

Abstract
Upon infection, pathogenic Leptospira species bind several complement regulators in order to overcome host innate immunity. We previously characterized a 20-kDa leptospiral surface protein which interacts with C4b binding protein (C4BP): leptospiral complement regulator-acquiring protein A (LcpA). Here we show that LcpA also interacts with human factor H (FH), which remains functionally active once bound to the protein. Antibodies directed against short consensus repeat 20 (SCR20) inhibited binding of FH to LcpA by approximately 90%, thus confirming that this particular domain is involved in the interaction. We have also shown for the first time that leptospires bind human vitronectin and that the interaction is mediated by LcpA. Coincubation with heparin blocked LcpA-vitronectin interaction in a dose-dependent manner, strongly suggesting that binding may occur through the heparin binding domains of vitronectin. LcpA also bound to the terminal pathway component C9 and inhibited Zn2+-induced polymerization and membrane attack complex (MAC) formation. Competitive binding assays indicated that LcpA interacts with C4BP, FH, and vitronectin through distinct sites. Taken together, our findings indicate that LcpA may play a role in leptospiral immune evasion.
INTRODUCTION
Leptospirosis is a spirochetal disease caused by pathogenic members of the genus Leptospira. Pathogenic leptospires persistently colonize the kidneys of reservoir animals, which eliminate the bacteria in the urine while presenting no clinical symptoms. Humans are incidental hosts who are susceptible to the disease (1). Upon infection, leptospires spread and propagate in susceptible hosts because they have evolved mechanisms to evade innate immune defense responses. A greater susceptibility to normal serum by nonpathogenic Leptospira strains was first reported by Johnson and Muschel in the mid-1960s (2). It became clear at that time that virulence correlated with the capacity of resisting complement-mediated killing (3). However, studies on the mechanisms underlying this resistance were only recently initiated. Acquisition of fluid-phase host complement regulators on the surfaces of pathogens is a common complement evasion mechanism, and it has been demonstrated that pathogenic Leptospira strains are able to bind factor H (FH), factor H-like 1 (FHL-1), factor H-related 1 (FHR-1), and C4b binding protein (C4BP) (4–7).
Over recent years, functional characterization of some Leptospira immune evasion proteins has been reported. Identification of specific host ligands and definition of the mechanism of complement inactivation have been accomplished for certain leptospiral outer membrane proteins present only in pathogenic species. In a previous work, we characterized a 20-kDa surface protein of Leptospira, LcpA (leptospiral complement regulator-acquiring protein A), which has been shown to interact with C4BP, a negative regulator of the classical and lectin pathways of complement (8). The cofactor activity of C4BP bound to immobilized LcpA was confirmed by detecting the C4d fragment obtained by factor I (FI)-mediated cleavage of C4b. LcpA was shown to be an outer membrane protein by use of immunoelectron microscopy, Triton X-114 fractionation, and cell surface proteolysis. The lcpA gene is conserved among pathogenic Leptospira spp., and the protein is expressed by serum-resistant and serum-intermediate strains (8). Moreover, it has been reported that LcpA is expressed during the course of human infection (9).
It is well known that a single pathogenic immune evasion protein is able to interact with more than one human complement molecule (reviewed in reference 10). By controlling multiple steps of the complement cascade, a pathogen can escape the host's innate immune responses more efficiently, thus being able to survive and establish an infection. Given the ability of certain bacterial surface proteins to bind multiple host molecules, we assessed in the present study if LcpA also interacts with FH, the main soluble regulator of the alternative pathway of complement, and vitronectin, a terminal pathway complement regulator.
Like C4BP, FH is composed of globular domains known as short consensus repeats (SCRs). The FH N terminus (SCRs 1 to 4 [SCR1–4]) exhibits regulatory activity. SCRs 5 to 7, 19, and 20 are the preferential binding sites for pathogenic microorganisms (reviewed in reference 11). FH regulates the alternative pathway of complement by acting as a cofactor for FI-mediated cleavage and inactivation of C3b and also by accelerating the decay of the C3 convertase (C3b,Bb) (12–14).
Vitronectin is a multifunctional glycoprotein that plays important roles in many biological processes, including tissue repair, cell migration, and regulation of the terminal pathway of complement by inhibition of C5b7 complex formation and C9 polymerization. Human vitronectin consists of an N-terminal somatomedin B domain, an RGD cell receptor binding site, four hemopexin-like domains, and three heparin binding domains (reviewed in reference 15). It circulates in the bloodstream at high concentrations (0.2 to 0.7 mg/ml) (16, 17) as monomers (65 and 75 kDa) and is also an important component of the extracellular matrix (ECM). Tissue and ECM vitronectin is a multimer that interacts with macromolecular ligands, including glycosaminoglycans and collagens (18, 19). Immunohistochemical studies allowed detection of vitronectin in a variety of normal human tissues, including the liver, lungs, kidneys, and blood vessel walls (15, 18). Previous reports have demonstrated that leptospires bind several extracellular matrix components (20, 21), but interaction of these particular spirochetes with vitronectin has never been evaluated.
In this study, we demonstrate that LcpA is an FH and vitronectin binding protein. Functional assays have shown that LcpA-bound FH retains cofactor activity. We have also shown that leptospires interact with the heparin binding domains of vitronectin via LcpA. Furthermore, LcpA also binds C9 and is capable of inhibiting C9 polymerization and membrane attack complex (MAC) formation. Our data suggest that LcpA may contribute to leptospiral serum resistance by interfering with multiple steps of the complement cascade.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Leptospira interrogans serovar Kennewicki strain Fromm, Leptospira interrogans serovar Copenhageni strain 10A, Leptospira interrogans serovar Pomona strain Pomona, Leptospira noguchi serovar Panama strain CZ214K, Leptospira borgpetersenii serovar Javanica strain Veldrat Batavia 46, Leptospira borgpetersenii serovar Tarassovi strain 17, Leptospira kirschneri serovar Cynopteri strain 3522C, and Leptospira santarosai serovar Shermani strain 1342K were used in the assays. The virulence of L. interrogans serovar Kennewicki strain Fromm is maintained by iterative passages in hamsters. Bacteria were cultured at 29°C under aerobic conditions as previously described (8).
Sera, purified proteins, and antibodies.
