seq analyses. A modest overlap was observed between our set of expression-positive intergenic Alus and the list of putative Pol III-transcribed Alus reported by a previous integrated analysis of ChIP seq studies of human Pol III machinery (19). A possible reason for this discrepancy could be the fact that, in contrast to that study, we also included in our analysis incomplete Alu elements, that turned out to be contributing to expression-positive Alu set. Another possibility is that the compilation in (19) was based on partial lists of potentially transcribed Alus that had already been pre-selected by the authors of the different ChIP-seq studies according to very stringent criteria, which could have led to the exclusion of expression-positive Alus.
The most evident features of Alu expression profiles as revealed by our analysis are: (i) the extremely low number of detectably expressed Alus in each cell line, in the order of hundreds, corresponding to less than 0.1% of all annotated Alus; (ii) the existence, among intergenic/antisense expression-positive Alus, of an unexpectedly large set of elements expressed in more than one cell line, suggesting that, in human cells, Alu transcript profiles result from the combined activities of very few transcription-prone Alu elements, that are thus reminiscent of the rare and elusive ‘source’ Alu elements possibly contributing to Alu expansion through retrotransposition (1); (iii) even though different cell lines share a significant number of expression-positive Alus, a marked cell-specificity of Alu transcriptomes is observed, thus suggesting that the Alu RNA expression profile in each cell line results form the expression of both commonly expressed and cell-specific Alu transcription units; (iv) Alu transcriptomes as revealed by ENCODE RNA-seq data analysis are composed of both full-length and incomplete Alu transcripts, some of which might be related to the previously described scAlu transcripts corresponding to the left Alu monomer (with the caveat that Alu RNA fragment detection in our case might also result from non-physiological RNA degradation).
An interesting outcome of our analysis is the identification of novel monomeric Alu elements whose RNA-seq signal profiles suggest a transcription unit organization similar to the one first reported for the BC200 RNA gene (23): a promoter-containing Alu left monomer directing Pol III to synthesize a ncRNA containing the Alu sequence itself followed by an Alu-unrelated RNA moiety. The so-generated, Alu-derived ncRNAs have the potential to play novel regulatory roles deriving from the combination of an Alu left arm with unique RNA sequences. An Alu left monomer-derived gene that we find of particular interest, and that we have called Ya5-lm, is located in multiple copies on chromosome 19, with each copy located very close to one of the snaR gene copies belonging to either of two snaR clusters on chromosome 19 (42). Such a close spatial relationship between Ya5-lm and snaR genes (that also likely evolved from Alu left monomers) suggests that Ya5-lms have been included in the same segmental duplication through which snaR genes are thought to have spread. The snaR clusters on chromosome 19 might thus host a chromatin environment favorable to Pol III transcription of different Alu-derived ncRNAs, possibly playing recently evolved functions in translation regulation (42,50).
Perhaps not surprisingly, given the relatively frequent occurrence of intronic nested genes in metazoan genomes (51), our data also suggest that a number of gene-hosted (and particularly intron-hosted), sense-oriented Alus are likely to represent autonomous transcription units that are recognized by the Pol III machinery and thus transcribed independently from Pol II transcription of the host gene. The possible interplay between Pol II and Pol III transcription of host and nested genes is an issue deserving further investigation, especially in light of recent evidence for the involvement of a Pol III–Pol II switch in the insulator activity of a mouse B1 SINE (52), and of the widespread association of Pol II factors with Pol III transcribed genes (44), including Alus as clearly confirmed by our results (see Supplementary Table S3). Related to this issue is the observation that gene-hosted sense-oriented Alus, revealed as expression-positive by our analysis, have a lesser tendency than intergenic/antisense Alus to be associated with the Pol III machinery. This leads to speculate that the synthesis of gene-hosted (mostly intron-hosted) Alus might occur either via the release of Alu RNAs from annotated Pol II-synthesized host transcripts [similarly to intron-derived microRNAs or snoRNAs (34,35)], of through the still uncharacterized production and processing of unannotated Alu-containing nc Pol II transcripts possibly related to Alu-associated Pol II and TFs revealed by ChIP-seq analyses. This possibility also applies to intergenic/antisense Alus found to be expression-positive but not Pol III-associated.
