

Abstract
Uropathogenic strains of Escherichia coli (UPEC) are the major cause of bacteremic urinary tract infections. Survival in the bloodstream is associated with different mechanisms that help to resist serum complement-mediated killing. While the phenotypic heterogeneity of bacteria has been shown to influence antibiotic tolerance, the possibility that it makes cells refractory to killing by the immune system has not been experimentally tested. In the present study we sought to determine whether the heterogeneity of bacterial cultures is relevant to bacterial targeting by the serum complement system. We monitored cell divisions in the UPEC strain CFT073 with fluorescent reporter protein. Stationary-phase cells were incubated in active or heat-inactivated human serum in the presence or absence of different antibiotics (ampicillin, norfloxacin, and amikacin), and cell division and complement protein C3 binding were measured by flow cytometry and immunofluorescence microscopy. Heterogeneity in the doubling times of CFT073 cells in serum enabled three phenotypically different subpopulations to be distinguished, all of them being recognized by the C3 component of the complement system. The population of rapidly growing cells resists serum complement-mediated lysis. The dominant subpopulation of cells with intermediate growth rate is susceptible to serum. The third population, which does not resume growth upon dilution from stationary phase, is simultaneously protected from serum complement and antibiotics.
INTRODUCTION
The immune system has several pathways for recognizing and killing pathogenic bacteria. However, some pathogenic bacteria can maintain infection in mammalian hosts despite inflammation, specific antimicrobial mechanisms, and a robust adaptive immune response and can therefore give rise to persistent infection (1). Uropathogenic Escherichia coli (UPEC) causes recurrent urinary tract infections that can progress from the lower to the upper urinary tract and can lead to the dissemination of bacteria into the bloodstream.
The complement system is part of the defense against invading pathogens, with an essential role in both innate and adaptive immunity (2). It is composed of more than 40 plasma and membrane proteins. It can be activated via three distinct routes: the classical (antibody dependent), lectin, and alternative pathways. Activation of complement cascades leads to the formation of the key component C3b on the bacterial surface, which stimulates phagocytosis. Late complement components (C5 to C9 proteins) are also activated via C3b, resulting in the formation of the membrane attack complex (MAC) causing cell lysis (2). The primary source of complement is blood, but complement proteins are also synthesized by a variety of other cell types and tissues (3).
Bacterial resistance to serum complement killing depends on the presence or absence of antigenic outer membrane proteins (4). Pathogens often resist recognition and subsequent complement activation owing to their surface capsular polysaccharide, which masks underlying structures and by itself activates complement poorly (5–7). In addition, secretion of the exopolysaccharide colanic acid protects UPEC from complement-mediated killing (8). Modification of lipopolysaccharide (LPS) is also important for complement evasion (9, 10). It has been shown that serum sensitivity depends on the bacterial growth phase; cells are more readily killed by serum during early logarithmic phase (11–14). However, there are examples showing that exponential-phase cells are more resistant to complement-mediated killing than stationary-phase cells (15, 16). That phenomenon is largely explained by the growth-phase-dependent expression of antigens, capsule, and LPS modification (14, 15). Cell size can also be an important determinant of complement-mediated killing, since larger or aggregated cells have more complement protein C3b on their membranes (17).
Some of the mechanisms that contribute to immune evasion can also help the bacteria survive antibiotic treatment. For example, the inability of many antibiotics to cross host membranes readily limits their effectiveness against intracellular bacteria (18). Also, changes in bacterial outer membrane composition can simultaneously influence the binding of complement proteins and the influx of antibiotics (4). Importantly, antibiotic-tolerant bacteria are not able to grow in the presence of drugs, but once the antibiotic concentration drops, surviving persister cells restore the population (19). Two clinical studies have demonstrated that patients with long-term infections had pathogen strains with elevated persister levels, indicating that persisters in a bacterial population might account for the failure of antibiotic treatment in chronic infections (20, 21). These studies investigated cystic fibrosis patients infected with Pseudomonas aeruginosa (20) and cancer patients with oral thrush caused by Candida albicans (21). In UPEC, it has been previously shown that recurrent community-acquired urinary tract infections are mainly due to persisting UPEC clones (22) and the presence of intracellular bladder reservoirs for UPEC in humans (23). Furthermore, the persistence of UPEC in bladder has been shown in a mouse urinary tract model, where the combined effect of antibiotics and immune system was investigated (18).
The question remains: why are these antibiotic persisters not killed by the immune system? It is generally believed that the immune system kills planktonic persisters, but biofilm persister cells are protected from the host defenses by the exopolymer matrix (24). It has also been proposed that dormant intracellular bacteria are simultaneously protected from antibiotics and the immune system (18). While the phenotypic heterogeneity of bacteria influences antibiotic tolerance, the possibility that it also makes cells refractory to the immune system has not been considered. We therefore sought to determine whether the heterogeneity of bacterial cultures manifested by the presence of persisters also influences bacterial targeting by the serum complement system. In addition, we determined whether the number of persisters is reduced by antibiotic treatment in human serum.
We demonstrate here that the complement system kills most dividing cells, but minor subpopulations of dormant and rapidly dividing cells evade complement killing. Importantly, stationary-phase dormancy protects intrinsically susceptible UPEC cells from killing by both antibiotics and the serum complement system.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth media.
E. coli strains CFT073 (25) and BW25113 [F− Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514] (26) were used in the experiments. To study cell division, the CFT073 strain was transformed with the plasmid pETgfp-mut2AGGAGG(3) carrying a green fluorescent protein (GFP) mut2 gene under the control of an IPTG (isopropyl-d-thiogalactopyranoside)-inducible promoter and a kanamycin resistance marker (27). For plasmid selection, kanamycin (25 μg/ml; Amresco) was added to the growth medium. To induce GFP, 1 mM IPTG was added when indicated. To test antibiotic sensitivity, ampicillin sodium (Sandoz), norfloxacin (Sigma), or amikacin (Sigma) was prepared as an aqueous solution and used at the indicated concentrations. To inhibit cell growth the bacteriostatic antibiotic chloramphenicol (AppliChem) was added at final concentration of 50 μg/ml. Bacteria were grown at 37°C in filtered Lennox lysogeny broth (LB) medium (Difco Laboratories). Sterile 1× phosphate-buffered saline (PBS) was used as a washing and incubation buffer when necessary.
Preparation of bacterial DMSO stocks.
Overnight cultures were diluted 1:100 in fresh LB medium and grown aerobically to exponential phase. At an optical density at 600 nm (OD600) of 0.8, dimethyl sulfoxide (DMSO) was added to a final concentration of 8%, and the culture was immediately frozen in 120-μl aliquots at −80°C. Stocks were stored up to 6 months.
Serum complement.
Blood was obtained by venipuncture from three healthy volunteers (after informed consent was obtained), allowed to clot at room temperature for 30 min, and then centrifuged at 1,700 × g for 10 min at 20°C. The serum layer was collected, divided into aliquots, and frozen at −80°C until use. A suitable volume of serum was thawed immediately before the experiment. Heat-inactivated serum (HIS) was prepared by incubating the serum at 56°C for 30 min to inactivate complement.
MIC measurements.
MIC tests were performed according to the broth microdilution method, except that LB medium was used (28). The MICs of ampicillin, norfloxacin, and amikacin for CFT073 strain were determined in 96-well microtiter plate. Growth was assessed after incubation for 18 h, and the MIC value was determined.