Normal human serum (NHS) and purified human FH, C4BP, FI, C3b, C5b6, C7, C8, and C9 were purchased from Complement Technology, and human vitronectin was purchased from Sigma-Aldrich. Recombinant FH fragments SCR8–14 and SCR15–20 were produced as described previously (7). Goat anti-human FH was purchased from Quidel, rabbit anti-human C4BP was purchased from Calbiochem, and rabbit anti-human vitronectin and goat anti-human C3 and C9 polyclonal antibodies were purchased from Complement Technology. The anti-human FH monoclonal antibody C18 (anti-SCR20) was purchased from Enzo Life Sciences, and the anti-human C5b9 monoclonal antibody (used as a negative control) was purchased from Dako. Secondary peroxidase-conjugated antibodies as well as antibodies labeled with colloidal gold particles were purchased from Sigma-Aldrich.
Cloning, expression, and purification of LcpA.
Cloning, expression, and purification of recombinant LcpA were described previously (8).
Western blot overlay.
Purified recombinant LcpA, whole-cell Leptospira lysates, and bovine serum albumin (BSA) were subjected to 15% SDS-PAGE under reducing conditions and then transferred to nitrocellulose membranes. Nonspecific binding sites were blocked by using 10% (wt/vol) dried milk in phosphate-buffered saline–0.05% Tween (pH 7.4) (PBST) for 16 h at 4°C. Subsequently, the membranes were rinsed three times in PBST and incubated for 90 min at room temperature (RT) with 10% NHS diluted in PBST. After five washes with PBST, membranes were incubated with polyclonal goat antibodies recognizing human FH (1:10,000) or with polyclonal rabbit antibodies recognizing human vitronectin (1:5,000), followed by peroxidase-conjugated secondary antibodies (1:5,000). Positive signals were detected by enhanced chemiluminescence (West Pico; Pierce).
SPR and kinetic analysis.
LcpA protein diluted in 10 mM sodium citrate, pH 3.5, was immobilized on a CM5 sensor chip by use of an amine coupling reagent kit (GE Healthcare) at 25°C. HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, and 0.005% Tween 20, pH 7.4) was used as running buffer. The carboxymethyl dextran surface was activated with 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC)–N-hydroxysuccinimide solution. As a reference, the same procedure was performed on another flow cell, without the protein (blank immobilization). Any remaining activated groups were blocked with 1 M ethanolamine. For the kinetic assay, we used the running buffer HBS-P (10 mM HEPES, 150 mM NaCl, and 0.05% Tween 20, pH 7.4) plus 2 mM CaCl2. FH protein was diluted in this buffer, to 0.1, 0.3, 0.5, or 0.7 μM, and injected at 10 μl/min for 120 s (association) over the immobilized LcpA protein and the blank reference. A Biacore T-100 instrument (GE Healthcare) was used for surface plasmon resonance (SPR) detection. Dissociation was monitored for 600 s. Regeneration was performed with 0.8 mM NaOH for 25 s at 30 μl/min. Kinetic constants were calculated by the global fitting method (1:1 Langmuir binding model).
Direct binding assays.
To map the FH domains involved in the interaction with LcpA, the recombinant protein (1 μg) was immobilized in enzyme-linked immunosorbent assay (ELISA) plate wells and, after blocking, incubated with 2 μg of SCR8–14 (middle region of FH) or SCR15–20 (C-terminal region of FH). Intact purified FH was included as a control. Bound proteins were detected with goat anti-human FH, which reacted similarly with all constructs (7). For the inhibition assay, purified FH (1 μg) was preincubated for 30 min at RT with the monoclonal antibody C18, directed against SCR20 (0.125 to 1 μg), or with anti-C5b9 (0.125 to 1 μg) as a negative control. FH was then allowed to interact with immobilized LcpA and was detected with goat anti-human FH as previously described (7). For investigation of the interference of heparin and the effect of ionic strength on LcpA-FH or LcpA-vitronectin interaction, microtiter plates were coated with recombinant LcpA (100 μl; 10 μg/ml) for 16 to 20 h at 4°C. The wells were washed with PBS, blocked with PBS-3% BSA for 2 h at 37°C, and incubated with purified FH or vitronectin (100 μl; 10 μg/ml) in the presence of increasing amounts of heparin (0.1 to 10 μg/ml) or NaCl (62.5 to 500 mM). Reaction mixtures were incubated for 60 min at 37°C. Unbound FH or vitronectin was removed by three washes with PBS-T, and bound FH or vitronectin was detected by use of goat anti-human FH (1:10,000) or rabbit anti-human vitronectin (1:5,000), respectively, followed by peroxidase-conjugated anti-goat IgG (1:10,000) or anti-rabbit IgG (1:5,000). Detection was performed with o-phenylenediamine dihydrochloride (Pierce), and absorbance was measured at 492 nm.
Cofactor assay.
The cofactor activity of FH bound to LcpA and rLIC10301 (negative control) was analyzed by measuring FI-mediated cleavage of C3b as previously described (7). Microtiter plate wells were coated with 1 μg of each recombinant protein diluted in PBS for 16 h at 4°C. After blocking with 3% BSA, 2 μg of FH was added to each well for 60 min at 37°C. After five washes with PBS, C3b (500 ng/reaction mixture) and FI (250 ng/reaction mixture) were added to the wells and incubated for up to 240 min at 37°C. Reaction mixtures were subjected to Western blotting, and cleavage fragments of C3b were detected with goat anti-human C3 polyclonal antibodies (1:10,000), followed by incubation with secondary peroxidase-conjugated antibodies. The cofactor activity of FH bound to L. interrogans serovar Kennewicki strain Fromm was assessed essentially as described above. In this case, 2 × 108 cells were used, and incubations were performed in 1.5-ml microcentrifuge tubes.
Detection of interaction of L. interrogans with vitronectin by Western blotting using whole-cell lysates.
Freshly harvested leptospires (1.0 × 109) were washed with PBS and incubated with 20% NHS for 16 h at RT with agitation. After five washes with PBS, pellets were subjected to 12% SDS-PAGE under nonreducing conditions and then transferred to nitrocellulose membranes. Nonspecific binding sites were blocked using 10% (wt/vol) dried milk in PBS-0.05% Tween (pH 7.4) overnight at 4°C. Subsequently, the membrane was incubated with polyclonal anti-human vitronectin at a 1:5,000 dilution, followed by peroxidase-conjugated anti-rabbit IgG (1:10,000). Positive signals were detected by enhanced chemiluminescence (West Pico; Pierce).
Detection of vitronectin bound to L. interrogans by immunogold labeling and negative staining.
