

Abstract
Cyclo(Phe-Pro) (cFP) is a secondary metabolite produced by certain bacteria and fungi. Although recent studies highlight the role of cFP in cell-to-cell communication by bacteria, its role in the context of the host immune response is poorly understood. In this study, we investigated the role of cFP produced by the human pathogen Vibrio vulnificus in the modulation of innate immune responses toward the pathogen. cFP suppressed the production of proinflammatory cytokines, nitric oxide, and reactive oxygen species in a lipopolysaccharide (LPS)-stimulated monocyte/macrophage cell line and in bone marrow-derived macrophages. Specifically, cFP inhibited inhibitory κB (IκB) kinase (IKK) phosphorylation, IκBα degradation, and nuclear factor κB (NF-κB) translocation to the cell nucleus, indicating that cFP affects the NF-κB pathway. We searched for genes that are responsible for cFP production in V. vulnificus and identified VVMO6_03017 as a causative gene. A deletion of VVMO6_03017 diminished cFP production and decreased virulence in subcutaneously inoculated mice. In summary, cFP produced by V. vulnificus actively suppresses the innate immune responses of the host, thereby facilitating its survival and propagation in the host environment.
INTRODUCTION
Vibrio vulnificus is a Gram-negative bacterium and an opportunistic pathogen that can cause septicemia through ingestion of raw contaminated seafood or through exposure of open wounds to salt water. The mortality rates for septicemia are >50%, and death can occur within 24 h after infection with the bacteria. V. vulnificus causes cellular damage by triggering the secretion of hemolysin and cytolysin and inhibits phagocytosis with a capsular polysaccharide to increase its survival in the host environment (1–3). Several virulence factors have been identified in this pathogen (1–3), and it appears that complex interactions among numerous factors, including many not yet identified, are required for its pathogenicity.
A diketopiperazine molecule, cyclo(l-phenylalanyl–l-proline) (cFP) (Fig. 1A), is a secondary metabolite produced by numerous fungi and bacteria that controls the expression of genes involved in pathogenicity (4). cFP was identified in Pseudomonas strains as a member of a new class of autoinducers by using lux-based acyl-homoserine lactone (AHL) biosensors (5, 6). We have previously shown that cFP production by V. vulnificus and Vibrio cholerae peaks when cell growth reaches the stationary phase and that cFP affects the expression of the ToxR-dependent gene ompU, which is important for the pathogenicity of Vibrio species (7). In addition, cFP has numerous effects on multiple hosts. On animal cells, it has antifungal (8) and antitumor (9–11) effects and also reverses irradiation damage (12). It also modulates auxin signaling in plants (13). Those reports suggest that cFP may have an important function in pathogenicity as well as in host-pathogen interactions. However, the effects of diketopiperazines, including cFP, on host immune responses have not yet been studied.
FIG 1.
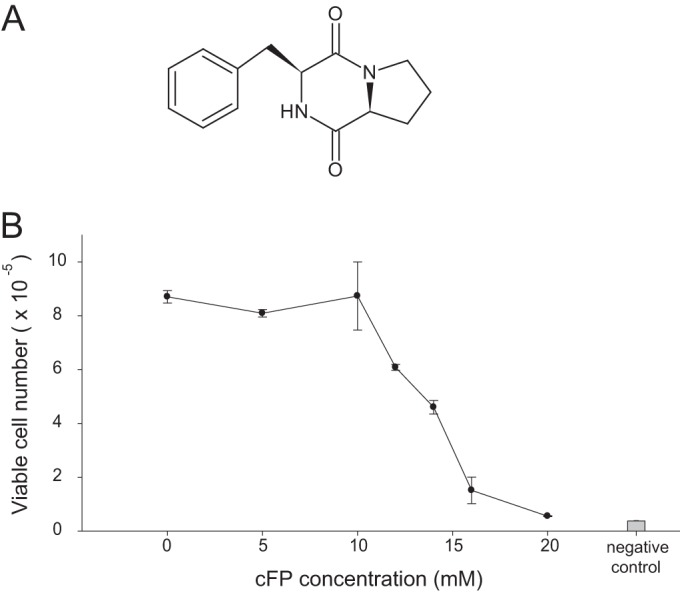
Structure of cFP and its effect on cell viability. (A) Structure of cyclo(Phe-Pro). (B) Cell viability was measured by an MTS assay after treatment with various concentrations of cFP for 48 h. Data represent means ± SD (n = 4). All data are representative of data from three independent experiments that yielded similar results.
