


Abstract
Androgen receptor (AR) is a ligand-activated transcription factor that plays a central role in the development and growth of prostate carcinoma. PIAS1 is an AR- and SUMO-interacting protein and a putative transcriptional coregulator overexpressed in prostate cancer. To study the importance of PIAS1 for the androgen-regulated transcriptome of VCaP prostate cancer cells, we silenced its expression by RNAi. Transcriptome analyses revealed that a subset of the AR-regulated genes is significantly influenced, either activated or repressed, by PIAS1 depletion. Interestingly, PIAS1 depletion also exposed a new set of genes to androgen regulation, suggesting that PIAS1 can mask distinct genomic loci from AR access. In keeping with gene expression data, silencing of PIAS1 attenuated VCaP cell proliferation. ChIP-seq analyses showed that PIAS1 interacts with AR at chromatin sites harboring also SUMO2/3 and surrounded by H3K4me2; androgen exposure increased the number of PIAS1-occupying sites, resulting in nearly complete overlap with AR chromatin binding events. PIAS1 interacted also with the pioneer factor FOXA1. Of note, PIAS1 depletion affected AR chromatin occupancy at binding sites enriched for HOXD13 and GATA motifs. Taken together, PIAS1 is a genuine chromatin-bound AR coregulator that functions in a target gene selective fashion to regulate prostate cancer cell growth.
INTRODUCTION
Androgen receptor (AR) acts as a hormone-controlled transcription factor that conveys the messages of both natural and synthetic androgens to the level of genome (1,2). In prostate, AR is needed as a differentiation factor for the normal development, function and maintenance of the gland. AR also plays a critical role in the pathogenesis of prostate cancer, the most commonly diagnosed cancer among Western men (3). Proliferation of cancerous prostate cells can initially be inhibited by androgen ablation. However, for reasons that are currently unclear, this therapy eventually fails leading to castration resistant prostate cancer (CRPC). The CRPC has been proposed to result from e.g. mutations in AR, AR variants, augmented expression of AR or its coregulator proteins, aberrant AR posttranslational modifications, gene fusions resulting in abnormal androgen regulation of oncogenic transcription factors and intracrine androgen production (4–6).
Binding of androgen to the AR monomer in the cytoplasm causes a conformational change in the receptor's ligand-binding domain (LBD), leading to dimerization and nuclear translocation of the AR. The dimeric AR binds to specific DNA elements, androgen response elements (AREs), resembling the sequence AGAACAnnnTGTTCT (n is any nucleotide) (2). According to recent genome-wide chromatin immunoprecipitation (ChIP-seq) analyses, the majority of genomic AR-binding sites contain at least one TGTTCT half-site flanked with a less well-conserved 3′ hexamer (7–10). In addition to the ARE itself, adjacent binding sites for other transcription factors seem to play an important role in the DNA binding, often governing the AR binding. These ‘AR-collaborating’ factors that often bind to sites in close proximity to AREs include GATA-binding protein 2 (GATA2), E twenty-six or E26 transformation-specific (ETS) transcription factors and pioneer factor forkhead box A1 (FOXA1) (7,11–14). In addition to facilitating AR binding to chromatin, FOXA1 can also mask chromatin binding sites of the receptor (7). However, the mechanisms underlying differential FOXA1 and subsequently AR binding are currently not known. In addition to the general transcription machinery and RNA polymerase II, AR requires a number of interacting coregulator proteins to regulate transcription. A large number, over 200 AR-interacting proteins have been reported to potentially influence the receptor's transcriptional activity (1,15). These include SWI/SNF chromatin-remodeling complexes, together with steroid receptor coactivators and corepressors that harbor or recruit enzymatic activities that catalyze histone acetylation or methylation and/or removal of these post-translational modifications (16). Some putative coregulators also promote ubiquitylation or SUMO (small ubiquitin-related modifier) modifications (SUMOylation) which all can also target nucleosomal histones and AR (1,17). Cell- and tissue-specific differences in the expression patterns of AR coregulators and collaborating transcription factors are thought to contribute significantly to the differences in AR target gene activities between different tissues (1). However, the physiological importance and functional relevance of the majority of potential AR coregulators have remained elusive, as they have in most cases been tested merely under ectopic overexpression conditions in reporter gene assays.
Protein inhibitor of activated STAT (PIAS) 1 is a candidate AR coregulator, as it interacts with AR and influences its activity in transactivation assays (18–20). Although PIAS1, like the other PIAS family proteins PIAS2 (x), PIAS3, PIAS4 (y), has the capacity to catalyze SUMOylation of interacting proteins, including AR, the SUMO ligase activity is not its sole function as is the case also with the other PIAS proteins (21,22). Of note, expression of PIAS1 protein is significantly elevated in malignant areas of prostate cancer compared to normal tissue, suggesting a pathophysiological role in prostate cancer cell growth (23,24). However, there is no information on the role of PIAS1 in the regulation of endogenous AR target genes in prostate cancer cells. In this study, we have used VCaP (Vertebral-Cancer of the Prostate) cells that are derived from a CRPC (25) to analyze the role of endogenous PIAS1 in the regulation of AR target genes in a genuine chromatin environment. The cell line contains amplified levels of AR, it is androgen sensitive and tumorigenic and it expresses enzymes involved in intratumoral androgen biosynthesis whose expression is induced in VCaP xenografts grown in castrated mice (26). It is thus an attractive in vitro model for CRPC. Our genome-wide approaches revealed that PIAS1 acts as a chromatin-bound AR coregulator that also interacts with FOXA1 and functions in a selective fashion in AR target gene expression. Expression of several genes was significantly affected, either activated or repressed by PIAS1, and these genes were associated with cellular growth and proliferation, gene expression and protein synthesis functions.
MATERIALS AND METHODS
Cell culture and hormones
VCaP cells was obtained from American Type Culture Collection (ATCC) and maintained as previously described (27). R1881 (methyltrienolone) and testosterone were from Steraloids Inc.
RNA interference
VCaP cells were seeded onto 12-well plates for RNA isolation (270 000 cells/well) and for proliferation assays onto six-well plates (500 000 cells/well) for 2 days before transfection. The transfection medium DMEM containing 2.5% charcoal-stripped FBS was changed to cells 3 h before transfection. For silencing of PIAS1, Dharmacon ON-TARGETplus SMARTpool for human PIAS1 (siPIAS1) (SMART pool of four individual siRNAs that were designed to minimize off-target effects) and non-targeting pool (siNON) were used at 100 nM final concentration using Trans-IT-siQUEST reagent (Mirus Bio Corp.) according to the manufacturer′s instructions. After 96 h, cells were exposed to vehicle (ethanol, EtOH) or androgen. For immunoblotting, RT-qPCR and Expression BeadChip analyses, cells were exposed to testosterone (100 nM) for 16 h and for proliferation assays to R1881 (10 nM) for 3 and 6 days. For ChIP-seq, cells were transfected by reverse transfection with siNON or siPIAS1 at 40 nM concentration using the Lipofectamine® RNAiMAX reagent (Invitrogen) according to the manufacturer′s instructions. VCaP cells were seeded in growth medium onto six-well plates (500 000–600 000 cells/well) or 10-cm plates (5 000 000 cells/plate) for AR and FOXA1 ChIP-seq, respectively. After 72 h, the medium was changed to transfection medium for 48 h, and prior to ChIP, the cells were exposed to R1881 (10 nM) for 2 h.
Immunoblotting and immunoprecipitation
VCaP cell samples were prepared as described (27) and analyzed by immunoblotting with anti-PIAS1 (Santa-Cruz Biotechnology, sc-8152), anti-AR (28), anti-p21 (Millipore, OP64) and anti-tubulin (Santa-Cruz Biotechnology, sc-5286) antibodies. The appropriate secondary antibody was from Invitrogen and chemiluminescence detection reagents from Pierce. PIAS1 was immunoprecipitated with anti-PIAS1 antibody (Abcam, ab109388) which was coupled to Magna ChIP™ Protein A Magnetic Beads (Millipore). Normal rabbit IgG (Santa-Cruz Biotechnology, sc-2027) was used as a control antibody and the immunoprecipitates were immunoblotted with anti-AR (Santa-Cruz Biotechnology, sc-7305) antibody. For FOXA1 and PIAS1 immunoprecipitation, anti-FOXA1 (Abcam, ab23738) antibody was used to immunoprecipitate FOXA1 and the immunoprecipitates were immunoblotted with anti-PIAS1 (Abcam, ab77231) antibody.