L. interrogans serovar Kennewicki strain Fromm (1 × 108 cells) was washed twice with PBS and then incubated with 20 μl of human purified vitronectin (1 μg/μl) or PBS (negative control) for 16 h with gentle agitation. After five washes, bacteria were fixed with 2% paraformaldehyde in PBS for 1 h at RT. After two washes with PBS, leptospires were applied to Formvar-coated nickel grids and incubated with rabbit anti-human vitronectin (1:10) in PBS-1.5% BSA. After washings with PBS, preparations were incubated with goat anti-rabbit antibody labeled with 10-nm colloidal gold particles (1:5) in PBS-1.5% BSA for 1 h at RT. After washings with PBS and distilled water, preparations were negatively stained with 2% uranyl acetate and observed by transmission electron microscopy (TEM) (LEO 906E; Leica Microsystems GmbH, Germany) at 80 kV.
Binding of C9 to immobilized LcpA.
Binding of the terminal complement component C9 to LcpA was assessed by ELISA. Microtiter plate wells were coated with 1 μg of recombinant LcpA, purified vitronectin (positive control), LIC10301, or BSA (negative controls) and blocked as described above. C9 (1 μg) was added, and incubation proceeded for 60 min. After washes, bound complement proteins were detected with goat anti-human C9 (1:5,000), followed by an incubation with peroxidase-conjugated secondary antibodies.
C9 polymerization assay.
The effect of LcpA on C9 polymerization was assessed according to a previously published protocol (22). Briefly, LcpA (1.25 to 5 μg) and the negative-control protein LIC103011 (2.5 μg) were preincubated with 3 μg of C9 at 37°C in 20 mM Tris-HCl (pH 7.2). After 40 min of incubation, 50 μM ZnCl2 in 20 mM Tris-HCl (pH 7.2) was added for 2 h at 37°C. The samples were separated in precast 8 to 16% gradient polyacrylamide gels (Bio-Rad, Hercules, CA), and C9 polymerization was visualized by silver staining.
Hemolytic assay.
The MAC-inhibitory activity of LcpA was analyzed in a hemolytic assay using sheep erythrocytes (23). The erythrocytes were resuspended to 1 × 108 cells/ml in Veronal-buffered saline (VBS) and were preincubated with 1 μg/ml C5b6 for 1 h at room temperature. In a separate preparation, 1 μg/ml C9 was preincubated with increasing concentrations of purified recombinant LcpA (6.25 to 50 μg/ml) or with LIC10301 (50 μg/ml) or FH (50 μg/ml) for 30 min at 37°C. Thereafter, the complement proteins C7 (1 μg/ml) and C8 (0.1 μg/ml) were added to the mixture for 15 min at 37°C. After preincubation, the C5b6-coated erythrocytes were added to the LcpA-C7-C8-C9 mixture and incubated for 30 min at 37°C. Erythrocytes were centrifuged, and the amount of hemoglobin, representing the lysed cells, was measured at 540 nm. The relative MAC-inhibitory activity is presented as the percentage of total hemolysis.
Competition assays.
Competitive binding assays were assessed by ELISA (7). Wells were coated with 1 μg of recombinant LcpA and blocked with 3% BSA for 2 h. One microgram of a given protein (FH or C4BP) mixed with a different amount of C4BP or vitronectin (0.25, 0.5, 1, 2, or 4 μg) was added to each well, and the bound proteins were detected with goat anti-human FH, rabbit anti-human C4BP, or rabbit anti-human vitronectin (diluted 1:5,000), followed by an incubation with peroxidase-conjugated secondary antibodies (1:10,000). Detection was performed with o-phenylenediamine dihydrochloride (Pierce), and absorbance was measured at 492 nm.
RESULTS
LcpA is an FH binding protein.
In a previous work by our group, we demonstrated that LcpA was able to interact with the complement regulator C4BP, which remained functional when bound to the protein (8). In order to assess binding of LcpA to FH, another soluble complement molecule known to negatively regulate the alternative pathway, LcpA and the negative-control protein BSA were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and then examined for the ability to bind soluble FH from normal human serum. As shown in Fig. 1A, LcpA interacted with FH. A band of approximately 20 kDa that possibly corresponds to LcpA was detected when the whole-cell lysate from L. interrogans strain Fromm was incubated with NHS and probed with anti-human FH (Fig. 1B, left panel). An immunoblot performed with the same whole-cell lysate and probed with anti-LcpA serum was included as a control (Fig. 1B, right panel). Biacore kinetic parameters describing the FH-LcpA interaction were as follows: ka = 5.7 × 104 M−1 s−1 and Kd (dissociation constant) = 2.2 × 103 s−1. These parameters resulted in an equilibrium dissociation constant (KD) of 3.9 × 10−8 M at 25°C (Fig. 1C). To map the binding sites within FH involved in this interaction, LcpA was immobilized and incubated with either full-length FH or the FH fragment SCR8–14 or SCR15–20. A considerable amount of binding of LcpA to SCR15–20 was observed (Fig. 1D). The monoclonal antibody C18, directed against SCR20, inhibited binding of FH to LcpA in a dose-dependent manner, strongly suggesting that this particular domain is relevant for the interaction (Fig. 1E). Binding of FH to LcpA was not blocked by the unrelated anti-C5b9 antibody, which was used as a negative control (Fig. 1E). FH is composed of 20 SCR domains, and it has been proposed that SCRs 7, 13 to 15, 19, and 20 contain heparin binding sites (reviewed in reference 24). We next assessed if LcpA binding sites would colocalize with the heparin binding domains on the FH molecule. No inhibition was observed even at the highest concentration of heparin tested (Fig. 1F). Thus, LcpA and heparin interact with FH through different binding sites. The effect of NaCl on the LcpA-FH interaction was also investigated. Binding was affected by increasing NaCl concentrations (Fig. 1G). At the lowest concentration tested (62.5 mM), the binding of FH to LcpA was already reduced by 45%. Taken together, the data show that LcpA is an FH binding protein and that the interaction is influenced by the ionic strength.
FIG 1.