Serum and antibiotic sensitivity.
Cells from DMSO stock were diluted 1:100 in filtered LB medium and grown aerobically to the stationary phase (20 h). Cells were harvested, washed, and diluted in 1× PBS or LB medium so that the final OD600 in the assays was 0.05. The final percentage of serum or HIS was 50% in PBS. Antibiotics were added at the indicated concentrations at time point zero. The cells were incubated at 37°C without shaking. Samples were withdrawn at the indicated times, diluted in sterile 1× PBS, and plated on LB plates. Colonies were counted on the following day after overnight incubation at 37°C.
Cell division assessed by flow cytometry.
To measure cell division, the CFT073 strain carrying GFP encoding plasmid pETgfp-mut2AGGAGG(3) was used. Experiments were carried out as described above for the serum and antibiotic sensitivity assay, except that the overnight cultures were supplemented with kanamycin (25 μg/ml) and 1 mM IPTG to induce GFP expression. Samples were mixed with an equal volume of 30% glycerol in 1× PBS and frozen at −80°C until further analysis with an LSR II flow cytometer (BD Biosciences). Populations of bacterial cells were gated on GFP fluorescence (GFP-H) and side scatter (SSC-H) plots. GFP was excited with a 488-nm laser, and emission was measured using a 530/30-nm band-pass filter. The software package Flowing Software was used to visualize and analyze the data. The distribution of the fluorescence level of events (single cells) is presented as a histogram consisting of 376 repartition bins. The number of events with the respective GFP fluorescence levels in 1 ml of cell culture grown under particular conditions is shown.
SEM of bacteria with or without serum treatment.
To investigate the morphology of intact and lysed bacteria, scanning electron microscopy (SEM) experiments were performed. The CFT073 strain carrying plasmid pETgfp-mut2AGGAGG(3) was used. Experiments were carried out as described above for the serum and antibiotic sensitivity assay, except that the overnight cultures were supplemented with kanamycin (25 μg/ml) and 1 mM IPTG to induce GFP expression. Bacterial suspensions (100 μl) were withdrawn at the indicated time points, and slides were prepared with Cytospin 4 (Thermo Scientific, USA). After a cytospinning step at 2,000 rpm for 3 min, the bacteria were fixed with freshly prepared 4% paraformaldehyde for 30 min at room temperature. All samples were washed three times with 1× PBS, dehydrated with a series of graded ethanol solutions, and dried in vacuum oven for 30 min. Samples were sputtered with 5 nm of gold using a high vacuum coater (EM ACE600; Leica, Germany) and examined with a scanning electron microscope (TM 3000; Hitachi, Japan).
Immunofluorescent detection of serum complement protein C3 binding.
To identify complement C3b deposition, the same experiments and sample preparation methods were used as for SEM analysis. However, after fixation with 4% paraformaldehyde, the slides were washed with 1× PBS for 5 min and blocked in PBS containing 2% bovine serum albumin for 15 min at room temperature. The samples were then incubated with 1:100-diluted mouse monoclonal complement C3b antibody (6C9; Thermo Scientific) in a blocking buffer for 1 h at room temperature in closed containers. After three washes with 1× PBS, the slides were incubated with secondary goat anti-mouse IgG antibodies labeled with red-fluorescent Alexa Fluor 568 dye (1:1,000 dilution in blocking buffer) for 1 h at room temperature in closed containers. Finally, the slides were washed three times with 1× PBS, moistened with 3 μl of 1× PBS, covered with slips, and sealed with nail polish. Samples were stored in covered closed containers at 4°C.
Fluorescence was observed, and images were acquired using the confocal fluorescence microscope LSM710 (Carl Zeiss, Munich, Germany) and Zen software (Zeiss). To detect GFP excitation, a 488-nm laser and a 484- to 571-nm emission filter were used. To detect Alexa Fluor 568 (C3 signal) excitation, a 561-nm laser and a 575- to 708-nm emission filter were used. The image resolution was 52.72 nm per pixel.
Photographed bacterial cells were examined using Cell Profiler (GPL, v2 [public license]) (29). At a first approximation, the cells were recognized using the GFP signal. Objects outside the diameter range (5 to 200 pixels) were ignored. The extent of C3 signal was limited to 10 pixels away from the edges of cells recognized according to the GFP. Approximately 7,000 cells were examined during C3 antibody-binding assays. In another analysis, C3 binding objects (single or clumped live cells or membranes of dead cells) were recognized using the C3 signal, and the GFP intensity of the objects was measured.
Statistics.
Since the distribution of the CFU and the percentages were skewed, the variables were log transformed to achieve normal distribution. To calculate the significance of differences between two conditions, a Student t test was performed. P values were calculated using a two-sample equal variance type of t test and two-tailed distribution. A Bonferroni correction for multiple testing was used, and P values of <0.0127 were considered statistically significant.
RESULTS
Subpopulation of CFT073 cells is tolerant to human serum.
In our study, we used the previously described UPEC strain CFT073 (O6:K2:H1) (25, 30). The CFT073 strain was isolated from the blood of a patient with acute pyelonephritis, so it is representative of UPEC strains that can cause bacteremia (25). CFT073 has been reported to be serum resistant (6, 8, 9, 31, 32). However, the experimental conditions for defining serum resistance are not standardized; the growth phase of the harvested bacteria, bacterial cell density, serum concentration, and incubation time can influence the number of cells that survive in the presence of serum (11–14, 33). We decided to test the survival of stationary-phase CFT073 cells (5 × 107/ml) in 50% serum, which is commonly used. We monitored cell survival over the time course to see whether CFT073 cells are always serum resistant or whether this depends on specific conditions.
Most of the stationary-phase CFT073 cells survived serum treatment during the first hour, when the cells were in lag phase and not yet dividing (Fig. 1). When CFT073 cells were incubated further (>1 h), cell growth was observed as indicated by the increased CFU counts in LB medium and heat-inactivated serum (HIS) (Fig. 1). Simultaneously, viable cell numbers started to decrease in the presence of serum (Fig. 1).
FIG 1.
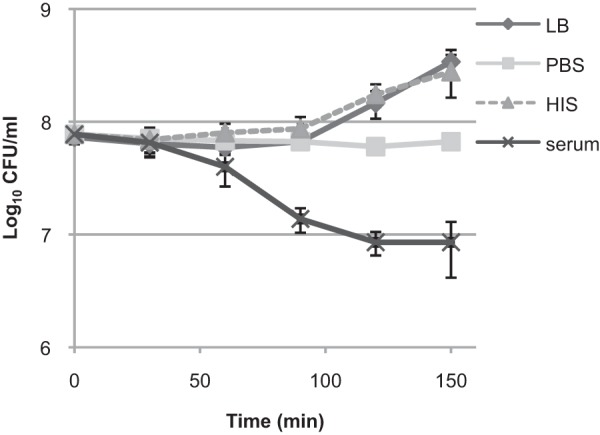
Serum killing coincides with the start of bacterial growth. E. coli CFT073 stationary-phase cells were diluted in either fresh LB medium, PBS, 50% HIS in 1× PBS, or 50% serum in 1× PBS and grown statically at 37°C without shaking. The CFU were determined at the indicated time points. The averages and standard deviations of at least three independent experiments are presented.