Macrophages play a central role in first-line host defense (14). They express proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) and antimicrobial agents such as reactive oxygen species (ROS) and nitric oxide (NO) when they recognize pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors such as the Toll-like receptors (TLRs) (15). When TLR4 recognizes bacterial lipopolysaccharide (LPS), it initiates a signaling cascade through the recruitment of proteins such as MyD88, IL-1 receptor-associated kinase (IRAK) family members, and TNF receptor-associated factor 6 (TRAF6). Ubiquitination of TRAF6 results in the sequential activation of transforming growth factor β-associated kinase 1 (TAK1) and the inhibitory κB (IκB) kinase (IKK) complex or the mitogen-activated protein kinase (MAPK) pathway through phosphorylation, which triggers downstream signaling and activates the transcription factor nuclear factor κB (NF-κB) (16, 17). NF-κB activation requires the phosphorylation and degradation of IκB, which is regulated by the IKK complex or by the MAPK pathway. IκB degradation allows NF-κB to translocate into the nucleus, where it binds target gene promoters and induces the expression of proinflammatory cytokines and antimicrobial agents involved in pathogen clearance (17).
In this study, we examined the role of cFP produced by the human pathogen V. vulnificus in the modulation of mammalian innate immune responses. Here we report that this small molecule suppresses innate immune responses by affecting the NF-κB pathway in macrophages, providing the bacteria with a survival advantage in the host environment. These results suggest an important role for cFP in the pathogenesis of bacteria.
MATERIALS AND METHODS
Mice and experimental reagents.
Four- to five-week-old ICR mice were purchased from Samkaco (South Korea). Experiments with live mice were approved by the Sogang University Institutional Animal Care and Use Committee. cFP (Bachem Inc., Switzerland) was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 M (stock solution).
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. Escherichia coli strains were cultured at 37°C in Luria-Bertani (LB) broth supplemented with the appropriate antibiotics. V. vulnificus strains were cultured in LB broth or thiosulfate-citrate-bile salt-sucrose (TCBS) agar at 30°C. Antibiotics were used at the following concentrations: 50 μg/ml ampicillin, 10 μg/ml tetracycline, and 25 μg/ml chloramphenicol for E. coli and 2 μg/ml tetracycline and 2 μg/ml chloramphenicol for V. vulnificus. All media used in this study were purchased from Difco/BD (Franklin Lakes, NJ). All reagents and antibiotics were purchased from Sigma-Aldrich (St. Louis, MO) and used according to the manufacturer's instructions.
Cell culture.
The J774A.1 cell line (mouse monocytes/macrophages) and the L929 cell line (NCTC 929; mouse connective tissue cells) were purchased from the Korean Cell Line Bank and maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Grand Island, NY) and 1% penicillin-streptomycin.
Bone marrow-derived macrophages (BMDMs) were prepared as described previously (18). L929 cell-conditioned medium was collected from the culture supernatant of L929 cells. Mouse bone marrow cells were obtained from mouse tibias and femurs by using a syringe filled with BMDM growth medium (RPMI 1640 supplemented with 10% FBS, 1% penicillin-streptomycin, and 30% L929 cell-conditioned medium). The collected cells were differentiated for 6 days in BMDM growth medium.
Cell viability assay.