Interaction of LcpA with FH. (A) (Left) Purified recombinant proteins were subjected to SDS–15% PAGE under reducing conditions, transferred to a nitrocellulose membrane, and stained with Ponceau S. (Right) The membrane was incubated with 10% NHS, and FH binding was detected with polyclonal goat antibodies recognizing human FH. WB, Western blot. (B) Whole-cell lysates from L. interrogans serovar Kennewicki strain Fromm were separated by SDS–15% PAGE and transferred to nitrocellulose membranes. (Left) One membrane was incubated with 10% NHS, and FH binding was detected with polyclonal goat antibodies recognizing human FH. (Right) The other membrane was probed with LcpA antiserum (5). (C) Globally fitted SPR data on FH binding to immobilized LcpA. FH was injected at concentrations ranging from 0.1 to 0.7 μM, at a flow rate of 10 μl/min. After a 120-s association phase, the dissociation phase was followed for an additional 600 s. The curve was plotted using a 1:1 Langmuir binding model, using Biacore T100 evaluation software. RU, relative units. (D) Recombinant LcpA (1 μg) was immobilized on microtiter plates and incubated with either full-length FH or recombinant FH fragment SCR15–20 or SCR8–14 (2 μg). Bound proteins were detected using goat anti-human FH followed by a peroxidase-conjugated secondary antibody. Each bar represents the mean absorbance value at 492 nm ± the standard deviation (SD) for 3 independent experiments, each performed in triplicate. Binding of LcpA to full-length FH was set as 100%. *, P ≤ 0.05; ***, P ≤ 0.001. (E) One microgram of purified FH was preincubated with 0 to 1 μg of the monoclonal antibody C18 (directed against SCR20) or with 0 to 1 μg of anti-C5b9 (negative control). Factor H was then allowed to interact with immobilized LcpA and was detected as described above. Each bar represents the mean absorbance value at 492 nm ± the SD for 3 independent experiments, each performed in triplicate. Binding of FH in the absence of antibody was set as 100%. ***, P ≤ 0.001. (F) The effect of heparin (0.1 to 10 μg/ml) on binding of FH (10 μg/ml) to immobilized LcpA (10 μg/ml) was assayed. (G) The effect of NaCl (62.5 to 500 mM) on binding of FH (10 μg/ml) to immobilized LcpA (10 μg/ml) was assayed. Each bar represents the mean absorbance value at 492 nm ± the SD for 3 independent experiments, each performed in triplicate. ***, P ≤ 0.001. Statistical analyses were performed using analysis of variance (ANOVA).
FH retains cofactor activity when bound to LcpA.
FH acts as a cofactor for FI, promoting cleavage and inactivation of C3b. Immobilized recombinant proteins were first incubated with purified FH, and after intensive washings to remove unbound FH, C3b and FI were added. Incubation proceeded for the indicated periods, and the cleavage fragments of C3b in the supernatant were subjected to Western blotting with anti-C3. Whole bacteria (L. interrogans serovar Kennewicki strain Fromm) were included as a positive control (Fig. 2, lane 3), since it was previously shown that surface-bound FH on this serum-resistant strain efficiently mediates cleavage of C3b (4). Three fragments, of 68, 46, and 43 kDa, were produced as a consequence of the degradation of the C3b α′ chain when LcpA was preincubated with FH. C3b cleavage was more efficient after 4 h of incubation (Fig. 2, lane 7). No cleavage fragments were detected when LcpA was incubated with C3b and FI in the absence of FH (Fig. 2, lanes 8 to 11). Recombinant LIC10301 was used as a negative control, since this Leptospira membrane protein does not bind FH (7) (Fig. 2, lanes 12 to 15). Our results indicate that FH bound to LcpA retains its complement-regulatory function.
FIG 2.

Cofactor activity of FH bound to LcpA or to intact L. interrogans. C3b (500 ng) and FI (250 ng) were added to immobilized recombinant proteins (1 μg) or to bacteria (2 × 108 cells) that had been preincubated with FH. The reaction mixtures were incubated for 1, 2, or 4 h at 37°C. The products were analyzed by SDS-PAGE, and the cleavage fragments of C3b were detected by Western blotting with anti-human C3 polyclonal antibodies. The presence of bands at 43, 46, and 68 kDa indicates that acquired FH was able to promote FI-mediated cleavage of C3b. Lanes: 1, purified C3b; 2, C3b, FH, and FI (positive control); 3, L. interrogans, FH, C3b, and FI; 4 to 7, LcpA, FH, C3b, and FI; 8 to 11, LcpA, C3b, and FI; 12 to 15, LIC10301, FH, C3b, and FI.
Leptospira interrogans binds human vitronectin.
We first assessed binding of vitronectin to a number of pathogenic Leptospira strains by incubating bacteria with normal human serum. After successive washes, whole-cell lysates were subjected to Western blotting, and bound vitronectin was detected by use of specific antibodies. As depicted in Fig. 3A, all strains bound serum vitronectin. Immunogold labeling of the virulent strain L. interrogans serovar Kennewicki strain Fromm preincubated with vitronectin showed a uniform labeling pattern, thus confirming that leptospires interact with this multifunctional protein (Fig. 3B). No binding was observed when bacteria were incubated with primary and secondary antibodies in the absence of vitronectin (Fig. 3B).
FIG 3.