The effect of serum on the bacteria was triphasic. The reaction started with a lag phase for 60 min during which the bacteria were not killed. The cells were then killed efficiently between 60 and 90 min and then at a slower rate between 120 and 150 min (Fig. 1). This raises the question of whether surviving cells are really more tolerant to serum killing or whether the complement proteins are exhausted and some cells survive only because there is not enough complement protein to kill all of the susceptible cells. To address this, we repeated our experiment with a 10-fold-lower bacterial CFU count and observed enhanced, but similarly reduced, killing at between 120 and 150 min (see Fig. S1A and B in the supplemental material). Furthermore, when serum-treated samples were supplemented with additional fresh serum after 90 min, the triphasic killing pattern remained (see Fig. S1A and B in the supplemental material). In addition, the nonpathogenic laboratory E. coli strain BW25113 (stationary-phase cells at a concentration of 109 CFU/ml) was readily killed (the CFU level was reduced by 6 orders of magnitude) by 20% serum over 30 min. These results suggest that in our experimental setup, the amount of serum protein per bacterial cell is somewhat limiting, but the CFT073 strain has a subpopulation of cells with enhanced serum tolerance.
Our results show that the CFT073 strain tolerates serum since the cells are not killed during lag phase. However, it is not strictly serum resistant because the cells can be killed during active growth. At the same time, a subpopulation of the cells is tolerant to serum killing.
The combined effect of antibiotics and serum depends on the antibiotic.
The existence of a serum-tolerant subpopulation raises the questions: (i) can the complement system kill antibiotic persisters, and (ii) do antibiotics eradicate serum persisters? To answer these questions, we counted the surviving cells after 150 min of incubation in the presence or absence of active serum in combination with the cell wall-targeting antibiotic ampicillin, the DNA replication-inhibiting antibiotic norfloxacin, and the translation-inhibiting antibiotic amikacin. These antibiotics were chosen for analysis, since they are all bactericidal and represent different antibiotic classes. Furthermore, these antibiotics have been used in several important studies investigating persisters and persistent infections (34–38). The MICs for CFT073 strain in LB medium were as follows: amikacin, 8 μg/ml; ampicillin, 2 μg/ml; and norfloxacin, 0.125 μg/ml.
Figure 2 shows the survival of bacterial cells at different concentrations of each antibiotic. The addition of ampicillin to the serum reduced the number of surviving cells by about 1 order of magnitude (Fig. 2A). This suggests that some serum-tolerant cells are killed by ampicillin. An additive effect of serum to ampicillin is apparent when the numbers of persisters in HIS and serum are compared, indicating that some ampicillin persisters are killed by serum (Fig. 2A). The bacteria are killed more efficiently in LB medium than in HIS (Fig. 2A). Growth resumption rates in different growth media affect the number of ampicillin persisters (39). Therefore, we can assume that the slower growth resumption in HIS compared to LB medium could result in more ampicillin persisters.
FIG 2.
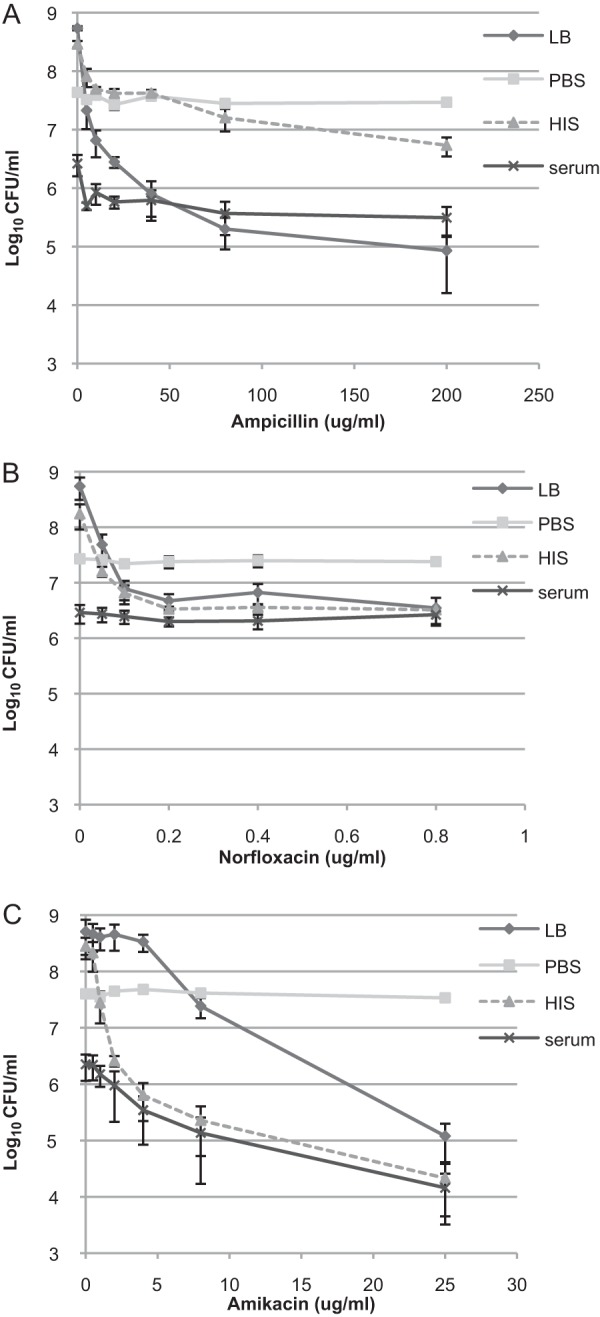
Subpopulation of cells surviving antibiotics and complement killing. Cells were grown to stationary phase in LB medium and diluted in fresh LB medium or PBS supplemented or not with HIS or serum (final concentration, 50%). Cells were incubated in the presence of ampicillin (A), norfloxacin (B), or amikacin (C). The number of surviving cells (CFU/ml) was determined after 150 min by plating. The averages and standard deviations of at least three independent experiments are presented.
Next, we measured the number of persisters after we incubated the cells with norfloxacin (Fig. 2B). The number of norfloxacin persisters was similar under all growth-promoting conditions: LB medium, HIS, and serum (Fig. 2B). This finding suggests that the same subpopulation survives treatment by both serum and norfloxacin.
Finally, we measured the number of amikacin persisters in different media (Fig. 2C). Surprisingly, in HIS and in serum the number of CFU started to decrease at lower amikacin concentrations than in LB medium (Fig. 2C). This indicates that both serum and HIS sensitize the bacteria to amikacin. At high amikacin concentrations, the numbers of surviving bacteria are similar in HIS and serum, suggesting that bacteria surviving the antibiotic treatment are also refractory to serum. We also confirmed that the antibiotics do not kill nongrowing cells since bacteria incubated in PBS were refractory to ampicillin, norfloxacin, and amikacin (Fig. 2A to C).
Serum preferentially kills cells with average growth rates.
The growth of bacteria in HIS demonstrated that this serum serves as a growth substrate (Fig. 1). Active serum has both killing- and growth-promoting effects, which are indistinguishable in experiments where only CFU counts are determined. The final CFU count in serum is the outcome of both the increase of CFU due to cell division and the decrease due to killing by complement. Therefore, we investigated the causes of the final CFU count to determine whether a random population survived serum treatment or whether these were nondividing cells as in the case of antibiotic persisters. We used CFT073 cells carrying plasmid pETgfp-mut2AGGAGG(3), which encodes GFP under the control of the IPTG-inducible promoter. This strain allowed us to monitor cell division using a previously established GFP dilution method (39, 40). The induction of GFP production using IPTG was performed only during the initial growth to the stationary phase.