Cell viability was determined by using the CellTiter 96 Aqueous Nonradioactive Cell Proliferation Assay kit (Promega, Madison, WI) according to the manufacturer's instructions. The assay kit contains of two solutions, MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] and PMS (phenazine methosulfate). MTS is soluble in tissue culture medium and is bioreduced by live cells into a formazan product that can be measured at 490 nm directly from 96-well plates. J774A.1 cells (1 × 104 cells/100 μl) were plated onto 96-well plates. After culture overnight, the cells were treated with various concentrations of cFP for 48 h. A combined MTS-PMS solution (20 μl) was added to each well of the 96-well plates, and the plates were further incubated for 3 h at 37°C in a humidified 5% CO2 atmosphere. The absorbance at 490 nm (A490) was measured by using a microplate reader (model 680; Bio-Rad, Hercules, CA). A standard curve was used to convert the absorbance to cell numbers.
Cytokine measurement.
For measurement of the expression of innate cytokines, cells were plated onto a 24-well culture plate and treated with 100 ng/ml LPS (catalog number S2325; Sigma-Aldrich) in the presence or absence of various concentrations of cFP for the indicated times. The amount of cytokine in the cell culture supernatant was measured by an enzyme-linked immunosorbent assay (ELISA). Each well of a 96-well Maxisorp plate (Nunc/Thermo-Fisher Scientific, Waltham, MA) was coated with 100 μl of a purified anticytokine antibody (for TNF-α [catalog number 4-7325-85; eBioscience, San Diego, CA], IL-6 [catalog number 554400; BD Biosciences, San Jose, CA], and IL-1β [catalog number 14-7012-85; eBioscience]) and incubated overnight at 4°C. The plate was washed with wash buffer (0.05% Triton X-100 in phosphate-buffered saline [PBS]), and nonspecific binding was blocked by incubation with 100 μl/well of ELISA blocking buffer (10% bovine serum in PBS) for 1 h at room temperature. Standards and samples were added (25 μl/well), and the plates were incubated overnight at 4°C. The plate was washed with wash buffer and then incubated with 100 μl/well of a biotinylated anticytokine antibody (for TNF-α [catalog number 4-7326-85; eBioscience], IL-6 [catalog number 554402; BD Biosciences], and IL-1β [catalog number 13-7112-85; eBioscience]) for 1 h at room temperature. The plate was washed with wash buffer, 100 μl of a horseradish peroxidase solution (catalog number 554058; BD Biosciences) was added to each well, and the plate was incubated for 30 min at room temperature. The plate was washed, and the color was developed with 150 μl of a TMB (3,3′,5,5′-tetramethylbenzidine) peroxidase solution. The reaction was stopped by the addition of 50 μl of TMB stop solution. The absorbance was recorded at 490 nm by using a microplate reader (model 680; Bio-Rad).
Measurement of nitric oxide production.
Cells were stimulated with 100 ng/ml LPS in the presence or absence of various concentrations of cFP. The culture supernatants were collected, and the concentration of nitrite was measured by using the Griess reagent system (Promega) according to the manufacturer's instructions.
Measurement of total cellular peroxides.
Intracellular oxidative stress was assayed by the oxidation of dichlorodihydrofluorescein diacetate (DCFH-DA). J774A.1 cells (1 × 106) were incubated with 100 ng/ml LPS in the presence or absence of 1 or 4 mM cFP at 37°C. Cells were then washed with PBS and incubated with 10 μM DCFH-DA for 30 min at 37°C. The cells were detached by scraping and washed with PBS. Cellular fluorescence was determined by using a FACSCalibur instrument.
Measurement of macrophage phagocytic activity.
Phagocytic activity was measured with a pHrodo Green E. coli BioParticles conjugate (catalog number P35366; Life Technologies). The particles are conjugated to pHrodo dye, the fluorescence of which increases in an acidic environment such as that of the phagosome. For measurement of phagocytic activity, 5 × 105 J774A.1 cells in 200 μl of culture medium were plated onto 24-well culture plates for 24 h, and the culture medium was then replaced with 200 μl of pHrodo Green E. coli BioParticles conjugate solution in the presence or absence of 4 mM cFP for the indicated times. Cells were detached by scraping and washed with PBS. Phagocytic activity was determined based on the fluorescence intensity of the cells by using a FACSCalibur instrument (BD Biosciences).