Reporter gene assays
VCaP cells were seeded onto 12-well plates (300 000 cells/well) for 2 days before transfection. The transfection medium DMEM containing 2.5% charcoal-stripped FBS was changed on cells 6 h before the cells were transfected with 600 ng of pARE2-TATA-LUC (29), and 100 ng pCMVβ (Clontech), 100–300 ng pFlag-PIAS1 (20) or pFlag-PIAS13CS. In the case of the latter plasmid, the three cysteine residues C346, C351 and C356 in the mPIAS1 RING domain were mutated to serines by QuikChange II Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. Point mutations were verified by sequencing. The total amount of DNA per well was balanced at 1000 ng with empty pFlag-CMV-2 (Sigma-Aldrich) and X-tremeGENE HP DNA Transfection Reagent (Roche) was used according to the manufacturer′s instructions. Twenty-four hours after transfection, cells were exposed to R1881 or vehicle for 17 h. Reporter gene activity was measured as described (29).
RNA isolation, real-time quantitative PCR (RT-qPCR) and microarray analysis
Total RNA in biological triplicates was extracted (TriPure isolation reagent, Roche) and converted to cDNA (Transcriptor First Strand cDNA synthesis Kit, Roche) according to the manufacturer's instructions. Expression of target genes with specific primers (Supplementary Table S1) was measured by RT-qPCR as described in (30) using GAPDH mRNA levels to normalize the amounts of total RNA between the samples. Illumina HumanHT-12 v4 Expression BeadChip analyses were carried out at the Finnish Microarray and Sequencing Centre (Turku, Finland) using protocols recommended by the manufacturer. The data were analyzed and visualized as described (31). The genes with adjusted P-value of <0.01 and fold change (FC) ≥1.5 and ≤0.7 were selected as significantly changed transcripts. To identify biological processes enriched for differentially expressed genes, the data were analyzed through the use of Ingenuity Pathway Analysis® (IPA, www.ingenuity.com). A core analysis was first performed with two distinct lists (androgen-regulated genes in siNON- and siPIAS1-treated cells) and the results were then compared to identify any distinct biological processes that were differentially regulated.
Cell proliferation assay
VCaP cells were grown and transfected with siNON and siPIAS1 as described above. After 72 h, the cells were seeded onto 96-well plates (5000 cells/well) in transfection DMEM containing 2.5% charcoal-stripped FBS. After 6 h, the cells were exposed to vehicle or R1881 for 3 and 6 days. The amount of viable cells was measured using CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to the manufacturer′s instructions. The results were calculated as percentage of cell growth using the first measurement (day 0) as reference point (100%).
Chromatin immunoprecipitation (ChIP), re-ChIP and ChIP-coupled to deep sequencing (ChIP-seq)
The protocol for ChIP experiments and analyses were performed as described (30). Briefly, the cells were seeded onto 10-cm plates (3 000 000 cells/dish) and grown for 72 h in growth medium. Steroid-depleted transfection medium was changed 48 h before the cells were exposed to vehicle or R1881 for 2 h. Cells were crosslinked with 1% (v/v) formaldehyde and harvested for immunoprecipitation with anti-AR (28), anti-PIAS1 (ab 77231), anti-FOXA1 (ab 23738), anti-H3K4me2 (ab 7766), anti-SUMO2/3 (MBL International 114-3), anti-RNA polymerase II (Covance, MMS-126R) or anti-IgG (sc-2027) antibody. Re-ChIP analyses were performed essentially as described (32). Chromatin was sequentially precipitated with anti-PIAS1 (ab 77231) and anti-FOXA1 (ab 23738) antibodies. qPCR analyses from chromatin templates were performed as described (30). Specific primers for different genomic regions are listed in Supplementary Table S2. Single-end AR (siNON), PIAS1, FOXA1 and SUMO2/3 (androgen) ChIP-seq data were generated using Solexa/Illumina 1.5 System in the Biomedicum Functional Genomics Unit (Helsinki, Finland) and AR (siPIAS1), FOXA1 (siNON and siPIAS1), H3K4me2 and SUMO2/3 (vehicle) with HiSeq 2000 in the EMBL Genomics Core Facility (Heidelberg, Germany). Single-end ChIP-seq data for RNA polymerase II (Pol II) were generated using Illumina 1.5 System in the BGI Tech Solutions (Shenzhen, China). Raw read quality check, preprocessing of reads (36-bp trimming) and Bowtie alignment were performed as described (31). Background (IgG) read set was scaled that an individual background was generated for each AR, PIAS1 and FOXA1 ChIP-seq sample. The read ratio between IgG and ChIP-seq sample was set to the same by randomly removing IgG reads the way that the ratio became the same for each ChIP-seq sample. The scaled IgG and unscaled AR, PIAS1 and FOXA1 reads were converted to sorted sam files using SAM tool (33). The same files were analyzed using HOMER software version 4.1 (34). Enriched binding sites were detected using findPeaks command with factor analysis strategy using 4-fold enrichment over IgG background read set. We sequenced single H3K4me2 sample and two independent biological replicates per sample for AR, PIAS1, FOXA1, SUMO2/3 and Pol II. Our biological replicates showed a good concordance (summary table of the peaks used in Supplementary Table S3). The samples met ENCODE guidelines; ≥10 million uniquely mapped reads per sample were gained, control IgG sample sequence depth was at the level of other ChIP-seq samples and replicate similarity matched the guidelines (35,36). Overlapped binding sites between each replicate pair were identified using BEDTools (37). Binding sites found in both replicates and normalized tag count ≥10 were used as final binding sites in analysis. To analyze unique binding sites for two chromatin binders, the non-overlapping binding sites were identified using getDifferentialPeaks command of HOMER using 3-fold difference in the occupancy of the chromatin binders. HOMER software was used as (31) to visualize data, for peak position annotation and for de novo motif analyses. Association of binding sites to the nearby genes (±50 kb from transcription start site, TSS) and androgen-regulated genes was done with Anduril as described (38). Genescores were calculated to nearby genes by taking into account the peak signal strength (normalized tags), distance between TSS and the peak and number of associated peaks to genes (with linear scoring function).
Accession numbers
Bead array and ChIP-seq data are submitted to the NCBI Gene Expression Omnibus database (39) (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession numbers GSE30316 and GSE56086, respectively.
RESULTS
PIAS1 and AR interact in prostate cancer cells
Based on mRNA expression, PIAS1 is the major PIAS1 family member in VCaP cells and the only PIAS protein whose mRNA expression is up-regulated by androgens (32) (Supplementary Figure S1A). Co-immunoprecipitation assays (immunoblotting of anti-PIAS1 immunoprecipitates with anti-AR antibody) confirmed the interaction between endogenous PIAS1 and AR in VCaP cells (Supplementary Figure S1B). Transfection of increasing doses of plasmids encoding PIAS1 or PIAS1 SP-RING mutant together with a minimal AR-regulated-promoter driven reporter gene (pARE2-TATA-LUC) further demonstrated that PIAS1 can influence the transcriptional activity of VCaP cell AR in a fashion that is dependent on the intact PIAS SP-RING structure (Supplementary Figure S1C).
PIAS1 activates or represses androgen-regulated genes in a target gene-selective fashion
We next investigated the role of PIAS1 for the androgen-regulated transcriptome in VCaP cells. PIAS1 was depleted from the cells using transfected PIAS1-specific siRNAs (siPIAS1) and cells transfected with non-targeting siRNAs (siNON) were used as a control. PIAS1 depletion was confirmed by immunoblotting with anti-PIAS1 antibody (Figure 1A). Gene expression profiling of cells treated with androgen or vehicle revealed that more genes were regulated by androgen in PIAS1-depleted than in control siNON cells (1 767 versus 1 341) (Figure 1B) (listed in Supplementary file S1). Expression of ∼10% of the AR-regulated genes in siNON cells (hereafter control cells) was significantly influenced, either activated (FC ≥1.5) or repressed (FC ≤0.7), by PIAS1 depletion. The majority of AR target genes in control cells remained androgen-responsive also in PIAS1-depleted cells, but ∼4% of the target genes lost their response to androgen upon PIAS1 depletion (e.g. KLK2). Notably, PIAS1 depletion exposed a large set of new genes (820) to androgen regulation, suggesting that PIAS1 can mask genes, such as NR5A2, from AR access (Figure 1B) (Supplementary file S1). The number of genes affected by PIAS1 depletion in the absence of androgen (138, Supplementary file S1) was much smaller than that in the presence of androgen, implying that PIAS1 is relatively selective to androgen-regulated genes in VCaP cells.