Stationary-phase cells carrying pETgfp-mut2AGGAGG(3) and filled with GFP were diluted in fresh IPTG-free medium and grown as in the previous experiment. The cells were analyzed at the beginning of the experiment, which allowed us to determine the initial GFP and side scatter (SSC) levels to gate the cells before they started dividing (P1 on Fig. 3A to E). When cells were kept in PBS for 150 min, most of the events identified by flow cytometry had similar high green fluorescence levels and low SSC, as at the zero time point (Fig. 3A and B). Flow cytometry measurements of cells grown in LB medium for 150 min showed high-SSC and low-green-fluorescence (GFP-H) events, indicating that most of the cells grown in LB medium had divided (Fig. 3C). These measurements allowed us to set the second region, P3, for actively dividing cells. Flow cytometry of cells grown in 50% HIS showed somewhat less division than in LB medium, and the extent of SSC was lower (Fig. 3D). Most cells grown in HIS had GFP and SSC values intermediate between those of the nondividing (P1) and actively dividing cells (P3) and were in the region P2 (Fig. 3D). Samples treated with serum showed events in regions P1 and P3; events with very low GFP fluorescence and low SSC also appeared, most likely representing dead cells and cell debris (Fig. 3E). The combined number of events detected by flow cytometry was comparable with CFU in all samples, indicating that all three regions (P1, P2, and P3) contain live cells (see Fig. S2 in the supplemental material).
FIG 3.

A bacterial population that survives serum treatment is enriched in rapidly dividing and nondividing cells. Stationary-phase CFT073 cells [carrying plasmid pETgfp-mut2AGGAGG(3)] were diluted in PBS, LB medium, 50% HIS, or 50% serum. The cells were incubated at 37°C without shaking for 150 min, and their fluorescence was measured by using flow cytometry. Density blots of GFP fluorescence (GFP-H) and the SSC parameter (SSC-H) are shown for cells in PBS at zero time (A), in PBS after 150 min (B), in LB medium (C), in HIS (D), or in serum (E). Each dot represents the fluorescence of a single event (particle). (F) The distribution of the fluorescence level of events (single cells) analyzed by flow cytometry is presented as a histogram consisting of 376 repartition bins. The number of events with the respective GFP fluorescence levels in 1-ml cell cultures grown under particular conditions is shown. (G and H) Scanning electron micrographs illustrate the morphology of the E. coli cells after 150 min of incubation in 50% HIS (G) and in 50% serum (H). Arrows indicate the different bacterial cells during lysis.
In terms of GFP levels, there are clear differences in distribution between cells surviving in serum and those grown in HIS (Fig. 3D to F). The numbers of low-GFP (rapidly dividing cells) and high-GFP (dormant cells) fluorescence events are comparable. However, events with intermediate GFP levels are missing for cells grown in serum (Fig. 3F). We can conclude that the complement system kills most of the dividing cells (P2), whereas minor subpopulations of dormant (P1) and rapidly dividing (P3) cells evade complement killing.
To confirm that the loss of subpopulation of CFT073 cells during serum treatment is due to cell lysis, bacterial cultures were inspected using SEM. Bacterial cells in HIS had the expected rod shape, but bacteria in serum had changed morphology (Fig. 3G and H). In addition to normal bacterial cells, different bacterial cells were observed in serum: (i) cells that had lost some of their integrity and were dented; (ii) cells that had lost their normal shape and were ruptured; and (iii) cells that were misshaped and completely lysed (Fig. 3H). These modified and lysed cells were not observed in HIS samples (Fig. 3G).
Nondividing cells preferentially survive combined treatment with serum and antibiotics.
We also analyzed the cell division profile after antibiotic treatment. Stationary-phase cells carrying the pETgfp-mut2AGGAGG(3) and filled with GFP were diluted in fresh IPTG-free medium and incubated in the presence or absence of antibiotics for 150 min. Antibiotics were used at the following concentrations: ampicillin, 200 μg/ml; norfloxacin, 5 μg/ml; and amikacin, 5 μg/ml. After the incubation, the numbers and fluorescence levels of cells were determined by flow cytometry, and the CFU counts were determined by plating.
First, we inspected cells incubated in PBS. As expected, these cells were not dividing, and the addition of antibiotics did not change the fluorescence level distribution or the number of events per volume of sample (see Fig. S3A in the supplemental material). The CFU counts and the numbers of GFP-positive cells (as determined by flow cytometry) were approximately equal in PBS in the presence or absence of antibiotics, indicating that all cells were viable and able to form colonies after the antibiotics were removed (see Fig. S3A and B in the supplemental material).
Next, we inspected the division profile of cells incubated in growth-supporting media. In HIS there was a decrease of ∼2 orders of magnitude in the number of dividing cells (events with low GFP fluorescence) when antibiotics were added (Fig. 4A). Ampicillin reduced the number of dividing cells in HIS, indicating that these cells have been lysed (Fig. 4A), although the number of nondividing (high GFP content) cells was not affected (Fig. 4A). In contrast to the ampicillin-treated cultures, the number of cells with high GFP content was increased after incubation in HIS, together with amikacin or norfloxacin (Fig. 4A). This indicates that norfloxacin and amikacin inhibited growth resumption.
FIG 4.

Cell division profile of persister cells indicates survival of nondividing cells. Cells were grown to stationary phase in LB medium and diluted in 50% HIS (A and B) or 50% serum (C and D). Cells were incubated without antibiotics (no AB) and in the presence of ampicillin (Amp; 200 μg/ml), norfloxacin (Nor; 5 μg/ml), or amikacin (Ami; 25 μg/ml).The distributions of the fluorescence levels of events (single cells) analyzed by flow cytometry are presented as histograms consisting of 376 repartition bins. The numbers of events with the respective GFP fluorescence levels in 1-ml cell cultures from HIS (A) or serum (C) are shown. Differently colored histograms represent conditions as follows: filled gray, without antibiotics; yellow, ampicillin; green, norfloxacin; and dashed red, amikacin. The norfloxacin and amikacin lines mostly overlap. (B and D) After 150 min, the CFU were determined by plating, and the number GFP-positive events was measured by flow cytometry (FACS events).
Treatment with a bactericidal antibiotic in growth-supporting media has been shown to leave a large fraction of cells in a nonrecoverable state; therefore, the number of events detected by flow cytometry can exceed the number of persister cells that form colonies after plating on a solid medium (41). In the case of norfloxacin, the ratio of CFU to GFP-positive events was almost 1 in HIS (Fig. 4B). We can therefore say that norfloxacin inhibited cell growth, but the cells retained their ability to form colonies. When they were incubated in the presence of amikacin in HIS, there was a difference of >2 orders of magnitude between CFU and GFP-positive events detected by flow cytometry (Fig. 4B). This means that amikacin killed most of the cells in HIS, but these were still visible by flow cytometry as cells containing high levels of GFP because they were not lysed as they were after ampicillin treatment (Fig. 4A). The division profiles of cells incubated with antibiotics in LB medium were similar to those in HIS (see Fig. S3C and D in the supplemental material).