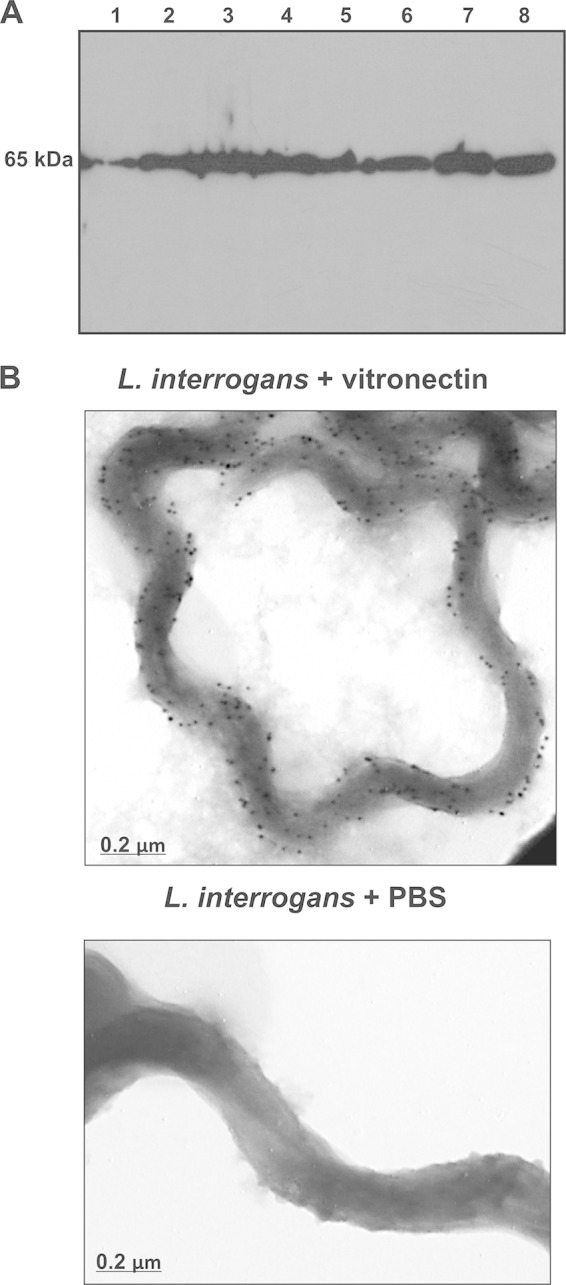
Binding of human vitronectin to Leptospira spp. (A) Leptospira strains were incubated with 20% NHS. Whole-cell lysates were subjected to Western blotting with rabbit anti-human vitronectin. Lanes: 1, L. interrogans serovar Copenhageni; 2, L. interrogans serovar Pomona; 3, L. noguchi serovar Panama; 4, L. borgpetersenii serovar Javanica; 5, L. borgpetersenii serovar Tarassovi; 6, L. kirshneri serovar Cynopteri; 7, L. santarosai serovar Shermani; 8, NHS (1:100). (B) L. interrogans serovar Kennewicki strain Fromm was incubated with purified human vitronectin or PBS. After extensive washing, leptospires were incubated with rabbit anti-human vitronectin, followed by incubation with goat anti-rabbit antibodies labeled with 10-nm colloidal gold particles. Bacteria were observed by TEM at 80 kV.
LcpA is a vitronectin binding protein.
Given that a single pathogenic protein can simultaneously bind several human complement proteins (reviewed in reference 10), we wondered if LcpA would also interact with vitronectin. Ligand affinity blotting data indicate that LcpA binds vitronectin from human serum (Fig. 4A). In the overlay assay using whole-cell lysate, a strong signal that possibly corresponds to LcpA and to additional Leptospira proteins with similar molecular masses was detected (Fig. 4B, left panel). An immunoblot performed with the same bacterial extract and probed with LcpA antiserum was included as a control (Fig. 4B, right panel). Since vitronectin possesses three heparin binding domains, we evaluated if LcpA binding sites on this particular regulatory protein would colocalize with the heparin binding domains. A dose-dependent inhibition was observed with increasing amounts of heparin added to the reaction mixtures (Fig. 4C). With regard to the role of ionic forces, NaCl at 250 to 500 mM could partially inhibit vitronectin binding to LcpA (Fig. 4D). These findings indicate that LcpA interacts with vitronectin through its heparin binding domains and that high salt concentrations may partially impair this interaction.
FIG 4.

Interaction of LcpA with vitronectin. (A) (Left) Purified recombinant proteins were subjected to SDS–15% PAGE under reducing conditions, transferred to a nitrocellulose membrane, and stained with Ponceau S. (Right) The membrane was incubated with 10% NHS, and vitronectin binding was detected with polyclonal goat antibodies recognizing human vitronectin. (B) Whole-cell lysates from L. interrogans serovar Kennewicki strain Fromm were separated by SDS–15% PAGE and transferred to nitrocellulose membranes. (Left) One membrane was incubated with 10% NHS, and vitronectin binding was detected with polyclonal goat antibodies recognizing human vitronectin. (Right) The other membrane was probed with LcpA antiserum (5). (C) The effect of heparin (0.1 to 10 μg/ml) on binding of vitronectin (10 μg/ml) to immobilized LcpA (10 μg/ml) was assayed. (D) The effect of NaCl (62.5 to 500 mM) on binding of vitronectin (10 μg/ml) to immobilized LcpA (10 μg/ml) was assayed. Each bar represents the mean absorbance value at 492 nm ± the SD for 3 independent experiments, each performed in triplicate. ***, P ≤ 0.001. Statistical analyses were performed using ANOVA.
LcpA binds to C9 and inhibits Zn2+-induced polymerization and MAC formation.
We further analyzed the role of LcpA in the complement terminal pathway by evaluating binding of C9 to the immobilized recombinant protein. A significant amount of binding of C9 to LcpA was observed. Purified human vitronectin was included as a positive control. No significant binding was detected with the negative-control proteins LIC10301 and BSA (Fig. 5A). Since LcpA binds to C9, we next investigated if this protein could impair C9 polymerization. LcpA was first incubated with purified C9, and polymerization was then induced by use of ZnCl2. In the presence of 2.5 μg of LcpA, polymerization was completely abolished. LIC10301 did not affect the formation of C9 polymers (Fig. 5B). The ability of LcpA to inhibit MAC formation was also evaluated using purified MAC components. LcpA was preincubated with C9 and then with C7 and C8. The LcpA-C7-C8-C9 mixture was added to C5b6-coated erythrocytes. LcpA significantly inhibited the lysis of erythrocytes in a dose-dependent manner (Fig. 5C). LcpA at 50 μg/ml inhibited lysis by 65%, whereas LIC10301 and FH, included as negative controls, did not block the cytolytic activity of the MAC (Fig. 5C).
FIG 5.