Figure 1.
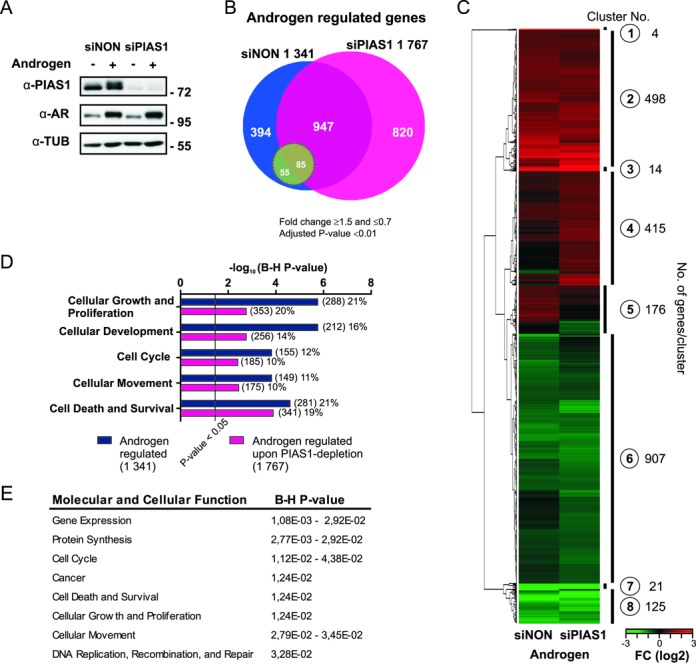
PIAS1 depletion affects androgen-regulated transcriptome of VCaP cells. VCaP cells were transfected with control siRNA (siNON) or PIAS1 siRNA (siPIAS1) and the cells were exposed to androgen (testosterone) or vehicle (ethanol) for 16 h, RNAs were isolated and microarray analysis was performed. (A) PIAS1 and AR protein levels in PIAS1-depleted VCaP cells. Cell lysates were immunoblotted with indicated antibodies. (B) Venn diagram showing androgen-regulated genes in siNON and siPIAS1 cells (1341 and 1767 genes, respectively). Green circle indicates AR-regulated genes significantly affected by PIAS1 depletion (when comparing fold changes of androgen-regulated genes in siNON- and siPIAS1-treated cells). (C) Heat map of the normalized expression values of differentially expressed transcripts in the BeadChip arrays based on unsupervised hierarchical clustering. (D) Biological processes of androgen-regulated transcripts in siNON and siPIAS1 cells as revealed by Ingenuity pathway analysis. Numbers in parentheses indicate the amount of transcripts linked to each biological process and percentages their relative portions among the androgen-regulated genes. Benjamini–Hochberg (B–H) multiple testing correlation was used for enrichment analysis. (E) Biological processes of the genes masked from androgen regulation by PIAS1 (820 genes) as revealed by IPA. B-H multiple testing correlation was used for enrichment analysis.
We next grouped all androgen-regulated genes with unsupervised hierarchical clustering (Figure 1C). Genes that were robustly up- or down-regulated by androgen, but did not markedly differ in their expression between the control and PIAS1-depleted cells were present in clusters 1 (e.g. FKBP5) and 7 (e.g. AR). Clusters 2 and 3 contained genes that were androgen-up-regulated in both control and PIAS1-depleted cells, but that had a difference in either fold-induction by androgen or both fold-induction and overall expression level with androgen (e.g. RASD1 and HPGD, respectively). Similarly, genes that were androgen-down-regulated both in control and PIAS1-depleted cells, but had a difference in their expression between the control and the PIAS1-depleted cells were clustered to clusters 6 (e.g. CSRP2) and 8 (e.g. TMEFF2). Clusters 4 and 5 were particularly interesting. Cluster 4 was enriched for genes that were up-regulated by androgen only in PIAS1-depleted cells (e.g. NR5A2) and genes whose androgen down-regulation was converted to up-regulation upon PIAS1 depletion (e.g. ATAD2). Cluster 5, in turn, was enriched for genes that were androgen up-regulated only in control cells (e.g. KLK2). In sum, these transcriptome profiles indicate that PIAS1 depletion affects, either activates or represses, in a gene selective manner expression of number of the androgen-regulated transcripts in VCaP cells. Moreover, a subset of genes becomes subject to androgen regulation upon PIAS1 depletion.
PIAS1 affects prostate cancer cell proliferation
All androgen-regulated genes in PIAS1-depleted and control cells were next subjected to Ingenuity Pathway Analysis® (IPA) to identify enriched molecular and/or cellular functions and pathways affected by PIAS1. Cellular growth and proliferation, cellular development, cellular movement, and cell cycle and cell survival were the top five molecular and cellular functions significantly enriched in both PIAS1-depleted and control cells (Figure 1D). Canonical pathways of cholesterol biosynthesis I–III and the superpathway of cholesterol biosynthesis showed the most significant differences in their enrichment between control and PIAS1-depleted cells, when all androgen-regulated genes were analyzed (Supplementary Figure S2A). In addition to these biosynthesis pathways, PIAS1 is predicted to be involved in the regulation of cell cycle-related events. Top canonical pathways enriched in the interesting cluster 4 (Figure 1C; androgen up-regulated only in PIAS1-depleted cells or androgen down-regulation converted to up-regulation upon PIAS1 depletion) included several signaling pathways, such as neuregulin, xenobiotic metabolism, PTEN, G beta gamma and PI3K/AKT signaling (Supplementary Figure S2B). In cluster 5 (genes androgen up-regulated only in control cells), the enriched pathways were the superpathway of cholesterol biosynthesis and purine nucleotides de novo biosynthesis II. The top five molecular and cellular functions enriched among the group of genes masked from androgen regulation by PIAS1 were gene expression, protein synthesis, cell cycle, cancer and cell death and survival (Figure 1E). An interaction map of the genes annotated to the top two molecular and cellular functions, gene expression and the protein synthesis, is shown in Supplementary Figure S3.
Since IPA analyses suggested involvement of PIAS1 in the regulation of expression of genes governing cellular proliferation and survival, we next studied the effect of PIAS1 depletion on cell growth. Cell growth assays indicated that PIAS1 depletion results in attenuated growth of VCaP cells both in the presence and absence of added androgen (Figure 2A). These results are in line with gene expression data showing that BCL6 (cluster 4), ATAD2 (cluster 4), NR5A2 (cluster 4), MAGEH1 (cluster 4), CLDN8 (cluster 5), MAFB (cluster 6), TNFSF10 (cluster 6), CSRP2 (cluster 6), IGFBP-3 (cluster 7) and TMEFF2 (cluster 8) that are androgen-regulated genes associated with cell growth and apoptosis or cancer (40–48) (Supplementary Figure S4) were significantly affected by PIAS1 depletion (Figure 2B).
Figure 2.
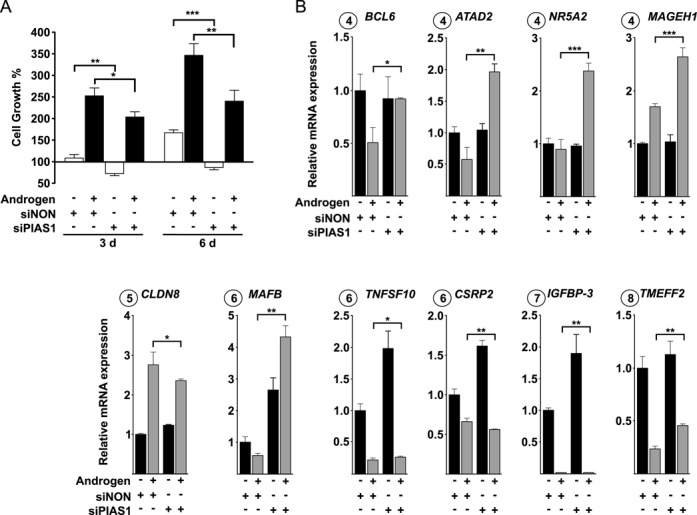
PIAS1 influences proliferation of VCaP cells. (A) VCaP cells were transfected with siNON (control) or siPIAS1 and cells were exposed to androgen or vehicle and cell numbers were measured by CellTiter96 Aqueous cell proliferation assay as described in ‘Materials and Methods’ section. Growth percentages represent the means of three independent experiments ± SDs. (B) Effect of PIAS1 depletion on the expression of select AR target genes linked to cell growth and apoptosis. Cells were treated as in Figure 1, but RNA quantification was carried out by RT-qPCR with specific primers for BCL6, ATAD2, NR5A2, MAGEH1, CLDN8, MAFB, TNFSF10, CSRP2, IGFBP-3 and TMEFF2 mRNAs. Measurements were normalized to GAPDH mRNA levels, and fold changes were calculated in reference to siNON and vehicle samples. Data points indicate the means of at least three biological replicates ± SDs. Student's t test was used to determine the significance of fold change differences between siNON and siPIAS1 cells when comparing androgen- and vehicle-exposure (***P < 0.001, **P < 0.01 and *P < 0.05).