Finally, the cell division profile in serum was determined. In serum alone there were two surviving subpopulations: nondividing and rapidly dividing cells (Fig. 3E and 4C). When serum and antibiotic treatment were applied simultaneously, the rapidly dividing subpopulation disappeared while the nondividing subpopulation survived (Fig. 4C). In serum, the number of nondividing cells was increased after treatment with norfloxacin or amikacin (Fig. 4C). Importantly, comparison of CFU and events detected by flow cytometry shows that in the presence of amikacin most of the cells detected by flow cytometry are not viable (Fig. 4D). In contrast, the nondividing cells induced by norfloxacin remained viable and were also refractory to serum (Fig. 4C and D). This result indicates that growth inhibition by an antibiotic can make bacteria resistant to lysis by serum. We conclude that the nondividing antibiotic persisters are not cleared by the complement system as efficiently as growing cells.
Differences in C3 deposition are not responsible for selective lysis of bacteria.
From the antibiotic and serum cotreatment it became evident that antibiotic persisters are not efficiently killed by human serum complement. It is not clear whether bacterial cells are refractory to complement killing because they are not recognized and subsequently opsonized, or whether they avoid complement-mediated lysis. In order to determine whether serum complement recognizes all of the CFT073 strain cells equally, C3 deposition was measured using sequential C3 antibody staining with Alexa 568 and immunofluorescence microscopy.
First, C3 deposition on CFT073 cells in serum, HIS, and PBS without antibiotics was investigated. Complement C3 deposition was observed on cells incubated in human serum after only 30 min compared to the cells incubated in HIS (data not shown). In order to discriminate between dividing and nondividing subpopulations, a 150-min time point was chosen for further analysis. The presence of GFP inside the intact cells enabled us to visualize nondividing cells with high GFP intensity, dividing cells with low GFP intensity, and lysed cells with no detectable GFP (Fig. 5A). In negative controls, cells incubated in HIS, or cells incubated in PBS, no specific C3 binding was detected (Fig. 5B), and the relative fluorescence of Alexa 568 was less than 0.05 U, which was taken as the threshold for determining C3-binding cells. After 150 min, almost all GFP-containing cells (99%) were recognized by human serum complement, and C3 protein was bound to them (Fig. 5A and B). In addition, C3-bound cells or cell membranes with no detectable GFP fluorescence were observed (Fig. 5A). This is consistent with CFU counts showing that most of the cells were lysed by serum (Fig. 1). In order to determine whether this C3 deposition level was sufficient for killing, we compared the C3 levels of lysed (no GFP) and intact cells. Figure 5C shows that the Alexa 568 intensities of lysed and intact cells were comparable; hence, unequal C3 deposition is not responsible for selective killing.
FIG 5.

Recognition and opsonization of CFT073 strain bacterial cells by serum complement. C3 deposition was measured by sequential C3 antibody staining and immunofluorescence microscopy. Cells were grown to stationary phase in LB medium and diluted in fresh medium (in PBS supplemented with either 50% HIS or 50% serum or in PBS alone) and incubated for 150 min at 37°C. (A) Immunofluorescence micrographs of GFP and C3-bound Alexa 568 fluorescence. Cells were grown in serum in the presence of ampicillin (200 μg/ml), norfloxacin (5 μg/ml), or amikacin (25 μg/ml), and C3 binding was detected by using immunofluorescence. From left to right is shown the GFP fluorescence inside the cells (visible GFP signal indicates intact cells) and the C3 deposition on cells (together with no GFP signal enables lysed cells to be identified); figure enlargements for GFP and C3 tile figures are also shown. Red dashed boxes show the exact locations of the figure enlargements on the tile figures. (B) Dot plot of C3-bound Alexa 568 and GFP fluorescence of cells incubated in HIS, PBS, or serum. Each dot represents the mean intensity (GFP and C3-bound Alexa 568) per cell measured. The dashed horizontal line indicates threshold value for cells with specifically bound C3. The dashed vertical line separates subpopulations of nondividing (high GFP levels) and dividing (low GFP levels) cells. (C) Relative distribution of cells by mean C3-bound Alexa 568 fluorescence. The objects examined were distinguished by the presence or absence of GFP. Objects with a mean GFP fluorescence higher than 0.01 (includes all cells shown in panel B) were considered live (GFP-positive cells). Objects with a GFP fluorescence ≤0.01 were considered lysed.
Next, we analyzed the C3 deposition on cells incubated in human serum in the presence of antibiotics. Intense C3 deposition was detected on cells incubated in serum in the presence of ampicillin, norfloxacin, or amikacin (Fig. 5A). Flow cytometry had revealed that in the presence of ampicillin, growing cells (expressing low levels of GFP) disappear (Fig. 4C). As expected, we found no low-GFP-level cells in ampicillin-treated samples. Instead, the number of lysed cells (manifested by the absence of GFP) had increased (Fig. 5A). The opposite effect was seen in norfloxacin- and amikacin-supplemented serum cultures: fewer lysed cells were observed (Fig. 5A). These results indicate that C3 binding is not enough for serum-dependent cell lysis. Even cells that are killed by amikacin remain intact (retain high GFP fluorescence) despite the presence of C3 on the surface (Fig. 4C and 5A).
Exponentially growing cells are susceptible to serum and require active protein synthesis for survival.
We observed that growth resumption coincides with serum-mediated killing (Fig. 1). To investigate how growth makes cells susceptible to complement-mediated lysis, the survival of exponential-phase cells was tested. We used the GFP dilution method as described in previous sections to follow cell division and the number of surviving cells simultaneously. Flow cytometry showed that after 60 min the cells in serum had divided as in HIS, but there were only half the number of cells (Fig. 6A). This indicated that some cells were lysed during cell division. The difference in the number of cells in HIS and serum was even more pronounced after 120 min; ca. 75% of cells were lysed in serum (Fig. 6A). Importantly, cell division profiles indicated that most cells surviving in serum had divided more actively than the population average in HIS (Fig. 6A).
FIG 6.
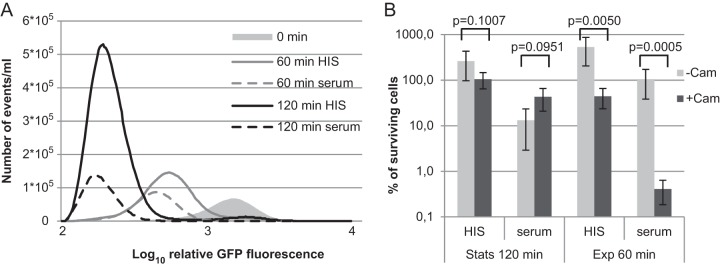
Exponentially growing cells are susceptible to serum. (A) Cells were grown to exponential phase in LB medium and diluted in 50% HIS or 50% serum. Cell division profiles of exponential-phase cells were determined at the start and 60 and 120 min after dilution. The relative number of events with particular GFP fluorescence levels in the same culture volume is shown. (B) Cells were grown to stationary (Stats) or exponential (Exp) phase in LB medium and diluted in 50% HIS or serum. To inhibit cell growth, chloramphenicol (Cam; 50 μg/ml) was added to one parallel sample. The CFU were counted after the indicated time by plating, and the percentage of surviving cells compared to the zero time point is shown. The averages and standard deviations of at least three independent experiments are presented.