With respect to mechanistic understanding of Alu transcription and its control, our study, by comparing in vivo expression levels with in vitro transcription rates of a number of Alu loci, confirms and extends previous knowledge about two peculiar features of Alu transcription by Pol III: (i) the stimulatory role of 5′-flanking sequences on Alu transcription; (ii) the strong epigenetic control on Alu expression in vivo. Of the 9 Alus whose transcription properties were analyzed in vitro in this study, 6 exhibited a 2-fold of higher reduction of transcription, and only one was unaffected, upon deletion of the 5′-flanking region. That upstream sequences may influence transcription by Pol III of its target genes, even when they display internal promoters, is a well documented possibility. For example, tRNA genes, whose internal promoter organization closely resembles the one found in Alus, tend to display a certain degree of upstream sequence conservation in the genomes of different eukaryotic lineages and, correspondingly, their transcription appears to be influenced by upstream sequence context both in vitro and in vivo (53). In the case of Alus, the internal Pol III promoter has been suggested not to be sufficiently strong to warrant their efficient transcription independently from favorable upstream sequences (1). With this respect, Alus resemble their 7SL progenitor, whose sub-optimal internal promoter requires upstream sequence elements to direct efficient transcription (9). If the general consensus sequences for A- and B-boxes, mainly deduced from tRNA gene sequence analysis (TRGYnnAnnnG and GWTCRAnnC, respectively (5)) are compared with the highly conserved Alu A- and B-box sequences (TGGCTCACGCC and GWTCGAGAC, (54)), a notable difference appears at the last position of the A box, which in Alu is C instead of G. Another difference is the distance between A and B boxes (50 and 35 bp in the case of Alu and of tRNA genes, respectively). Both of these peculiar features might contribute to the intrinsic weakness of Alu internal promoter, especially if one considers that A box acts as a fundamental core promoter element in Pol III transcription, frequently in synergy with upstream elements (5,55). Interestingly, the A box (TGGCGCGTGCC) and B box (GTTCTGGGC) recognizable within the human 7SL genes differ from tRNA gene consensus even more than Alu internal control regions do, in line with the severe requirement of upstream control elements in 7SL gene transcription (9,56).
The existence of a strong epigenetic control on Alu expression in vivo has previously been proposed and widely accepted to explain the discrepancy between the extremely high number of genomic Alus and the paucity of their overall expression level (reviewed in (26,57)). In our study, in vivo epigenetic silencing can be easily deduced from the similar in vitro transcription rates of Alu elements which profoundly differ from each other for their expression properties in cell lines. DNA methylation is generally proposed as the main factor responsible for widespread Alu downregulation (26), which may also involve H3K9 methylation (58), even though more recent investigation on Alu histone modification patterns, based on ChIP-seq, revealed that, somehow unexpectedly, Alus tend to be possess histone modifications (such as H3K4me1/2) generally associated with open chromatin and enhancers (48). Clearly, we are still missing important information on the mechanisms of general Alu silencing and local derepression and their relationship with DNA methylation and chromatin organization. An initial contribution to this issue is represented by our finding that the P300 histone acetyltransferase is enriched at expressed Alu loci, whose upstream regions also tend to be associated with JunD and C/EBP beta TFs. In principle, these Pol II TFs that were found enriched in the 500-bp region upstream of expression-positive Alus might be involved in the modulation of Alu Pol III transcription by 5′-flanking region, and they might also possibly favor hypothetical Pol II transcription at Alu loci which might contribute to Alu RNA biogenesis.
The ability to determine Alu expression profiles at single-locus resolution represents a key step toward a better understanding of Alu transcriptional control, a largely unexplored issue in spite of its high relevance for human genome stability. The possible cellular functions of SINE RNAs are just starting to be discerned (22); it is thus presently difficult to interpret Alu RNA profiles in terms of their significance in cell physiology. However, as suggested by the present work together with previous studies, Alu RNA profiles are likely determined by a tiny subset of loci particularly responsive to DNA methylation and chromatin status. Alu RNA profiling through RNAseq might thus represent a novel, extremely subtle and sensitive way to monitor epigenome alterations accompanying physiological and pathological states. Our work opens the possibility to easily profile the human transcriptome in any human cell line or tissue, under any condition compatible with RNAseq. We anticipate that our pipeline will be widely exploited to extract unprecedented information on Alu expression profiles from the plethora of available human RNA-seq data sets. Of particular interest with this respect will be Alu RNA profiling in relation to development, malignant transformation, cellular alteration in various diseases and inter-individual differences in gene expression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Arianna Molinari for assistance in plasmid construction, and Martin Teichmann (IECB, Pessac, France) for HeLa cell nuclear extract.
Footnotes
These authors contributed equally to the paper as first authors.
FUNDING
Italian Association for Cancer Research [AIRC, IG13383]. Funding for open access charge: Italian Association for Cancer Research [AIRC, IG13383].
Conflict of interest statement. None declared.