Androgen-enhanced co-occupancy of PIAS1 and AR on chromatin
To complement the above analyses, we next investigated whether PIAS1 is able to bind chromatin and interact with AR also on VCaP cell chromatin. Genome-wide ChIP-seq analysis was performed also for AR in androgen-treated cells. Comparison of recently reported VCaP cell AR cistromes (7,8) with our AR data identifying 41 469 AR-binding sites (ARBs), showed a good (>70%) overlap with the ARBs in all data sets (Supplementary Figure S5). Interestingly, PIAS1 ChIP-seq revealed 3904 PIAS1-binding sites that overlapped almost completely (>97%) with the ARBs (Figure 3A and B). Of note, the ARBs with the strongest signals were the ones enriched at PIAS1-occupying sites, as illustrated with the correlation of AR versus PIAS1 tag signals of within the ARBs (Figure 3C). ChIP-seq of H3K4me2, a mark of active enhancers and chromatin, showed similar enrichment adjacent to both AR/PIAS1-shared and AR sites (Figure 3B). In addition, RNA polymerase II (Pol II) at promoter regions and two other active enhancer/promoter marks, H3K4me3 and acetylated H3 (panH3ac) (data from GEO), were also enriched at AR/PIAS1-shared genomic regions, further supporting the notion that these genomic regions are transcriptionally potent regions (Supplementary Figure S6). The overall distribution of AR and AR/PIAS1-shared sites to gene structures was very similar, mostly in intergenic and intronic in both cases (Supplementary Figure S7A).
Figure 3.
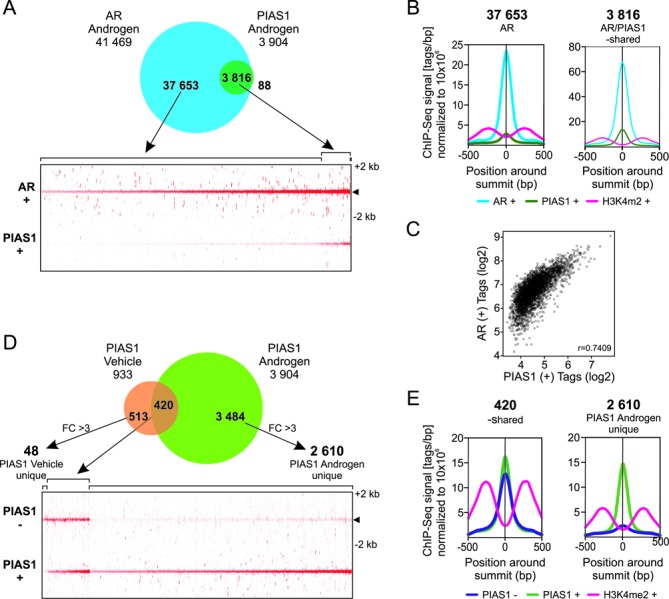
ChIP-seq analysis of AR- and PIAS1-binding sites on VCaP cell chromatin. (A) Venn diagram showing overlap of AR and PIAS1 cistromes in cells exposed to androgen (10 nM R1881) for 2 h (upper panel). Heat map showing AR- and PIAS1-binding site tag densities in ±2 kb window (lower panel). (B) Comparison of AR, PIAS1 and H3K4me2 average tag counts in ±500 bp from the centers of AR- and PIAS1-binding sites in unique ARBs and AR/PIAS1-shared sites. (C) Scatter plot of AR and PIAS1 tag signals around ±500 bp of the AR/PIAS1-shared binding sites. (D) Venn diagram showing overlap of PIAS1 cistromes in presence and absence of androgen (upper panel). Non-overlapping sites were further analyzed using get-DifferentialPeak tool to achieve final categories. Heat map showing PIAS1-binding site tag densities for vehicle/androgen-shared and unique androgen regions in a window ±2 kb (lower panel). (E) Comparison of PIAS1 and H3K4me2 average tag counts in ±500 bp from the centers of PIAS1-binding sites in vehicle/androgen-shared and androgen unique sites.
The majority (>70%) of the androgen-regulated genes in both control and PIAS1-depeleted cells had at least one ARB and ≥25% of the genes had at least one AR/PIAS1-shared-binding site within ±50 kb of their TSS (Supplementary Figure S7B). To further analyze association of genes to the nearby peaks, a gene score that takes into account the peak signal strength (normalized tags), distance between the peak and TSS and number of associated peaks was calculated for each gene. Both the ARBs and AR/PIAS1-shared sites showed stronger association with androgen-regulated genes than with non-regulated genes, with the AR/PIAS1-shared sites showing higher gene scores than the AR sites (Supplementary Figure S7C). PIAS1 depletion did not, however, alter the gene scores.
Comparison of the PIAS1 cistrome in VCaP cells in the presence of androgen to that in the absence of the hormone revealed that androgen exposure increased the number of PIAS1-occupying chromatin sites >4-fold (Figure 3D), with about half (45%) of the sites in vehicle-exposed cells overlapping with those in androgen-treated cells. Based on tag densities of PIAS1 occupancy and average tag profiles, the majority of PIAS1-binding sites (67%) were considered unique to androgen exposure, i.e. they showed at least 3-fold difference (FC > 3) between the occupancy in the presence and absence of androgen (Figure 3D). Genomic location analyses showed that PIAS1 binds by and large to intronic and intergenic regions irrespectively of androgen treatment (Supplementary Figure S8A). H3K4me2 marks showed somewhat stronger enrichment in the surroundings of vehicle/androgen-shared than androgen unique PIAS1 sites (Figure 3E).
PIAS1 interacts with FOXA1 and SUMO2/3 on chromatin
We next performed de novo motif analyses of all PIAS1-binding sites to identify transcription factor-binding motifs enriched within them. Interestingly, FOXA1 motif was the most enriched motif in both vehicle/androgen-shared and androgen unique sites (Figure 4A). Approximately 70% of the PIAS1 sites shared by vehicle- and androgen-exposed cells showed a FOXA1 motif and <30% of them had an ARE, whereas both a FOXA1 and an ARE motif were found at about the same frequency (∼50% and ∼55%, respectively) in the PIAS1 sites unique to androgen exposure (Figure 4B). At least one ARE and FOXA1 motif occurred together ∼1.5-times more frequently in the androgen unique sites (41%) than in the vehicle/androgen-shared sites (28%). Comparison of the average tag counts in ±300 bp from the centers of PIAS1 peaks further showed a higher occurrence of FOXA1 motifs at the summits of the vehicle/androgen-shared sites than at those of the androgen exposure unique sites (Supplementary Figure S8B). Also NKX3.1 and GATA motifs were enriched in the PIAS1-occupying chromatin sites, with the GATA motifs being clearly more prevalent in the vehicle/androgen-shared sites than androgen unique sites (Figure 4B).
Figure 4.
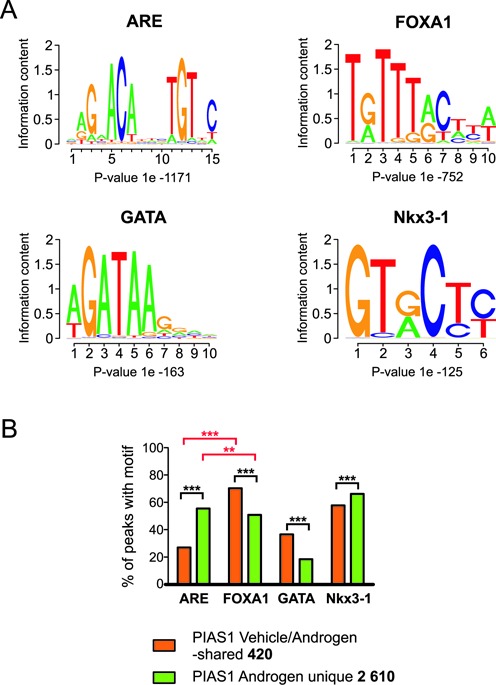
Androgen enhances PIAS1-binding to ARE motif-containing chromatin sites. (A) The top four motifs predicted by de novo motif analysis of all PIAS1-binding sites. De novo motif analysis was performed on ±100 bp of the PIAS1 (vehicle and R1881 sites) peak center. (B) Distribution of the most enriched de novo motifs in vehicle/androgen-shared and androgen unique sites. Statistical significances were calculated by χ2-test (***P < 0.001 and **P < 0.01).