Next, we used a bacteriostatic antibiotic, chloramphenicol, to inhibit growth and division without killing the cells. Parallel experiments were performed with stationary- and exponential-phase cells. Stationary-phase E. coli was transferred to fresh medium and chloramphenicol added at time point zero. After 120 min, the CFU were counted. As expected, the addition of chloramphenicol to HIS inhibited the growth of stationary-phase cells but did not kill them (Fig. 6B). Inhibition of cell growth with chloramphenicol slightly increased the number of cells surviving in the presence of serum (Fig. 6B). However, the effect of chloramphenicol on stationary-phase cells was not statistically significant (Fig. 6B). Next, we repeated the experiment with exponential-phase cells. Here, a 60-min time point was chosen because there is no lag phase, and the cells start to divide rapidly. Addition of chloramphenicol to exponential-phase cells growing in HIS stopped cell growth efficiently and even reduced the CFU count (Fig. 6B). When chloramphenicol was added to exponential-phase cells in serum, there was a decrease of >2 orders of magnitude in the number of CFU (Fig. 6B). This suggests that exponential-phase cells need active protein synthesis to counteract serum-mediated killing. The survival of nondividing cells in the presence of serum is characteristic only of stationary-phase persisters. Inhibition of cell growth in exponential-phase does not rescue them from complement-mediated lysis (Fig. 6B).
DISCUSSION
In nature, bacteria are constantly confronted with changes in nutrient availability and the presence of antimicrobials or other damaging agents. Stochastic differentiation of a genetically identical population into subpopulations with distinct phenotypes provides a strong advantage in an unpredictable and fluctuating environment (42, 43). One example of phenotypically different subpopulation is the formation of persisters, i.e., cells that survive treatment with bactericidal antibiotics (36, 44).
It is clear that during antibiotic treatment the immune system contributes heavily to the outcome of therapy. Therefore, in order to persist, bacteria have to evade killing by both antibiotics and the immune system. It has been proposed that biofilm-embedded persisters are the ones that survive antibiotic treatment, but planktonic persisters are killed by the immune system (24). We sought to determine whether the heterogeneity of cultures affects bacterial targeting by the immune system and, if so, what happens to the persisters when antibiotic treatment takes place in the presence of the serum complement system.
First, we determined the serum susceptibility of E. coli strain CFT073. Our results show that CFT073 is not strictly serum resistant as previously reported (6, 8, 9, 31, 32). Rather, only a fraction of cells survived serum treatment (Fig. 1 and Fig. 3H; see also Fig. S1 in the supplemental material). We observed that two subpopulations survived in serum, in contrast to cells grown in HIS: cells with low GFP levels (growing most actively) and cells with high GFP levels (dormant) (Fig. 3D and E). It is important to note that most cells, with moderate GFP levels, were lysed by serum.
To test the hypothesis that active growth makes cells more vulnerable to serum, the survival of exponential-phase cells was tested. CFU counting and flow cytometry revealed the number of GFP-positive cells in serum to be half that in HIS after 60 min (Fig. 6A and B). However, analysis of the GFP levels showed that the cells had divided as rapidly in serum as in HIS, indicating that most of them had been lysed during active growth (Fig. 6A). It has been shown preciously by Miajlovic et al. that when exponential-phase CFT073 cells were incubated in the presence of 50% serum in LB medium, the CFU count did not decrease over 180 min (8). In that study, ∼50 times more cells were incubated with serum than in our experiment (Fig. 6) (8). We repeated the experiment with the same initial cell concentration as in the study by Miajlovic et al. (3 × 108 CFU/ml), and no killing of cells by serum was detected (data not shown). This suggests that complement proteins can become limiting when large numbers of exponential-phase cells are treated with serum. However, clinical data from adult bacteremic patients indicated that bacterial cell numbers in blood are relatively low, typically fewer than 10 CFU/ml (45).
We also determined the serum tolerance of exponential-phase cells in the presence of the bacteriostatic antibiotic chloramphenicol. The addition of chloramphenicol to HIS efficiently stopped cell growth with <3-fold killing of the exponential-phase cells (Fig. 6B). Chloramphenicol increased the killing of exponential-phase cells by serum by >2 orders of magnitude (Fig. 6B). Thus, the survival of nondividing cells in the presence of serum is characteristic only of stationary-phase persisters, and inhibition of growth does not rescue exponential-phase cells from complement-mediated lysis. A transcriptome study of serum-incubated exponential-phase CFT073 cells demonstrated that many RcsB-regulated genes are induced (8). RcsB, either alone or in combination with other regulators, activates transcription of a wide range of genes, including those for colanic acid capsule synthesis, cell division, and some membrane proteins, and it regulates flagellum synthesis negatively (46). The importance of the RcsB regulon in serum survival has been confirmed by the observation that an rcsB mutant survives less well than wild-type CFT073 in serum (8). It has been shown that E. coli K1 cells can resist complement attack by OmpA-dependent binding of complement protein C4bp, which subsequently prevents MAC formation (15). Interestingly, exponential-phase E. coli K1 cells were more resistant to complement attack than stationary-phase cells, suggesting an additional role of growth-phase-dependent expression of the capsule and LPS (15). Our results, together with literature data, suggest that active adaptation mechanisms are needed for the survival of rapidly dividing (exponential phase) cells in serum.
Our first characterization of serum-surviving cells revealed both dormant and dividing cells. Since antibiotic persisters are usually nondividing cells, they might not be killed by serum either. We measured the survival of CFT073 cells in the presence of different antibiotics in a standard laboratory medium, as well as in human serum.
Our results demonstrate an additive effect of serum to ampicillin if the numbers of persisters in HIS and serum are compared, indicating that some ampicillin persisters are killed by serum complement (Fig. 2A). However, norfloxacin persisters were not killed by serum complement (Fig. 2B). Amikacin had the strongest effect on the killing of bacteria in HIS and serum (Fig. 2). Nevertheless, at high (>2 μg/ml) amikacin concentrations the numbers of persisters were similar in serum and HIS, indicating that amikacin persisters were also not killed by serum complement (Fig. 2C). Thus, antibiotic persisters are considerably more resistant to serum complement than the rest of the population. Our study therefore reveals that planktonic antibiotic persisters are not as efficiently cleared by the immune system as the antibiotic-susceptible population. This is different from the current view that antibiotic persisters need to be embedded in biofilm in order to escape the immune system (47).
Several studies have shown that stationary-phase CFT073 cells are resistant to serum complement-mediated killing (6, 9, 31, 32). Importantly, there was no significant growth of cells in HIS during the treatment time (6, 31) (or the data on growth of bacteria in HIS were not presented [9, 32]). This suggests that stationary-phase dormancy protects CFT073 cells from serum complement-mediated killing. A study by Buckles et al. (6) demonstrated that most CFT073 wild-type cells were phagocytosed in the presence of human serum within 90 min. Thus, the results of Buckles et al. support our observation that CFT073 cells are not killed in the absence of growth but are marked with the C3 protein (Fig. 4C and Fig. 5).
The presence of C3 on the bacterial cell membrane and resistance to lysis might constitute an important mechanism for establishing infection. Some UPEC strains have been shown to use C3 opsonization to facilitate uptake by uroepithelial cells (48). Importantly, UPEC can also survive within mouse and human macrophages (49). It has been suggested that chronically infected epithelial cells could thus provide a constant source for reinfection of macrophages, which are unable to eliminate the bacteria completely, thus helping the bacteria to spread (49).