Nuclear protein extraction and Western blot analysis.
J774A.1 cells (2 × 106) were harvested by scraping and washed with 1× PBS. For whole-cell lysate extraction, the cells were mixed with 0.5 ml of radioimmunoprecipitation assay (RIPA) lysis buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Nonidet P-40 [NP-40], 1% sodium deoxycholate, and 0.1% SDS) and pipetted 20 times to form a homogeneous lysate. The mixture was allowed to stand for 5 min at 4°C and centrifuged at 12,000 × g for 10 min at 4°C, and the supernatants were used for Western blotting.
For the preparation of nuclear extracts, cells were resuspended in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM dithiothreitol [DTT], and 0.5 mM phenylmethylsulfonyl fluoride [PMSF]), mixed by vortexing, and incubated on ice for 15 min. NP-40 was then added to a final concentration of 10%, and the extracts were vortexed for 10 s. Pellets were obtained by centrifugation at 15,000 × g for 1 min at 4°C. The pellets were resuspended by vortexing in buffer B (20 mM HEPES [pH 7.9], 0.4 M NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, and 10% NP-40) for 10 min. The mixture was then centrifuged at 15,000 × g for 10 min at 4°C, and the supernatants were used for Western blot analysis.
The whole-cell lysates or nuclear lysates were electrophoresed on an SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride (PVDF) membrane for 1 h. The membranes were blocked for 1 h in Tris-buffered saline with 0.05% Tween 20 (TBST) and 5% skim milk. Anti-phospho-IKK (catalog number 2078; Cell Signaling Technology, Manassas, VA), anti-IκBα (catalog number 9242; Cell Signaling Technology), anti-p65 (catalog number sc-8008; Santa Cruz Biotechnology, Santa Cruz, CA), anti-β-actin (catalog number sc-47778; Santa Cruz Biotechnology), and anti-lamin B (catalog number ab16048; Abcam, Cambridge, MA) antibodies were diluted 1:1,000 in TBST and incubated with the membrane overnight at 4°C. Blots were washed with TBST three times for 10 min each and incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (catalog number A90-116P; Bethyl, Montgomery, TX) or chicken anti-goat IgG (catalog number A50-110P; Bethyl) for 1 h at room temperature. Protein bands were detected by incubation with enhanced chemiluminescence reagent and by exposure to an X-ray film. Densitometric analyses were performed by using ImageJ software (NCBI freeware).
NF-κB activity assay.
The pNF-κB luciferase vector (containing the NF-κB binding sites) and a Renilla luciferase reporter plasmid were transfected into HEK293T cells. A total of 2 ×105 cells were used for each transfection. Transfected cells were allowed to recover in complete medium for 24 h and then incubated with TNF-α in the presence or absence of 4 mM cFP for 0, 1, 3, or 6 h. Cells were then harvested and washed with PBS. The luciferase assay was performed by using the Dual-Luciferase Reporter Assay system (Promega) according to the manufacturer's instructions. Transfection efficiency was normalized by dividing firefly luciferase activity by Renilla luciferase activity.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR) analysis.