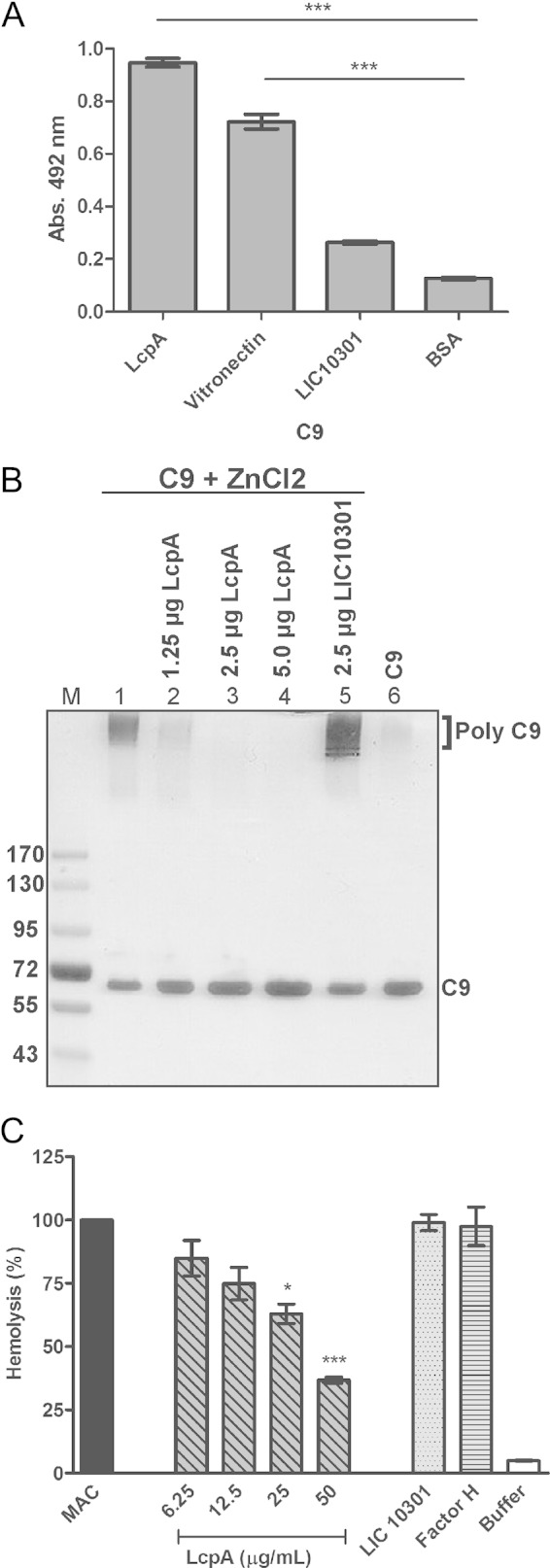
LcpA binds to C9 and inhibits Zn2+-induced polymerization and MAC formation. (A) LcpA, vitronectin, and the negative-control proteins LIC10301 and BSA were immobilized (10 μg/ml), and binding of the terminal complement component C9 (10 μg/ml) was assessed. Bound protein was detected with goat anti-human C9, followed by incubation with secondary peroxidase-conjugated antibodies. Each bar represents the mean absorbance value at 492 nm ± the SD for 3 independent experiments, each performed in triplicate. ***, P ≤ 0.001. (B) C9 was incubated with LcpA (1.25 to 5 μg) or LIC10301 (2.5 μg) at 37°C for 40 min before the addition of 50 μM ZnCl2 for 2 h at 37°C. Samples were subjected to SDS-PAGE on a gradient gel (8 to 16%), and C9 polymerization was visualized by silver staining. (C) LcpA (0 to 50 μg/ml), LIC10301 (50 μg/ml), and FH (50 μg/ml) were preincubated with C7, C8, and C9 and then added to C5b6-coated sheep erythrocytes. After incubation, cell lysis was measured, and the lysis in the absence of inhibitor (MAC) was set to 100%. Statistical analyses were performed using ANOVA. *, P ≤ 0.05; ***, P ≤ 0.001.
FH, C4BP, and vitronectin have distinct binding sites on LcpA.
Because LcpA interacts with three distinct complement regulators (C4BP, FH, and vitronectin), we then analyzed whether these regulators would compete for the same binding sites. The recombinant LcpA protein was immobilized in microtiter wells, and competitive binding assays were performed by fixing the amount of a particular complement regulator and adding increasing amounts of a second regulator, resulting in different molar ratios. The wells were probed with either anti-FH or anti-C4BP (Fig. 6A), anti-FH or anti-vitronectin (Fig. 6B), or anti-C4BP or anti-vitronectin (Fig. 6C). In all cases, no competition for binding to LcpA was observed, even at molar ratios beyond those observed under physiological conditions. Taken together, our findings indicate that FH, C4BP, and vitronectin interact with LcpA through different sites.
FIG 6.

FH, C4BP, and vitronectin have distinct binding sites on LcpA. Competition inhibition assays were performed in which different amounts of C4BP (0 to 40 μg/ml) and a constant amount of FH (10 μg/ml) (A), different amounts of FH (0 to 40 μg/ml) and a constant amount of vitronectin (10 μg/ml) (B), or different amounts of C4BP (0 to 40 μg/ml) and a constant amount of vitronectin (10 μg/ml) (C) were added to immobilized LcpA (10 μg/ml). Molar ratios are indicated below the graphs. Bound molecules were detected using specific antibodies followed by peroxidase-conjugated secondary antibodies. Optical densities were determined at 492 nm. Data represent the means ± SD for 3 independent experiments, each performed in triplicate. Molar ratios found under physiological conditions are underlined.
DISCUSSION
Successful colonization of hosts by pathogenic microorganisms can be attributed to the ability of these microorganisms to disrupt complement effector functions, thus compromising the first line of defense of the host innate immune response. Control of complement activation on a pathogen's surface or in its surrounding microenvironment is achieved by the interaction of surface-exposed proteins with complement-regulatory molecules, by the expression of membrane-associated proteins that may modulate/inhibit complement activation, such as the CD59-like protein from Borrelia burgdorferi (25), or by the secretion of proteases capable of degrading key complement molecules into nonfunctional fragments (reviewed in reference 26). Another prerequisite for colonization of a human host is the expression of multiple surface adhesins, allowing attachment of a given pathogen to extracellular matrix components and host cells, as well as infection at distinct niches.
Pathogenic Leptospira strains have been shown to bind the soluble complement-regulatory proteins FH, FHL-1, FHR-1, and C4BP (4–7) and several ECM and plasma molecules, including laminin, collagens, fibronectin, elastin, tropoelastin, proteoglycans, fibrinogen, and plasminogen (20, 21, 27, 28). Here we demonstrate that this spirochete also interacts with human vitronectin. This multifunctional glycoprotein harbors distinct binding sites for pathogens and eukaryotic cells and may serve as a bridging molecule between the bacterial surface and the host cell membrane, facilitating adhesion and invasion (15, 29). In human plasma, vitronectin acts as a regulator of the terminal pathway of complement. A number of vitronectin binding proteins supposed to have a role in serum resistance and/or adhesion have been described for Gram-negative bacteria (reviewed in reference 15). Once this protein is bound to the bacterial surface, MAC formation is inhibited and the pathogen is protected against lysis.