A simplified model based on our results is shown in Fig. 7. We propose that heterogeneous growth resumption and different growth rates of CFT073 cells (in serum) incorporate three phenotypically different subpopulations. Rapidly growing cells are susceptible to antibiotics but resist serum complement-mediated lysis. These cells might be important during the acute phase of infection. Cells that do not resume growth are simultaneously protected from serum complement and antibiotics. They might be important in establishing persistent infection. The dominant subpopulation of cells, with intermediate growth rate, is susceptible to both antibiotics and serum. Although the medium growth-rate population is killed by the complement system, it might still have a role in establishing infection by synthesizing and secreting virulence factors. In Salmonella, the virulence factors needed for host manipulation are expressed in a bistable fashion, leading to a slowly growing subpopulation that expresses virulence genes and a rapidly growing subpopulation that is phenotypically avirulent (50).
FIG 7.
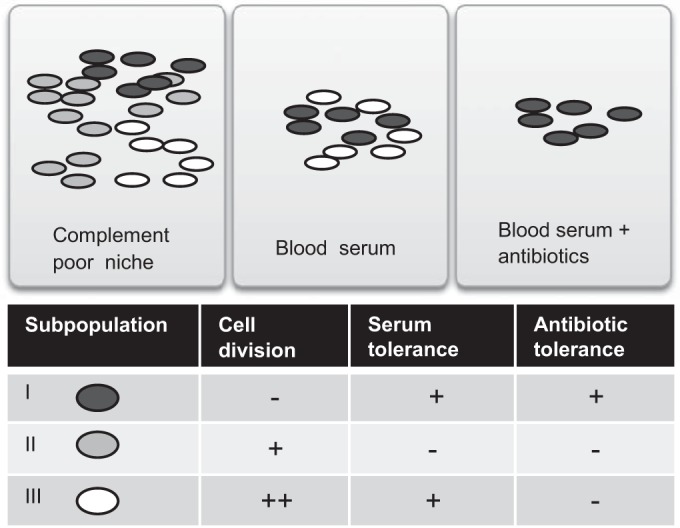
Model of phenotypic heterogeneity of UPEC cells during infection. Phenotypic heterogeneity leads to the formation of three different subpopulations. Dormant nondividing cells are simultaneously protected from serum and antibiotics. The dominant subpopulation of cells with an intermediate growth rate is susceptible to antibiotics and serum. Rapidly dividing cells are susceptible to antibiotics but can resist serum complement-mediated lysis. Bacteria are depicted as dark gray ovals, and lighter gray ovals indicate dilution of the color by cell division.
Several studies have focused on the interplay between antibiotic and serum susceptibility. For example, it has been shown that overexpression of antibiotic efflux pump MexCD-OprJ reduces P. aeruginosa virulence by increasing its susceptibility to complement-mediated killing (51). At the same time, growth in serum has been shown to induce expression of antibiotic efflux pumps in P. aeruginosa and Acinetobacter, suggesting enhanced antibiotic tolerance during infection (52). Beside induction of efflux pumps, the growth in serum has been shown to enhance expression of OprD porin in P. aeruginosa (53). Recent study has shown the mutation in oprD gene in P. aeruginosa conferred resistance to carbapenem antibiotics, as well as enhanced resistance to killing by human serum (54). The relationship between antibiotic and serum sensitivity can be even more complex when population heterogeneity during infection is considered. Recent work on persistent Salmonella infection in a mouse model has revealed that internalization by macrophages can induce phenotypic heterogeneity, leading to either bacterial growth or the formation of nondividing persisters that could provide a reservoir for relapsing infection (55). Another study has shown that nondividing Salmonella survived antibiotic treatment the best, but overall clearance of infection was delayed primarily by abundant subsets of moderately growing Salmonella with partial tolerance (56).
The complex phenotypic heterogeneity described in the present study could be regulated by molecular mechanisms, which could become new targets for treating acute and persistent infections caused by UPEC. Further work is needed to establish the potential role of the phenotypic heterogeneity of UPEC described here in in vivo infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Harry Mobley for generously providing the E. coli CFT073 strain. We thank Hans Priks and Tarmo Tamm for assistance with the SEM. We are grateful to Niilo Kaldalu and Ülo Maiväli for discussing the manuscript.
This study was supported by grant IUT2-22 from the Estonian Research Council and European Regional Development Fund through the Center of Excellence in Chemical Biology. K.K. is supported by the European Social Fund through the Estonian Science Foundation program Mobilitas (MJD321).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02725-14.
REFERENCES
- 1.Young D, Hussell T, Dougan G. 2002. Chronic bacterial infections: living with unwanted guests. Nat Immunol 3:1026–1032. doi: 10.1038/ni1102-1026. [DOI] [PubMed] [Google Scholar]
- 2.Sarma JV, Ward PA. 2011. The complement system. Cell Tissue Res 343:227–235. doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Timar KK, Dallos A, Kiss M, Husz S, Bos JD, Asghar SS. 2007. Expression of terminal complement components by human keratinocytes. Mol Immunol 44:2578–2586. doi: 10.1016/j.molimm.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Liu YF, Yan JJ, Lei HY, Teng CH, Wang MC, Tseng CC, Wu JJ. 2012. Loss of outer membrane protein C in Escherichia coli contributes to both antibiotic resistance and escaping antibody-dependent bactericidal activity. Infect Immun 80:1815–1822. doi: 10.1128/IAI.06395-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merino S, Tomas J. 2010. Bacterial capsules and evasion of immune responses. In Encyclopedia of life sciences. John Wiley & Sons, Ltd., Chichester, United Kingdom. doi: 10.1002/9780470015902.a0000957.pub3. [DOI] [Google Scholar]
- 6.Buckles EL, Wang X, lane MC, Lockatell CV, Johnson DE, Rasko DA, Mobley HL, Donnenberg MS. 2009. Role of the K2 capsule in Escherichia coli urinary tract infection and serum resistance. J Infect Dis 199:1689–1697. doi: 10.1086/598524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miajlovic H, Smith SG. 2014. Bacterial self-defense: how Escherichia coli evades serum killing. FEMS Microbiol Lett 354:1–9. doi: 10.1111/1574-6968.12419. [DOI] [PubMed] [Google Scholar]
- 8.Miajlovic H, Cooke NM, Moran GP, Rogers TR, Smith SG. 2014. Response of extraintestinal pathogenic Escherichia coli to human serum reveals a protective role for Rcs-regulated exopolysaccharide colanic acid. Infect Immun 82:298–305. doi: 10.1128/IAI.00800-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarkar S, Ulett GC, Totsika M, Phan MD, Schembri MA. 2014. Role of capsule and O antigen in the virulence of uropathogenic Escherichia coli. PLoS One 9:e94786. doi: 10.1371/journal.pone.0094786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuura M. 2013. Structural modifications of bacterial lipopolysaccharide that facilitate gram-negative bacteria evasion of host innate immunity. Front Immunol 4:109. doi: 10.3389/fimmu.2013.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis SD, Wedgwood RJ. 1965. Kinetics of the bactericidal action of normal serum on gram-negative bacteria. J Immunol 95:75–79. [PubMed] [Google Scholar]
- 12.Rowley D, Wardlaw AC. 1958. Lysis of gram-negative bacteria by serum. J Gen Microbiol 18:529–533. doi: 10.1099/00221287-18-2-529. [DOI] [PubMed] [Google Scholar]
- 13.DeMatteo CS, Hammer MC, Baltch AL, Smith RP, Sutphen NT, Michelsen PB. 1981. Susceptibility of Pseudomonas aeruginosa to serum bactericidal activity. A comparison of three methods with clinical correlations. J Lab Clin Med 98:511–518. [PubMed] [Google Scholar]
- 14.Tee GL, Scott GK. 1980. Analysis of outer membrane components of Escherichia coli ML308 225 and of a serum-resistant mutant. Infect Immun 28:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wooster DG, Maruvada R, Blom AM, Prasadarao NV. 2006. Logarithmic phase Escherichia coli K1 efficiently avoids serum killing by promoting C4bp-mediated C3b and C4b degradation. Immunology 117:482–493. doi: 10.1111/j.1365-2567.2006.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biedzka-Sarek M, Venho R, Skurnik M. 2005. Role of YadA, Ail, and lipopolysaccharide in serum resistance of Yersinia enterocolitica serotype O:3. Infect Immun 73:2232–2244. doi: 10.1128/IAI.73.4.2232-2244.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalia AB, Weiser JN. 2011. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe 10:486–496. doi: 10.1016/j.chom.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blango MG, Mulvey MA. 2010. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob Agents Chemother 54:1855–1863. doi: 10.1128/AAC.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis K. 2005. Persister cells and the riddle of biofilm survival. Biochemistry 70:267–274. doi: 10.1007/s10541-005-0111-6. [DOI] [PubMed] [Google Scholar]