J774A.1 cells or BMDMs (5 × 105 cells) in 200 μl of culture medium were plated onto 24-well plates. After culture overnight, the cells were treated with 0.1 μg/ml LPS in the presence or absence of cFP for the indicated times. The cells were resuspended in 1 ml of TRIzol reagent (Life Technologies) and 200 μl of chloroform and centrifuged at 15,000 × g for 15 min at 4°C. The aqueous phase (supernatant) was transferred into a new microtube, mixed with 500 μl of isopropanol, and incubated at room temperature for 10 min. Total RNA was precipitated by centrifugation at 15,000 × g for 10 min at 4°C, and the RNA pellet was washed with 70% ethanol by centrifugation at 7,500 × g for 5 min at 4°C. cDNA was synthesized by using the TOPscript reverse transcriptase kit (catalog number RT0025; Enzynomics, Japan) according to the manufacturer's instructions. Briefly, total RNA was mixed with 100 μM oligo(dT)18 and heated for 5 min at 70°C. Reverse transcription was performed at final concentrations of 50 mM Tris-HCl (pH 7.5), 3 mM MgCl2, 10 mM DTT, 75 mM KCl, 2 mM each deoxynucleoside triphosphate (dNTP), 20 units RNase inhibitor, and 200 units Topscript reverse transcriptase at 50°C for 60 min. The reaction was terminated at 95°C for 5 min.
Real-time PCRs were carried out with the HiFast Probe Lo-Rox kit (catalog number Q200220; Genepole, South Korea) by using the primers listed in Table S2 in the supplemental material (400 nM each) on an Applied Biosystems 7500 real-time PCR system. Real-time PCR was performed as follows: a starting reaction at 95°C for 2 min followed by 40 cycles of denaturation at 95°C for 10 s and annealing/extension at 60°C for 34 s.
Flow cytometry analysis.
J774A.1 cells (5 × 105) in 200 μl of culture medium were plated onto a 24-well plate for 24 h, and the cells were treated with LPS (0.1 μg/ml) in the presence or absence of various concentrations of cFP for the indicated times. Cells were detached by scraping and washed two times with PBS. The cells were treated with an Fc blocker (catalog number 101302; BioLegend, San Diego, CA) for 20 min on ice. For the measurement of TLR4 internalization, cells were stained with an anti-TLR4-phycoerythrin (PE) antibody (catalog number 145403; BioLegend) for 30 min at 4°C. After washing with PBS, cellular fluorescence was determined by using a FACSCalibur instrument. For intracellular cytokine staining, cells were fixed/permeabilized by using an intracellular cytokine staining kit (BD Biosciences) for 20 min. The fixed cells were sequentially stained with a biotin-conjugated anti-TNF-α antibody (catalog number 13-7326-85; eBioscience) and PE-streptavidin (catalog number 554061; BD Biosciences) for 30 min at 4°C and washed with PBS, and cellular fluorescence was determined on a FACSCalibur instrument.
Isothermal titration calorimetry analysis.
To determine the binding affinity of cFP and LPS, isothermal titration calorimetry (ITC) was performed by using an iTC200 isothermal titration calorimeter (Microcal), which enables the direct measurement of the enthalpy change when two molecules interact (19). cFP (1 mM) was titrated into a solution of LPS (12 μM) at 37°C, and the resulting temperature changes were measured by the input of power required to maintain an equal temperature between the sample and reference cells.
Construction of the genomic DNA library of V. vulnificus and transposon mutagenesis.
To construct the V. vulnificus MO6-24/O (20) genomic library, chromosomal DNA was extracted and then partially digested with the restriction enzyme Sau3AI. Digested fragments were size fractionated after sucrose gradient centrifugation, and fragments with an average size of 25 kb were isolated. The fragments were cloned into the BamHI site of pCP13/B (21) and transformed into LE392 cells. Transposon insertion mutagenesis was performed using by the transposon mini-Tn5 lacZ1 as described previously (22).
Quantitative analyses of cFP production.