The above ChIP-seq data suggest that PIAS1 interacts with FOXA1. Co-immunoprecipitation assays (immunoblotting of anti-FOXA1-immunoprecipitates with anti-PIAS1 antibody) confirmed the interaction of the two proteins in VCaP cells (Figure 5A). These data prompted us to map the FOXA1 cistrome in VCaP cells and relate it to the PIAS1 cistrome. We identified 58 364 and 69 780 FOXA1-binding sites in vehicle- and androgen-treated cells, respectively (Figure 5B). Compared to PIAS1, the effect of androgen on the formation of new FOXA1 chromatin binding sites was minor (plus 20%), albeit the hormone generally enhanced FOXA1′s chromatin binding events on the sites co-occupied by the AR (Figure 5B). Interestingly, the overlap of PIAS1 with FOXA1 in the presence and absence of androgen was nearly complete, being in both cases ≥93% (Figure 5C). Practically all PIAS1-binding sites overlapped with both FOXA1 and AR in androgen-exposed cells (Figure 5D). To investigate whether PIAS1 and FOXA1 are directly interacting on chromatin, we utilized re-ChIP assays. Chromatin was sequentially immunoprecipitated with anti-PIAS1 and anti-FOXA1 antibody and analyzed using qPCR with primers for selected genomic regions exhibiting (based on ChIP-seq data) co-occupancy of PIAS1 and FOXA1 (ATAD2 and FKBP5) or only FOXA1 occupancy (S100P) (Supplementary Figure S9). These experiments confirmed simultaneous presence of PIAS1 and FOXA1 at ATAD2 and FKBP5 regions, but not at the S100P region, thus providing further proof for a direct interaction between PIAS1 and FOXA1 proteins on chromatin.
Figure 5.
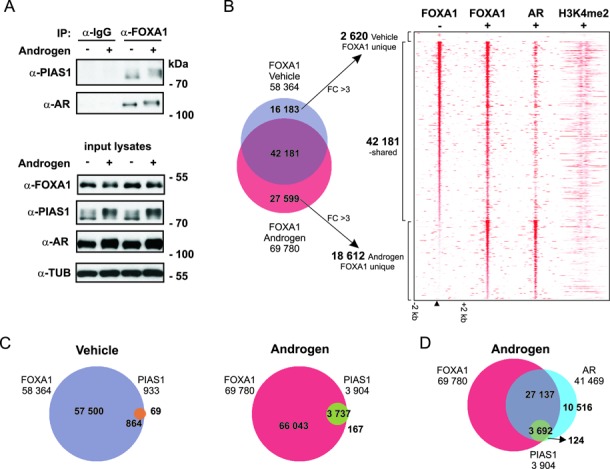
Interaction of PIAS1 with FOXA1. (A) PIAS1 and AR co-immunoprecipitate with FOXA1. VCaP cells exposed to vehicle (−) or androgen (+, R1881) for 2 h were immunoprecipitated with anti-FOXA1 antibody or normal rabbit IgG (used as a control antibody). The immunoprecipitates and the input cell lysates were analyzed by immunoblotting with indicated antibodies. (B–D) ChIP-seq analyses of FOXA1-, PIAS1- and AR-binding sites in VCaP cells. (B) Venn diagram showing overlap of FOXA1 cistromes after vehicle and androgen exposure (2 h) and heat map showing FOXA1, AR and H3K4me2-binding site tag densities in a window ±2 kb (−, vehicle; +, androgen exposure). (C) Venn diagrams showing overlap of FOXA1- and PIAS1-binding sites in vehicle- and androgen-exposed cells. (D) Venn diagram showing overlap of AR-binding sites with FOXA1- and PIAS1-binding sites in androgen-exposed cells.
We also performed ChIP-seq analyses with anti-SUMO2/3 antibody in the presence and absence of androgen. In keeping with the role of PIAS1 as a SUMOylation pathway component, PIAS1, AR, and FOXA1 co-occurring genomic sites were also generally bound by SUMO2/3 and the binding of SUMO2/3 was markedly enhanced by androgen exposure (Figure 6). Figure 7 shows snapshots of MAFB, ATAD2, CLDN8, RASD1, BCL6, and IGFBP-3 loci as examples of chromatin co-occupancy of AR and PIAS1 together with FOXA1 and SUMO2/3. Their androgen-regulated expression is significantly affected by PIAS1 depletion (cf. Figure 2). Except for ATAD2 and BCL6, PIAS1 or FOXA1 binding was not detected in the co-occupying chromatin regions prior to androgen exposure.
Figure 6.
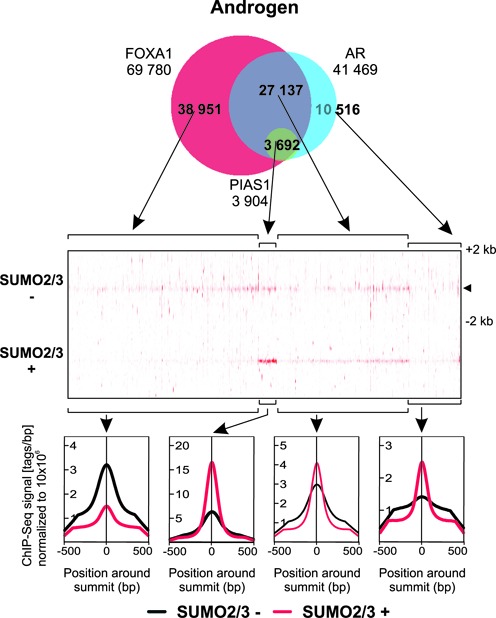
Androgen-enhanced co-occurrence of SUMO2/3 at AR-, PIAS1- and FOXA1-shared genomic locations. Venn diagram showing the overlap of FOXA1, AR, and PIAS1 cistromes in cells exposed to androgen (upper panel). Heat map showing SUMO2/3 tag densities for indicated Venn sectors in a window ±2 kb (−, vehicle; +, androgen exposure) (middle panel). Comparison of SUMO2/3 average tag counts in ±500 bp from the centers of the indicated sites (lower panel).
Figure 7.
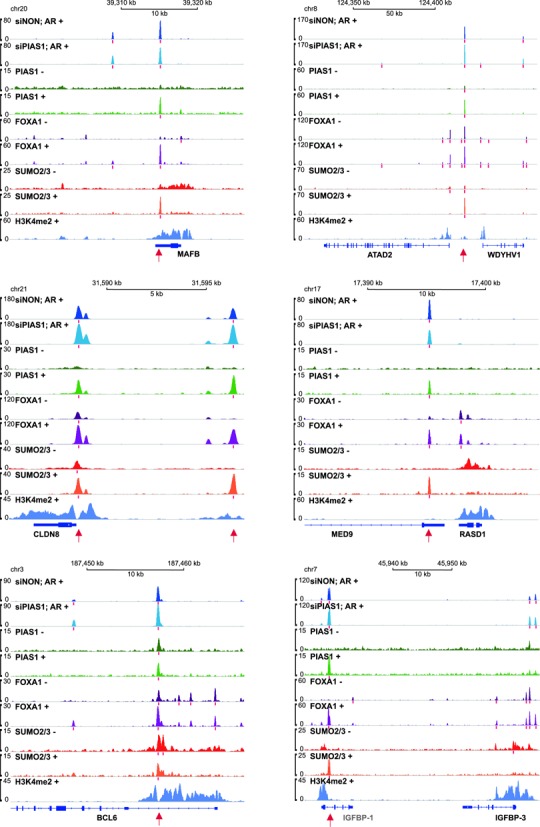
ChIP-seq track examples of AR-, PIAS1-, FOXA1- and SUMO2/3-binding events in growth-associated AR target loci in VCaP cells. The occupancy of AR (dark blue in siNON-treated cells and light blue in siPIAS1-treated cells) and H3K4me2 (violet) in the presence of androgen (+, R1881 2 h) and that of PIAS1 (green), FOXA1 (purple) and SUMO2/3 (red) in the absence (−) and presence (+) of androgen. Red bars below the tracks depict the positions of the identified peak sites and red arrows sites co-occupied by AR, PIAS1, FOXA1 and SUMO2/3. Except for IGFBP-1, the expression of all loci was affected by androgen exposure.