- 20.Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol 192:6191–6199. doi: 10.1128/JB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lafleur MD, Qi Q, Lewis K. 2010. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob Agents Chemother 54:39–44. doi: 10.1128/AAC.00860-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skjot-Rasmussen L, Hammerum AM, Jakobsen L, Lester CH, Larsen P, Frimodt-Moller N. 2011. Persisting clones of Escherichia coli isolates from recurrent urinary tract infection in men and women. J Med Microbiol 60:550–554. doi: 10.1099/jmm.0.026963-0. [DOI] [PubMed] [Google Scholar]
- 23.Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med 4:e329. doi: 10.1371/journal.pmed.0040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322:107–131. doi: 10.1007/978-3-540-75418-3_6. [DOI] [PubMed] [Google Scholar]
- 25.Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. 1990. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun 58:1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vimberg V, Tats A, Remm M, Tenson T. 2007. Translation initiation region sequence preferences in Escherichia coli. BMC Mol Biol 8:100. doi: 10.1186/1471-2199-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrews JM. 2001. Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48(Suppl 1):S5–S16. doi: 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- 29.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. 2011. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welch RA, Burland V, Plunkett G III, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HL, Donnenberg MS, Blattner FR. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A 99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Debnath I, Norton JP, Barber AE, Ott EM, Dhakal BK, Kulesus RR, Mulvey MA. 2013. The Cpx stress response system potentiates the fitness and virulence of uropathogenic Escherichia coli. Infect Immun 81:1450–1459. doi: 10.1128/IAI.01213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diard M, Baeriswyl S, Clermont O, Gouriou S, Picard B, Taddei F, Denamur E, Matic I. 2007. Caenorhabditis elegans as a simple model to study phenotypic and genetic virulence determinants of extraintestinal pathogenic Escherichia coli. Microbes Infect 9:214–223. doi: 10.1016/j.micinf.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Kilpi MK, Atosuo JT, Lilius EM. 2009. Bacteriolytic activity of the alternative pathway of complement differs kinetically from the classical pathway. Dev Comp Immunol 33:1102–1110. doi: 10.1016/j.dci.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 34.Bigger J. 1944. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244:497–500. doi: 10.1016/S0140-6736(00)74210-3. [DOI] [Google Scholar]
- 35.Moyed HS, Bertrand KP. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J Bacteriol 155:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 37.Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allison KR, Brynildsen MP, Collins JJ. 2011. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jõers A, Kaldalu N, Tenson T. 2010. The frequency of persisters in Escherichia coli reflects the kinetics of awakening from dormancy. J Bacteriol 192:3379–3384. doi: 10.1128/JB.00056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roostalu J, Joers A, Luidalepp H, Kaldalu N, Tenson T. 2008. Cell division in Escherichia coli cultures monitored at single cell resolution. BMC Microbiol 8:68. doi: 10.1186/1471-2180-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orman MA, Brynildsen MP. 2013. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob Agents Chemother 57:3230–3239. doi: 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Acar M, Mettetal JT, van Oudenaarden A. 2008. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet 40:471–475. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- 43.Geisel N, Vilar JM, Rubi JM. 2011. Optimal resting-growth strategies of microbial populations in fluctuating environments. PLoS One 6:e18622. doi: 10.1371/journal.pone.0018622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol 6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reimer LG, Wilson ML, Weinstein MP. 1997. Update on detection of bacteremia and fungemia. Clin Microbiol Rev 10:444–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarke DJ. 2010. The Rcs phosphorelay: more than just a two-component pathway. Future Microbiol 5:1173–1184. doi: 10.2217/fmb.10.83. [DOI] [PubMed] [Google Scholar]
- 47.Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 48.Li K, Zhou W, Hong Y, Sacks SH, Sheerin NS. 2009. Synergy between type 1 fimbriae expression and C3 opsonisation increases internalisation of Escherichia coli by human tubular epithelial cells. BMC Microbiol 9:64. doi: 10.1186/1471-2180-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bokil NJ, Totsika M, Carey AJ, Stacey KJ, Hancock V, Saunders BM, Ravasi T, Ulett GC, Schembri MA, Sweet MJ. 2011. Intramacrophage survival of uropathogenic Escherichia coli: differences between diverse clinical isolates and between mouse and human macrophages. Immunobiology 216:1164–1171. doi: 10.1016/j.imbio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 50.Sturm A, Heinemann M, Arnoldini M, Benecke A, Ackermann M, Benz M, Dormann J, Hardt WD. 2011. The cost of virulence: retarded growth of Salmonella typhimurium cells expressing type III secretion system 1. PLoS Pathog 7:e1002143. doi: 10.1371/journal.ppat.1002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez-Ramos I, Mulet X, Moya B, Barbier M, Oliver A, Alberti S. 2014. Overexpression of MexCD-OprJ reduces Pseudomonas aeruginosa virulence by increasing its susceptibility to complement-mediated killing. Antimicrob Agents Chemother 58:2426–2429. doi: 10.1128/AAC.02012-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blanchard C, Barnett P, Perlmutter J, Dunman PM. 2014. Identification of Acinetobacter baumannii serum-associated antibiotic efflux pump inhibitors. Antimicrob Agents Chemother 58:6360–6370. doi: 10.1128/AAC.03535-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukuoka T, Ohya S, Narita T, Katsuta M, Iijima M, Masuda N, Yasuda H, Trias J, Nikaido H. 1993. Activity of the carbapenem panipenem and role of the OprD (D2) protein in its diffusion through the Pseudomonas aeruginosa outer membrane. Antimicrob Agents Chemother 37:322–327. doi: 10.1128/AAC.37.2.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skurnik D, Roux D, Cattoir V, Danilchanka O, Lu X, Yoder-Himes DR, Han K, Guillard T, Jiang D, Gaultier C, Guerin F, Aschard H, Leclercq R, Mekalanos JJ, Lory S, Pier GB. 2014. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc Natl Acad Sci U S A 110:20747–20752. doi: 10.1073/pnas.1221552110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW. 2014. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343:204–208. doi: 10.1126/science.1244705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Claudi B, Sprote P, Chirkova A, Personnic N, Zankl J, Schurmann N, Schmidt A, Bumann D. 2014. Phenotypic variation of salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 158:722–733. doi: 10.1016/j.cell.2014.06.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.