In this study, we demonstrated that LcpA is a Leptospira protein capable of binding several human complement molecules simultaneously. This surface-exposed protein is able to recruit the soluble regulator FH, allowing downregulation of the alternative pathway of complement. Interaction of LcpA with FH is mediated by the carboxy-terminal FH SCR20. Numerous bacteria, including Leptospira, have the capacity to interact with FH through SCRs 19 and 20 (11, 30). When bound to LcpA, FH remains functionally active, as indicated by the presence of C3b cleavage fragments upon incubation with FI. We have also shown that LcpA is one of the leptospiral ligands that mediate interaction with vitronectin. Previous reports have shown that the binding sites on vitronectin for certain bacterial surface proteins, such as Neisseria Opc, Haemophilus PE, and Moraxella UspA-2, include the heparin binding domain 3 (reviewed in reference 15), which prompted us to investigate if the heparin domains would also be involved in LcpA-vitronectin interaction. Indeed, heparin inhibited LcpA-vitronectin association in a concentration-dependent manner. Ionic forces also seem to play a role in LcpA-vitronectin and LcpA-FH interactions. LcpA also binds to the terminal pathway component C9, inhibits Zn2+-induced polymerization in a dose-dependent manner, and hampers MAC formation.
Since LcpA is able to interact with three different complement regulators, we wondered if these components would share overlapping regions and compete for binding on LcpA. Our competition assays involving FH, C4BP, and vitronectin clearly demonstrate that they interact simultaneously with the protein through separate sites.
Successful evasion of the human immune system seems to be crucial for Leptospira infection. Nonpathogenic strains can be distinguished from pathogenic ones by a greater susceptibility to normal serum, which can be attributed to the capacity to resist complement-mediated killing. Pathogenic Leptospira strains have multiple complement evasion strategies, including the acquisition of host regulators of complement activation (4, 5; this study) and the secretion of proteases that cleave complement proteins of all three pathways (31). To date, well-characterized complement evasion molecules from Leptospira include the LenA, LenB, LigA, LigB, and LcpA proteins. Interestingly, all of them share a feature: they bind multiple host molecules (7, 8, 27, 30). The same is true for the functionally related immune evasion proteins CspA, CspZ, ErpP, ErpC, and ErpA from Lyme disease Borrelia species, which bind FH and plasminogen but differ in binding FHL-1 and complement factor H-related proteins (CFHRs) (32–35). CspA also interacts with multiple ECM components (36).
Taken together, our findings show that LcpA is a multiligand binding molecule for distinct complement regulators. Through interaction with FH and C4BP, this leptospiral membrane protein may contribute to downregulation of the alternative, classical, and lectin pathways of complement, and by interacting with vitronectin and C9, LcpA may interfere with the terminal complement pathway, preventing MAC deposition on the bacterial surface. Control of multiple steps of the complement cascade certainly contributes to successful colonization by pathogenic Leptospira. As already mentioned, the human glycoprotein vitronectin is also part of the ECM, and it has been demonstrated that leptospires interact with several ECM components. The possibility that surface-bound vitronectin may help in Leptospira adhesion to host tissues is currently being addressed.
ACKNOWLEDGMENTS
We thank Sílvio de Arruda Vasconcellos and his group (Zenáide Moraes, Gisele Oliveira, and Amane Paldês), Faculty of Veterinary Medicine and Zootechnics of the University of São Paulo, for technical assistance.
This work was supported by the São Paulo Research Foundation (FAPESP) (grants 2011/07297-3 and 2010/50043-0), CNPq (A.M.M. is the recipient of productivity fellowship 311934/2013-7), and the Hungarian Academy of Sciences (grant LP2012-43).
REFERENCES
- 1.Faine S, Adler B, Bolin C, Perolat P. 1999. Leptospira and leptospirosis, 2nd ed. MediScience, Melbourne, Australia. [Google Scholar]
- 2.Johnson RC, Muschel LH. 1965. Antileptospiral activity of normal serum. J Bacteriol 89:1625–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson RC, Harris VG. 1967. Antileptospiral activity of serum. II. Leptospiral virulence factor. J Bacteriol 93:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meri T, Murgia R, Stefanel P, Meri S, Cinco M. 2005. Regulation of complement activation at the C3-level by serum resistant leptospires. Microb Pathog 39:139–147. doi: 10.1016/j.micpath.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Verma A, Hellwage J, Artiushin S, Zipfel PF, Kraiczy P, Timoney JF, Stevenson B. 2006. LfhA, a novel factor H-binding protein of Leptospira interrogans. Infect Immun 74:2659–2666. doi: 10.1128/IAI.74.5.2659-2666.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbosa AS, Abreu PA, Vasconcellos SA, Morais ZM, Gonçales AP, Silva AS, Daha MR, Isaac L. 2009. Immune evasion of Leptospira species by acquisition of human complement regulator C4BP. Infect Immun 77:1137–1143. doi: 10.1128/IAI.01310-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castiblanco-Valencia MM, Fraga TR, Silva LB, Monaris D, Abreu PA, Strobel S, Józsi M, Isaac L, Barbosa AS. 2012. Leptospiral immunoglobulin-like proteins interact with human complement regulators factor H, FHL-1, FHR-1, and C4BP. J Infect Dis 205:995–1004. doi: 10.1093/infdis/jir875. [DOI] [PubMed] [Google Scholar]
- 8.Barbosa AS, Monaris D, Silva LB, Morais ZM, Vasconcellos SA, Cianciarullo AM, Isaac L, Abreu PA. 2010. Functional characterization of LcpA, a surface-exposed protein of Leptospira spp. that binds the human complement regulator C4BP. Infect Immun 78:3207–3216. doi: 10.1128/IAI.00279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gamberini M, Gómez RM, Atzingen MV, Martins EA, Vasconcellos AS, Romero EC, Leite LC, Ho PL, Nascimento AL. 2005. Whole-genome analysis of Leptospira interrogans to identify potential vaccine candidates against leptospirosis. FEMS Microbiol Lett 244:305–313. doi: 10.1016/j.femsle.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Zipfel PF, Hallström T, Riesbeck K. 2013. Human complement control and complement evasion by pathogenic microbes—tipping the balance. Mol Immunol 56:152–160. doi: 10.1016/j.molimm.2013.05.222. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira VP, Pangburn MK, Cortés C. 2010. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol 47:2187–2197. doi: 10.1016/j.molimm.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whaley K, Ruddy S. 1976. Modulation of the alternative complement pathways by beta 1 H globulin. J Exp Med 144:1147–1163. doi: 10.1084/jem.144.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiler JM, Daha MR, Austen KF, Fearon DT. 1976. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U S A 73:3268–3272. doi: 10.1073/pnas.73.9.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. 1977. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J Exp Med 146:257–270. doi: 10.1084/jem.146.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh B, Su YC, Riesbeck K. 2010. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol Microbiol 78:545–560. doi: 10.1111/j.1365-2958.2010.07373.x. [DOI] [PubMed] [Google Scholar]