Influence of PIAS1 on AR and FOXA1 chromatin occupancy
FOXA1 depletion has been shown to elicit extensive redistribution of ARBs on prostate cancer cell chromatin (7). Also PIAS1 depletion influenced the number of ARBs, albeit most of the AR cistrome remained unaffected (Figure 8A). Based on ARB tag densities and average tag profiles in siNON- and siPIAS1-treated cells (Figure 8A and B), PIAS1 depletion resulted in emergence of nearly three thousand (2 772) new ARBs, and a concomitant loss of less than thousand (1 067) sites. Even though the average signals of these binding events, especially the PIAS1-dependent ones, were generally weaker than those of the PIAS1 level-unaffected ones, they were reproducible and seen in both replicates. Snapshots of ten loci displaying these binding events are shown as examples in Supplementary Figure S10. Interestingly, de novo motif analyses of the unaffected and affected sites revealed that the new ARBs contained more often HOXD13 and GATA motifs, but less frequently AREs, than the PIAS1 depletion insensitive sites or the sites from which AR binding was lost (Figure 8C and D). The new ARBs on chromatin did not however generally overlap with the PIAS1 or FOXA1 cistromes (Supplementary Figure S11). The effect of PIAS1 depletion on the FOXA1-occupying chromatin sites was smaller than that on the AR cistrome: less than one thousand (906) new FOXA1 sites appeared and a negligible number of sites (28) were lost (Supplementary Figure S12A). According to de novo motif analyses, the new FOXA1-binding sites harbored less frequently FOXA1 and HOXD13 motifs, but more often AREs than the PIAS1 depletion insensitive sites (Supplementary Figure S12). These results suggest that in addition to interacting with AR and FOXA1 on chromatin, PIAS1 can indirectly regulate chromatin occupancy of AR or FOXA1, but this does not seem to be its main mechanism of action.
Figure 8.
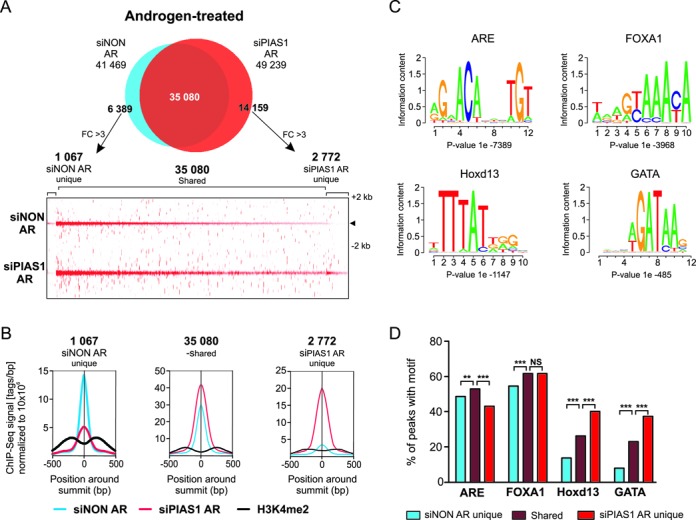
Influence of PIAS1 depletion on the AR cistrome. VCaP cells were treated as in Figure 7. (A) Venn diagram showing overlap of AR cistromes in siNON and siPIAS1 cells (upper panel). Non-overlapping sites were further analyzed using get-DifferentialPeak tool to achieve final categories. Heat map showing AR tag densities for siNON unique, siNON/siPIAS1-shared and siPIAS1 unique sites in a window ±2 kb (lower panel). (B) Comparison of AR and H3K4me2 average tag counts in ±500 bp from the centers of the shared and unique sites. (C) The four most enriched motifs of AR-binding sites in VCaP cells. Initial de novo motif discovery was performed on all AR-binding sites (siNON) on ±100 bp of the peak center. (D) Distribution of the most enriched de novo motifs in siNON unique, siNON/siPIAS1-shared and siPIAS1 unique sites. Statistical significances were calculated by χ2-test (***P < 0.001).
DISCUSSION
Androgen-regulated gene programs require in addition to androgen, the AR and RNA polymerase II machinery, a number coregulator proteins, coactivators and corepressor (1). Expression patterns of coregulator proteins are thought to underlie tissue-specific differences in AR-regulated gene programs. Deregulated expression of AR coregulators has been, in turn, suggested to contribute to CRPC (49). PIAS1 is one of the putative AR coregulators overexpressed in clinical prostate cancer samples (23,24), albeit its expression appears to be lower in hormone-refractory prostate tumors than in untreated prostate tumors (50). In this work, we have employed unbiased approaches to reveal the genome-wide role of PIAS1 in the regulation of AR target gene expression in VCaP cell line. Silencing of PIAS1 in VCaP cells affected expression of AR target genes. However, it did not have a general repressing or activating effect on all AR targets, but PIAS1 depletion resulted in significant activation or repression of about one tenth of all genes. This implies that the ability of PIAS1 to regulate AR target genes is intriguingly target-specific, and that it exhibits characteristics of both a coactivator and a corepressor.
According to the pathway analyses of all androgen-regulated genes in control vs. PIAS1-depleted cells, PIAS1 is involved in the regulation of canonical pathways of cholesterol biosynthesis which potentially supports intratumoral androgen biosynthesis enhanced in CRPC (5,6). Gene expression analyses also indicated that PIAS1 can mask a large group of genes, such as NR5A2, VLDLR and CAMK1, from androgen regulation. The most enriched molecular and cellular functions among the latter gene group were gene expression, protein synthesis, cell cycle, cancer, cell death and survival. In keeping with the role of PIAS1 in cell cycle and growth, silencing of PIAS1 attenuated growth of VCaP cells, also in the absence of added androgen. Our results are in agreement with two recent studies utilizing both AR negative and positive cell lines (23,51). According to Hoefer et al. (23), PIAS1 depletion increased the proportion of cells in the GO/G1 phase and decreases the portion in the S-phase, which they reported in AR negative PC3 cells to be mediated by increased p21 protein levels. In VCaP cells, the silencing did not however result in up-regulation, but slight down-regulation, of p21 mRNA accumulation (Supplementary Figure S13), suggesting that the mechanism underlying the attenuated cell growth is more complex. Interestingly, many AR targets that are linked to cell growth and apoptosis, such as ATAD2, BCL6, MAFB, TNFSF10 (TRAIL), IGFBP-3, and TMEFF2 were regulated by PIAS1 (41–48). TMEFF2 has previously been shown exhibit antiproliferative effects in prostate cancer cells. ATAD2 and MAFB are interesting examples of AR target genes whose androgen-mediated repression was converted to activation upon PIAS1 depletion in VCaP cells. ATAD2 has previously been implicated as a coactivator for AR in prostate cancer (44) and for c-MYC in many aggressive tumors (52). Both ATAD2 and MYC are overexpressed in prostate tumors and their expression is critical for prostate cell proliferation and survival. TNFSF10 and IGFBP-3 are androgen-repressed genes with pro-apoptotic effects (43,47). In VCaP cells, PIAS1 attenuated their expression especially in the absence of androgens.
PIAS1 was able to bind to chromatin, mainly at FOXA1 motif-containing sites in the absence of added androgen, but androgen exposure increased the number of PIAS1-binding sites markedly, resulting in practically complete overlap with about one tenth of the AR cistrome. PIAS1 sites resulting from the androgen exposure showed enrichment for AREs and FOXA1 motifs at about the same frequency of 50%. In general, the ARBs with the strongest signals were co-enriched with the PIAS1-binding chromatin sites, and more than one fourth of the genes regulated by androgen were associated with relatively nearby (± 50 kb from their TSSs) AR/PIAS1-shared sites. The AR- and PIAS1-shared sites were proximal to an active enhancer mark of H3K4me2, implying their active involvement in transcriptional regulation. The PIAS1 cistrome also showed almost complete overlap with the FOXA1 cistrome, but again, <10% FOXA1 binding sites was shared with PIAS1. These data are in line with our gene expression data revealing AR target gene selective action of PIAS1. The genes associated with cell growth, apoptosis and cancer listed in the previous paragraph are prominent examples of AR-, PIAS1- and FOXA1-co-occupied enhancers.