REFERENCES
- 1.Deininger P. Alu elements: know the SINEs. Genome Biol. 2011;12:236. doi: 10.1186/gb-2011-12-12-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dieci G., Conti A., Pagano A., Carnevali D. Identification of RNA polymerase III-transcribed genes in eukaryotic genomes. Biochim. Biophys. Acta. 2013;1829:296–305. doi: 10.1016/j.bbagrm.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Elder J.T., Pan J., Duncan C.H., Weissman S.M. Transcriptional analysis of interspersed repetitive polymerase III transcription units in human DNA. Nucleic Acids Res. 1981;9:1171–1189. doi: 10.1093/nar/9.5.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuhrman S.A., Deininger P.L., LaPorte P., Friedmann T., Geiduschek E.P. Analysis of transcription of the human Alu family ubiquitous repeating element by eukaryotic RNA polymerase III. Nucleic Acids Res. 1981;9:6439–6456. doi: 10.1093/nar/9.23.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orioli A., Pascali C., Pagano A., Teichmann M., Dieci G. RNA polymerase III transcription control elements: themes and variations. Gene. 2012;493:185–194. doi: 10.1016/j.gene.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Paolella G., Lucero M.A., Murphy M.H., Baralle F.E. The Alu family repeat promoter has a tRNA-like bipartite structure. EMBO J. 1983;2:691–696. doi: 10.1002/j.1460-2075.1983.tb01486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chesnokov I., Schmid C.W. Flanking sequences of an Alu source stimulate transcription in vitro by interacting with sequence-specific transcription factors. J. Mol. Evol. 1996;42:30–36. doi: 10.1007/BF00163208. [DOI] [PubMed] [Google Scholar]
- 8.Roy A.M., West N.C., Rao A., Adhikari P., Aleman C., Barnes A.P., Deininger P.L. Upstream flanking sequences and transcription of SINEs. J. Mol. Biol. 2000;302:17–25. doi: 10.1006/jmbi.2000.4027. [DOI] [PubMed] [Google Scholar]
- 9.Ullu E., Weiner A.M. Upstream sequences modulate the internal promoter of the human 7SL RNA gene. Nature. 1985;318:371–374. doi: 10.1038/318371a0. [DOI] [PubMed] [Google Scholar]
- 10.Paulson K.E., Schmid C.W. Transcriptional inactivity of Alu repeats in HeLa cells. Nucleic Acids Res. 1986;14:6145–6158. doi: 10.1093/nar/14.15.6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panning B., Smiley J.R. Activation of RNA polymerase III transcription of human Alu repetitive elements by adenovirus type 5: requirement for the E1b 58-kilodalton protein and the products of E4 open reading frames 3 and 6. Mol. Cell. Biol. 1993;13:3231–3244. doi: 10.1128/mcb.13.6.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu W.M., Chu W.M., Choudary P.V., Schmid C.W. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res. 1995;23:1758–1765. doi: 10.1093/nar/23.10.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kroutter E.N., Belancio V.P., Wagstaff B.J., Roy-Engel A.M. The RNA polymerase dictates ORF1 requirement and timing of LINE and SINE retrotransposition. PLoS Genet. 2009;5:e1000458. doi: 10.1371/journal.pgen.1000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaikh T.H., Roy A.M., Kim J., Batzer M.A., Deininger P.L. cDNAs derived from primary and small cytoplasmic Alu (scAlu) transcripts. J. Mol. Biol. 1997;271:222–234. doi: 10.1006/jmbi.1997.1161. [DOI] [PubMed] [Google Scholar]
- 15.Sinnett D., Richer C., Deragon J.M., Labuda D. Alu RNA transcripts in human embryonal carcinoma cells. Model of post-transcriptional selection of master sequences. J. Mol. Biol. 1992;226:689–706. doi: 10.1016/0022-2836(92)90626-u. [DOI] [PubMed] [Google Scholar]
- 16.Canella D., Praz V., Reina J.H., Cousin P., Hernandez N. Defining the RNA polymerase III transcriptome: genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 2010;20:710–721. doi: 10.1101/gr.101337.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oler A.J., Alla R.K., Roberts D.N., Wong A., Hollenhorst P.C., Chandler K.J., Cassiday P.A., Nelson C.A., Hagedorn C.H., Graves B.J., et al. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat. Struct. Mol. Biol. 2010;17:620–628. doi: 10.1038/nsmb.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moqtaderi Z., Wang J., Raha D., White R.J., Snyder M., Weng Z., Struhl K. Genomic binding profiles of functionally distinct RNA polymerase III transcription complexes in human cells. Nat. Struct. Mol. Biol. 2010;17:635–640. doi: 10.1038/nsmb.