- 16.Boyd NA, Bradwell AR, Thompson RA. 1993. Quantitation of vitronectin in serum: evaluation of its usefulness in routine clinical practice. J Clin Pathol 46:1042–1045. doi: 10.1136/jcp.46.11.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chauhan AK, Moore TL. 2006. Presence of plasma complement regulatory proteins clusterin (Apo J) and vitronectin (S40) on circulating immune complexes (CIC). Clin Exp Immunol 145:398–406. doi: 10.1111/j.1365-2249.2006.03135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preissner KT, Seiffert D. 1998. Role of vitronectin and its receptors in haemostasis and vascular remodeling. Thromb Res 89:1–21. doi: 10.1016/S0049-3848(97)00298-3. [DOI] [PubMed] [Google Scholar]
- 19.Stockmann A, Hess S, Declerck P, Timpl R, Preissner KT. 1993. Multimeric vitronectin. Identification and characterization of conformation-dependent self-association of the adhesive protein. J Biol Chem 268:22874–22882. [PubMed] [Google Scholar]
- 20.Barbosa AS, Abreu PA, Neves FO, Atzingen MV, Watanabe MM, Vieira ML, Morais ZM, Vasconcellos SA, Nascimento AL. 2006. A newly identified leptospiral adhesion mediates attachment to laminin. Infect Immun 74:6356–6364. doi: 10.1128/IAI.00460-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breiner DD, Fahey M, Salvador R, Novakova J, Coburn J. 2009. Leptospira interrogans binds to human cell surface receptors including proteoglycans. Infect Immun 77:5528–5536. doi: 10.1128/IAI.00546-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Yang J, Wei J, Yang Y, Chen X, Zhao X, Gu Y, Cui S, Zhu X. 2011. Trichinella spiralis paramyosin binds to C8 and C9 and protects the tissue-dwelling nematode from being attacked by host complement. PLoS Negl Trop Dis 5:e1225. doi: 10.1371/journal.pntd.0001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hallström T, Blom AM, Zipfel PF, Riesbeck K. 2009. Nontypeable Haemophilus influenzae protein E binds vitronectin and is important for serum resistance. J Immunol 183:2593–2601. doi: 10.4049/jimmunol.0803226. [DOI] [PubMed] [Google Scholar]
- 24.Józsi M, Zipfel PF. 2008. Factor H family proteins and human diseases. Trends Immunol 29:380–387. doi: 10.1016/j.it.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Pausa M, Pellis V, Cinco M, Giulianini PG, Presani G, Perticarari S, Murgia R, Tedesco F. 2003. Serum-resistant strains of Borrelia burgdorferi evade complement-mediated killing by expressing a CD59-like complement inhibitory molecule. J Immunol 170:3214–3222. doi: 10.4049/jimmunol.170.6.3214. [DOI] [PubMed] [Google Scholar]
- 26.Lambris JD, Ricklin D, Geisbrecht BV. 2008. Complement evasion by human pathogens. Nat Rev Microbiol 6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choy HA, Kelley MM, Chen TL, Møller AK, Matsunaga J, Haake DA. 2007. Physiological osmotic induction of Leptospira interrogans adhesion: LigA and LigB bind extracellular matrix proteins and fibrinogen. Infect Immun 75:2441–2450. doi: 10.1128/IAI.01635-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin YP, Lee DW, McDonough SP, Nicholson LK, Sharma Y, Chang YF. 2009. Repeated domains of leptospira immunoglobulin-like proteins interact with elastin and tropoelastin. J Biol Chem 284:19380–19391. doi: 10.1074/jbc.M109.004531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergmann S, Lang A, Rohde M, Agarwal V, Rennemeier C, Grashoff C, Preissner KT, Hammerschmidt S. 2009. Integrin-linked kinase is required for vitronectin-mediated internalization of Streptococcus pneumoniae by host cells. J Cell Sci 122:256–267. doi: 10.1242/jcs.035600. [DOI] [PubMed] [Google Scholar]
- 30.Stevenson B, Choy HA, Pinne M, Rotondi ML, Miller MC, Demoll E, Kraiczy P, Cooley AE, Creamer TP, Suchard MA, Brissette CA, Verma A, Haake DA. 2007. Leptospira interrogans endostatin-like outer membrane proteins bind host fibronectin, laminin and regulators of complement. PLoS One 2:e1188. doi: 10.1371/journal.pone.0001188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fraga TR, Courrol D, Castiblanco-Valencia MM, Hirata IY, Vasconcellos SA, Juliano L, Barbosa AS, Isaac L. 2014. Immune evasion by pathogenic Leptospira strains: the secretion of proteases that directly cleave complement proteins. J Infect Dis 209:876–886. doi: 10.1093/infdis/jit569. [DOI] [PubMed] [Google Scholar]
- 32.Kraiczy P, Stevenson B. 2013. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: structure, function and regulation of gene expression. Ticks Tick Borne Dis 4:26–34. doi: 10.1016/j.ttbdis.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bykowski T, Woodman ME, Cooley AE, Brissette CA, Brade V, Wallich R, Kraiczy P, Stevenson B. 2007. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete's mammal-tick infection cycle. Infect Immun 75:4227–4236. doi: 10.1128/IAI.00604-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seling A, Siegel C, Fingerle V, Jutras BL, Brissette CA, Skerka C, Wallich R, Zipfel PF, Stevenson B, Kraiczy P. 2010. Functional characterization of Borrelia spielmanii outer surface proteins that interact with distinct members of the human factor H protein family and with plasminogen. Infect Immun 78:39–48. doi: 10.1128/IAI.00691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hallström T, Siegel C, Mörgelin M, Kraiczy P, Skerka C, Zipfel PF. 2013. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 4:e00481-13. doi: 10.1128/mBio.00481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hallström T, Haupt K, Kraiczy P, Hortschansky P, Wallich R, Skerka C, Zipfel PF. 2010. Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. J Infect Dis 202:490–498. doi: 10.1086/653825. [DOI] [PubMed] [Google Scholar]