Recent comparison of steroid receptor coactivator 3 (SRC-3) cistrome to estrogen receptor (ER) α and FOXA1 cistromes in breast cancer cells revealed that estrogen increases only modestly (∼30%) the co-occupancy of the coactivator with the receptor or FOXA1 and that only 30% of the SRC-3 cistrome overlaps with the ERα and FOXA1 cistromes (53). In comparison to the situation of SRC-3, the overlap of the PIAS1 cistrome with the AR and the FOXA1 cistromes is rather remarkable, suggesting that the PIAS1 actively recruited to the AR target gene-regulating enhancers. This is likely to occur via complex formation of PIAS1 with both holo-AR and FOXA1 either prior to or after chromatin binding of the latter two factors. PIAS1 has been also suggested to be capable of binding on its own to DNA via its amino terminal scaffold attachment factor-A/B, acinus and PIAS (SAP) domain (22).
In comparison to FOXA1 depletion which elicits extensive redistribution of ARBs on prostate cancer cell chromatin (7), PIAS1 depletion had only relatively small effects on AR chromatin occupancy. The new ARBs uncovered by PIAS1 depletion harbored more often HOXD13 and GATA motifs than the other AR-binding sites. Interestingly, the homeodomain protein HOXB13 that recognizes the HOXD13 motif has been shown to interact with AR and behave as a bifunctional regulator of AR transcriptional activity, showing characteristics of both a coactivator and corepressor (54). Moreover, germline mutations in HOXB13 are associated with a significantly increased risk of hereditary prostate cancer (14). Since the new ARBs did not however generally overlap with the PIAS1 cistrome, it is possible that PIAS1 is regulating SUMOylation of HOXD13 and GATA factors, but this is likely to take place off-chromatin.
In line with the interaction of FOXA1 with PIAS1 as a SUMOylation pathway component, we have recently shown that FOXA1 is SUMOylated and that the modification modulates its activity and interaction with AR (55). Given that SUMO2/3 co-occurred with PIAS1 at FOXA1- and AR-binding enhancer sites, we propose in our schematic model (Figure 9) that the regulatory effects of PIAS1 at these genomic loci are, at least in part, mediated via enhanced SUMOylation of FOXA1 and/or AR or other factors, which influences their interactions with coregulator proteins. In conclusion, our genome-wide analyses strongly suggest that PIAS1 functions as a genuine and chromatin-bound AR coregulator and that it cooperates with the pioneer factor FOXA1 in an AR target gene selective fashion to regulate gene programs relevant to prostate cancer cell growth and signaling. Further studies are required to decipher the role of PIAS1′s SUMO ligase function on chromatin and assess whether PIAS1 could be utilized as a therapeutic target for inhibition of prostate cancer cell growth.
Figure 9.
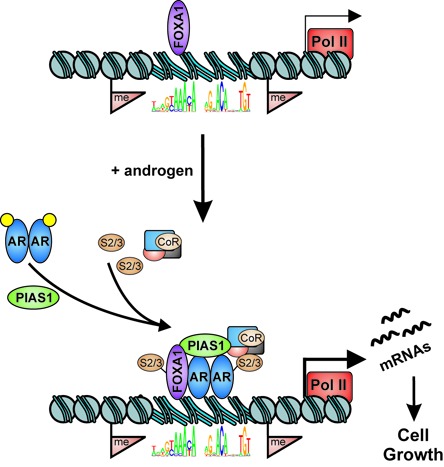
A schematic model of PIAS1 function on AR target genes regulating prostate cancer cell growth. Androgen-bound ARs recruit PIAS1 to chromatin enhancer sites that are neighbored by FOXA1 and surrounded by active chromatin H3 methyl marks. PIAS1 subsequently promotes SUMO2/3 (S2/3) modification of FOXA1 and/or AR and other factors, which also influences interactions with coregulator proteins (CoR). Pol II; RNA polymerase II and basal transcription machinery.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank Merja Räsänen and Eija Korhonen for their assistance with cell culture, Merja Heinäniemi for her advice with microarray analyses and Sami Heikkinen for his help with the construction of deep sequencing pipeline. The EMBL GeneCore sequencing team and Finnish Microarray and Sequencing Center are also acknowledged for deep sequencing and microarray analyses, respectively.
FUNDING
The Academy of Finland, the Finnish Cancer Organisations, UEF Doctoral Programme in Molecular Medicine, the strategic funding of the University of Eastern Finland, and the Sigrid Jusélius Foundation. Funding for open access charge: The Academy of Finland.
Conflict of interest statement. None declared.
REFERENCES
- 1.Heemers H.V., Tindall D.J. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 2.Palvimo J.J. The androgen receptor. Mol. Cell. Endocrinol. 2012;352:1–3. doi: 10.1016/j.mce.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Bray F., Lortet-Tieulent J., Ferlay J., Forman D., Auvinen A. Prostate cancer incidence and mortality trends in 37 european countries: an overview. Eur. J. Cancer. 2010;46:3040–3052. doi: 10.1016/j.ejca.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013;154:4010–4017. doi: 10.1210/en.2013-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai C., Balk S.P. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer. 2011;18:R175–82. doi: 10.1530/ERC-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan X., Cai C., Chen S., Chen S., Yu Z., Balk S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33:2815–2825. doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahu B., Laakso M., Ovaska K., Mirtti T., Lundin J., Rannikko A., Sankila A., Turunen J.P., Lundin M., Konsti J., et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962–3976. doi: 10.1038/emboj.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massie C.E., Lynch A., Ramos-Montoya A., Boren J., Stark R., Fazli L., Warren A., Scott H., Madhu B., Sharma N., et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J., Yu J., Mani R.S., Cao Q., Brenner C.J., Cao X., Wang X., Wu L., Li J., Hu M., et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer. Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahu B., Pihlajamaa P., Dubois V., Kerkhofs S., Claessens F., Jänne O.A. Androgen receptor uses relaxed response element stringency for selective chromatin binding and transcriptional regulation in vivo. Nucleic Acids Res. 2014;42:4230–4240. doi: 10.1093/nar/gkt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao N., Zhang J., Rao M.A., Case T.C., Mirosevich J., Wang Y., Jin R., Gupta A., Rennie P.S., Matusik R.J. The role of hepatocyte nuclear factor-3 alpha (forkhead box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003;17:1484–1507. doi: 10.1210/me.2003-0020. [DOI] [PubMed] [Google Scholar]
- 12.Wu D., Sunkel B., Chen Z., Liu X., Ye Z., Li Q., Grenade C., Ke J., Zhang C., Chen H., et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014;42:3607–3622. doi: 10.1093/nar/gkt1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q., Li W., Liu X.S., Carroll J.S., Jänne O.A., Keeton E.K., Chinnaiyan A.M., Pienta K.J., Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mills I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer. 2014;14:187–198. doi: 10.1038/nrc3678. [DOI] [PubMed] [Google Scholar]
- 15.van de Wijngaart D.J., Dubbink H.J., van Royen M.E., Trapman J., Jenster G. Androgen receptor coregulators: recruitment via the coactivator binding groove. Mol. Cell. Endocrinol. 2012;352:57–69. doi: 10.1016/j.mce.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Rosenfeld M.G., Lunyak V.V., Glass C.K. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 17.Coffey K., Robson C.N. Regulation of the androgen receptor by post-translational modifications. J. Endocrinol. 2012;215:221–237. doi: 10.1530/JOE-12-0238. [DOI] [PubMed] [Google Scholar]
- 18.Gross M., Liu B., Tan J., French F.S., Carey M., Shuai K. Distinct effects of PIAS proteins on androgen-mediated gene activation in prostate cancer cells. Oncogene. 2001;20:3880–3887. doi: 10.1038/sj.onc.1204489. [DOI] [PubMed] [Google Scholar]
- 19.Nishida T., Yasuda H. PIAS1 and PIASxalpha function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem. 2002;277:41311–41317. doi: 10.1074/jbc.M206741200. [DOI] [PubMed] [Google Scholar]
- 20.Kotaja N., Karvonen U., Jänne O.A., Palvimo J.J. PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol. Cell. Biol. 2002;22:5222–5234. doi: 10.1128/MCB.22.14.5222-5234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharrocks A.D. PIAS proteins and transcriptional regulation—more than just SUMO E3 ligases. Genes Dev. 2006;20:754–758. doi: 10.1101/gad.1421006. [DOI] [PubMed] [Google Scholar]