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oler A.J., Traina-Dorge S., Derbes R.S., Canella D., Cairns B.R., Roy-Engel A.M. Alu expression in human cell lines and their retrotranspositional potential. Mob. DNA. 2012;3:11. doi: 10.1186/1759-8753-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett E.A., Keller H., Mills R.E., Schmidt S., Moran J.V., Weichenrieder O., Devine S.E. Active Alu retrotransposons in the human genome. Genome Res. 2008;18:1875–1883. doi: 10.1101/gr.081737.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger A., Strub K. Multiple roles of Alu-related noncoding RNAs. Prog. Mol. Subcell. Biol. 2011;51:119–146. doi: 10.1007/978-3-642-16502-3_6. [DOI] [PubMed] [Google Scholar]
- 23.Martignetti J.A., Brosius J. BC200 RNA: a neural RNA polymerase III product encoded by a monomeric Alu element. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11563–11567. doi: 10.1073/pnas.90.24.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castelnuovo M., Massone S., Tasso R., Fiorino G., Gatti M., Robello M., Gatta E., Berger A., Strub K., Florio T., et al. An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. FASEB J. 2010;24:4033–4046. doi: 10.1096/fj.10-157032. [DOI] [PubMed] [Google Scholar]
- 25.Orioli A., Pascali C., Quartararo J., Diebel K.W., Praz V., Romascano D., Percudani R., van Dyk L.F., Hernandez N., Teichmann M., et al. Widespread occurrence of non-canonical transcription termination by human RNA polymerase III. Nucleic Acids Res. 2011;39:5499–5512. doi: 10.1093/nar/gkr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy-Engel A.M. LINEs, SINEs and other retroelements: do birds of a feather flock together? Front Biosci. (Landmark Ed) 2012;17:1345–1361. doi: 10.2741/3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umylny B., Presting G., Efird J.T., Klimovitsky B.I., Ward W.S. Most human Alu and murine B1 repeats are unique. J. Cell. Biochem. 2007;102:110–121. doi: 10.1002/jcb.21278. [DOI] [PubMed] [Google Scholar]
- 28.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Treangen T.J., Salzberg S.L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anders S., Pyl T.P., Huber W. HTSeq — a Python framework to work with high-throughput sequencing data. 2014 doi: 10.1093/bioinformatics/btu638. doi:10.1101/004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Preti M., Ribeyre C., Pascali C., Bosio M.C., Cortelazzi B., Rougemont J., Guarnera E., Naef F., Shore D., Dieci G. The telomere-binding protein Tbf1 demarcates snoRNA gene promoters in Saccharomyces cerevisiae. Mol. Cell. 2010;38:614–620. doi: 10.1016/j.molcel.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 33.Dignam J.D., Lebovitz R.M., Roeder R.G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dieci G., Preti M., Montanini B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics. 2009;94:83–88. doi: 10.1016/j.ygeno.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 35.Hesselberth J.R. Lives that introns lead after splicing. Wiley Interdiscip. Rev. RNA. 2013;4:677–691. doi: 10.1002/wrna.1187. [DOI] [PubMed] [Google Scholar]
- 36.Liu W.M., Maraia R.J., Rubin C.M., Schmid C.W. Alu transcripts: cytoplasmic localisation and regulation by DNA methylation. Nucleic Acids Res. 1994;22:1087–1095. doi: 10.1093/nar/22.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Comeaux M.S., Roy-Engel A.M., Hedges D.J., Deininger P.L. Diverse cis factors controlling Alu retrotransposition: what causes Alu elements to die. Genome Res. 2009;19:545–555. doi: 10.1101/gr.089789.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maraia R.J., Driscoll C.T., Bilyeu T., Hsu K., Darlington G.J. Multiple dispersed loci produce small cytoplasmic Alu RNA. Mol. Cell. Biol. 1993;13:4233–4241. doi: 10.1128/mcb.13.7.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kojima K.K. Alu monomer revisited: recent generation of Alu monomers. Mol. Biol. Evol. 2011;28:13–15. doi: 10.1093/molbev/msq218. [DOI] [PubMed] [Google Scholar]
- 40.Sassa T. The role of human-specific gene duplications during brain development and evolution. J. Neurogenet. 2013;27:86–96. doi: 10.3109/01677063.2013.789512. [DOI] [PubMed] [Google Scholar]