- 22.Rytinki M.M., Kaikkonen S., Pehkonen P., Jääskeläinen T., Palvimo J.J. PIAS proteins: pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 2009;66:3029–3041. doi: 10.1007/s00018-009-0061-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoefer J., Schafer G., Klocker H., Erb H.H., Mills I.G., Hengst L., Puhr M., Culig Z. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am. J. Pathol. 2012;180:2097–2107. doi: 10.1016/j.ajpath.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 24.Li P., Yu X., Ge K., Melamed J., Roeder R.G., Wang Z. Heterogeneous expression and functions of androgen receptor co-factors in primary prostate cancer. Am. J. Pathol. 2002;161:1467–1474. doi: 10.1016/S0002-9440(10)64422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korenchuk S., Lehr J.E., MClean L., Lee Y.G., Whitney S., Vessella R., Lin D.L., Pienta K.J. VCaP, a cell-based model system of human prostate cancer. In Vivo. 2001;15:163–168. [PubMed] [Google Scholar]
- 26.Knuuttila M., Yatkin E., Kallio J., Savolainen S., Laajala T.D., Aittokallio T., Oksala R., Häkkinen M., Keski-Rahkonen P., Auriola S., et al. Castration induces up-regulation of intratumoral androgen biosynthesis and androgen receptor expression in an orthotopic VCaP human prostate cancer xenograft model. Am. J. Pathol. 2014;184:2163–2173. doi: 10.1016/j.ajpath.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Makkonen H., Kauhanen M., Paakinaho V., Jääskeläinen T., Palvimo J.J. Long-range activation of FKBP51 transcription by the androgen receptor via distal intronic enhancers. Nucleic Acids Res. 2009;37:4135–4148. doi: 10.1093/nar/gkp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karvonen U., Kallio P.J., Jänne O.A., Palvimo J.J. Interaction of androgen receptors with androgen response element in intact cells. roles of amino- and carboxyl-terminal regions and the ligand. J. Biol. Chem. 1997;272:15973–15979. doi: 10.1074/jbc.272.25.15973. [DOI] [PubMed] [Google Scholar]
- 29.Makkonen H., Jääskeläinen T., Rytinki M.M., Palvimo J.J. Analysis of androgen receptor activity by reporter gene assays. Methods Mol. Biol. 2011;776:71–80. doi: 10.1007/978-1-61779-243-4_5. [DOI] [PubMed] [Google Scholar]
- 30.Paakinaho V., Kaikkonen S., Makkonen H., Benes V., Palvimo J.J. SUMOylation regulates the chromatin occupancy and anti-proliferative gene programs of glucocorticoid receptor. Nucleic Acids Res. 2014;42:1575–1592. doi: 10.1093/nar/gkt1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sutinen P., Malinen M., Heikkinen S., Palvimo J.J. SUMOylation modulates the transcriptional activity of androgen receptor in a target gene and pathway selective manner. Nucleic Acids Res. 2014;42:8310–8319. doi: 10.1093/nar/gku543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rytinki M., Kaikkonen S., Sutinen P., Paakinaho V., Rahkama V., Palvimo J.J. Dynamic SUMOylation is linked to the activity cycles of androgen receptor in the cell nucleus. Mol. Cell. Biol. 2012;32:4195–4205. doi: 10.1128/MCB.00753-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furey T.S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ovaska K., Laakso M., Haapa-Paananen S., Louhimo R., Chen P., Aittomäki V., Valo E., Nunez-Fontarnau J., Rantanen V., Karinen S., et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010;2:65. doi: 10.1186/gm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar R., Domrachev M., Lash A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benod C., Vinogradova M.V., Jouravel N., Kim G.E., Fletterick R.J., Sablin E.P. Nuclear receptor liver receptor homologue 1 (LRH-1) regulates pancreatic cancer cell growth and proliferation. Proc. Natl. Acad. Sci. U.S.A. 2011;108:16927–16931. doi: 10.1073/pnas.1112047108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kataoka K., Noda M., Nishizawa M. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both fos and jun. Mol. Cell. Biol. 1994;14:700–712. doi: 10.1128/mcb.14.1.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker S.R., Liu S., Xiang M., Nicolais M., Hatzi K., Giannopoulou E., Elemento O., Cerchietti L., Melnick A., Frank D.A. The transcriptional modulator BCL6 as a molecular target for breast cancer therapy. Oncogene. 2014 doi: 10.1038/onc.2014.61. doi:10.1038/onc.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehta H.H., Gao Q., Galet C., Paharkova V., Wan J., Said J., Sohn J.J., Lawson G., Cohen P., Cobb L.J., et al. IGFBP-3 is a metastasis suppression gene in prostate cancer. Cancer Res. 2011;71:5154–5163. doi: 10.1158/0008-5472.CAN-10-4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zou J.X., Guo L., Revenko A.S., Tepper C.G., Gemo A.T., Kung H.J., Chen H.W. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009;69:3339–3346. doi: 10.1158/0008-5472.CAN-08-3440. [DOI] [PubMed] [Google Scholar]
- 45.Gery S., Sawyers C.L., Agus D.B., Said J.W., Koeffler H.P. TMEFF2 is an androgen-regulated gene exhibiting antiproliferative effects in prostate cancer cells. Oncogene. 2002;21:4739–4746. doi: 10.1038/sj.onc.1205142. [DOI] [PubMed] [Google Scholar]
- 46.Selimovic D., Sprenger A., Hannig M., Haikel Y., Hassan M. Apoptosis related protein-1 triggers melanoma cell death via interaction with the juxtamembrane region of p75 neurotrophin receptor. J. Cell. Mol. Med. 2012;16:349–361. doi: 10.1111/j.1582-4934.2011.01304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang D., Lu J., Tindall D.J. Androgens regulate TRAIL-induced cell death in prostate cancer cells via multiple mechanisms. Cancer Lett. 2013;335:136–144. doi: 10.1016/j.canlet.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiskirchen R., Moser M., Weiskirchen S., Erdel M., Dahmen S., Buettner R., Gressner A.M. LIM-domain protein cysteine- and glycine-rich protein 2 (CRP2) is a novel marker of hepatic stellate cells and binding partner of the protein inhibitor of activated STAT1. Biochem. J. 2001;359:485–496. doi: 10.1042/0264-6021:3590485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sung Y.Y., Cheung E. Androgen receptor co-regulatory networks in castration-resistant prostate cancer. Endocr. Relat. Cancer. 2013;21:R1–R11. doi: 10.1530/ERC-13-0326. [DOI] [PubMed] [Google Scholar]
- 50.Linja M.J., Porkka K.P., Kang Z., Savinainen K.J., Jänne O.A., Tammela T.L., Vessella R.L., Palvimo J.J., Visakorpi T. Expression of androgen receptor coregulators in prostate cancer. Clin. Cancer Res. 2004;10:1032–1040. doi: 10.1158/1078-0432.ccr-0990-3. [DOI] [PubMed] [Google Scholar]
- 51.Heemers H.V., Regan K.M., Schmidt L.J., Anderson S.K., Ballman K.V., Tindall D.J. Androgen modulation of coregulator expression in prostate cancer cells. Mol. Endocrinol. 2009;23:572–583. doi: 10.1210/me.2008-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciro M., Prosperini E., Quarto M., Grazini U., Walfridsson J., McBlane F., Nucifero P., Pacchiana G., Capra M., Christensen J., et al. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 2009;69:8491–8498. doi: 10.1158/0008-5472.CAN-09-2131. [DOI] [PubMed] [Google Scholar]
- 53.Lanz R.B., Bulynko Y., Malovannaya A., Labhart P., Wang L., Li W., Qin J., Harper M., O'Malley B.W. Global characterization of transcriptional impact of the SRC-3 coregulator. Mol. Endocrinol. 2010;24:859–872. doi: 10.1210/me.2009-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Norris J.D., Chang C.Y., Wittmann B.M., Kunder R.S., Cui H., Fan D., Joseph J.D., McDonnell D.P. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol. Cell. 2009;36:405–416. doi: 10.1016/j.molcel.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sutinen P., Rahkama V., Rytinki M., Palvimo J.J. Nuclear mobility and activity of FOXA1 with androgen receptor are regulated by SUMOylation. Mol. Endocrinol. 2014;28:1719–1728. doi: 10.1210/me.2014-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.