- 41.Parrott A.M., Mathews M.B. snaR genes: recent descendants of Alu involved in the evolution of chorionic gonadotropins. Cold Spring Harb. Symp. Quant. Biol. 2009;74:363–373. doi: 10.1101/sqb.2009.74.038. [DOI] [PubMed] [Google Scholar]
- 42.Parrott A.M., Tsai M., Batchu P., Ryan K., Ozer H.L., Tian B., Mathews M.B. The evolution and expression of the snaR family of small non-coding RNAs. Nucleic Acids Res. 2011;39:1485–1500. doi: 10.1093/nar/gkq856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Canella D., Bernasconi D., Gilardi F., LeMartelot G., Migliavacca E., Praz V., Cousin P., Delorenzi M., Hernandez N., Cycli X.C. A multiplicity of factors contributes to selective RNA polymerase III occupancy of a subset of RNA polymerase III genes in mouse liver. Genome Res. 2012;22:666–680. doi: 10.1101/gr.130286.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White R.J. Transcription by RNA polymerase III: more complex than we thought. Nat. Rev. Genet. 2011;12:459–463. doi: 10.1038/nrg3001. [DOI] [PubMed] [Google Scholar]
- 45.Donze D. Extra-transcriptional functions of RNA Polymerase III complexes: TFIIIC as a potential global chromatin bookmark. Gene. 2012;493:169–175. doi: 10.1016/j.gene.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 46.Pascali C., Teichmann M. RNA polymerase III transcription - regulated by chromatin structure and regulator of nuclear chromatin organization. Subcell. Biochem. 2013;61:261–287. doi: 10.1007/978-94-007-4525-4_12. [DOI] [PubMed] [Google Scholar]
- 47.Raha D., Wang Z., Moqtaderi Z., Wu L., Zhong G., Gerstein M., Struhl K., Snyder M. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc. Natl. Acad. Sci. U.S.A. 2010;107:3639–3644. doi: 10.1073/pnas.0911315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su M., Han D., Boyd-Kirkup J., Yu X., Han J.D. Evolution of Alu elements toward enhancers. Cell Rep. 2014;7:376–385. doi: 10.1016/j.celrep.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 49.Chang D.Y., Maraia R.J. A cellular protein binds B1 and Alu small cytoplasmic RNAs in vitro. J. Biol. Chem. 1993;268:6423–6428. [PubMed] [Google Scholar]
- 50.Eom T., Muslimov I.A., Tsokas P., Berardi V., Zhong J., Sacktor T.C., Tiedge H. Neuronal BC RNAs cooperate with eIF4B to mediate activity-dependent translational control. J. Cell. Biol. 2014;207:237–252. doi: 10.1083/jcb.201401005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar A. An overview of nested genes in eukaryotic genomes. Eukaryot. Cell. 2009;8:1321–1329. doi: 10.1128/EC.00143-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roman A.C., Gonzalez-Rico F.J., Molto E., Hernando H., Neto A., Vicente-Garcia C., Ballestar E., Gomez-Skarmeta J.L., Vavrova-Anderson J., White R.J., et al. Dioxin receptor and SLUG transcription factors regulate the insulator activity of B1 SINE retrotransposons via an RNA polymerase switch. Genome Res. 2011;21:422–432. doi: 10.1101/gr.111203.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giuliodori S., Percudani R., Braglia P., Ferrari R., Guffanti E., Ottonello S., Dieci G. A composite upstream sequence motif potentiates tRNA gene transcription in yeast. J. Mol. Biol. 2003;333:1–20. doi: 10.1016/j.jmb.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 54.Batzer M.A., Deininger P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 55.Guffanti E., Ferrari R., Preti M., Forloni M., Harismendy O., Lefebvre O., Dieci G. A minimal promoter for TFIIIC-dependent in vitro transcription of snoRNA and tRNA genes by RNA polymerase III. J. Biol. Chem. 2006;281:23945–23957. doi: 10.1074/jbc.M513814200. [DOI] [PubMed] [Google Scholar]
- 56.Englert M., Felis M., Junker V., Beier H. Novel upstream and intragenic control elements for the RNA polymerase III-dependent transcription of human 7SL RNA genes. Biochimie. 2004;86:867–874. doi: 10.1016/j.biochi.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 57.Ichiyanagi K. Epigenetic regulation of transcription and possible functions of mammalian short interspersed elements, SINEs. Genes Genet. Syst. 2013;88:19–29. doi: 10.1266/ggs.88.19. [DOI] [PubMed] [Google Scholar]
- 58.Kondo Y., Issa J.P. Enrichment for histone H3 lysine 9 methylation at Alu repeats in human cells. J. Biol. Chem. 2003;278:27658–27662. doi: 10.1074/jbc.M304072200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.