For quantitative analyses of cFP production, bioluminescence assays and high-performance liquid chromatography (HPLC) analyses were performed as previously described (7). Briefly, cultures of bacterial cells grown overnight in LB broth were inoculated into fresh LB medium. After 6 h of incubation, cell-free culture supernatants were collected by centrifugation, and 200 μl of each supernatant was plated onto a 96-well plate. For quantitative measurement of cFP production in the supernatants, E. coli strain MT102 harboring sensor plasmid pSB403 (23) was used as a bioindicator. Cultures of MT102(pSB403) grown overnight in LB medium were washed and diluted to an A600 value of 0.1 into fresh LB medium, and 200 μl of the diluted MT102(pSB403) cells was added to a 96-well plate. After 4 h of incubation, the cFP level was calculated as luminescence units normalized to the cell density (relative luminescence units [RLU]). For HPLC analysis, 50 ml of the cell-free culture supernatant from V. vulnificus or E. coli cells grown to stationary phase in LB medium was extracted twice with the same volume of ethyl acetate. The ethyl acetate extract was vaporized and dissolved in 50% methanol. Samples were applied to a Mightysil RP-18 GP 250-4.6 column (Kanto Chemical, Japan) that had been washed with 100% methanol and equilibrated with 30% methanol in water. The flow rate was 1.0 ml/min, and the cFP peak was detected at 256 nm.
Construction of an llcA deletion mutant and cloning of the llcA gene.
To construct the llcA gene deletion mutant, a 777-bp fragment of the upstream region and a 724-bp fragment of the downstream region of the llcA gene of V. vulnificus MO6-24/O were amplified by using primer sets llcKO_F1/llcKO_B1 and llcKO_F2/llcKO_B2, respectively (see Table S3 in the supplemental material). Each fragment was digested with the restriction enzyme StyI and then ligated into the pGEM-T Easy vector (Promega) to generate plasmid pGEM-llcKO. The resulting construct has a 462-bp deletion in the llcA gene. Plasmid pGEM-llcKO was digested with the restriction enzymes SalI and SphI and cloned into the vector pDM4 to generate pDM4-llcKO, which was then introduced into E. coli S17-1λpir (24), from which it was mobilized into V. vulnificus MO6-24/O by conjugation. Exconjugants were grown on TCBS agar containing chloramphenicol (2 μg/ml) to select for a double crossover, as previously described (25). The resulting V. vulnificus MO6-24/O strain with a deletion in the chromosomal copy of llcA was confirmed by DNA sequencing, and the mutant was named the ΔllcA mutant. For the llcA complementary assay, the 830-bp DNA fragment comprising the promoter region and the coding region of llcA was amplified by PCR using primers llcA_comF and llcA_comB (see Table S3 in the supplemental material). The resulting product was cloned into pRK415 (26) to construct pRK-llcA. This plasmid was conjugated into wild-type (WT) V. vulnificus MO6-24/O and the ΔllcA strain.
Bacteria and mouse infection.
For the mouse infection experiment, wild-type V. vulnificus (MO6-24/O) and the ΔllcA mutant were cultured in LB medium, harvested, washed with PBS, and resuspended in PBS. The bacteria (5 × 104, 1 × 105, or 1 × 106 CFU in 100 μl of PBS) were subcutaneously injected into 4- to 5-week-old ICR mice.
For the mouse infection experiment with V. vulnificus conjugated with either pRK415 or pRK-llcA, the WT strain harboring pRK415 [WT(pRK415)], the ΔllcA mutant harboring pRK415 [ΔllcA(pRK415) strain], and the ΔllcA(pRK-llcA) strain were cultured in LB medium containing 2 μg/ml tetracycline, harvested, washed with PBS, and resuspended in 100 μl of PBS containing 2 μg/ml tetracycline. The bacteria (2 × 106, 1 × 106, or 1 × 105 CFU) were subcutaneously injected into 4- to 5-week-old ICR mice.
Statistical analysis.
Results are expressed as means ± standard deviations (SD). Statistical differences between groups were analyzed by using Student's t test or Pearson's chi-squared test.
RESULTS
cFP is not cytotoxic to J774A.1 cells in the range of 0 to 10 mM.
To determine the effect of cFP on the viability of cells, a cytotoxicity assay was performed. J774A.1 mouse monocyte/macrophage cells were treated with cFP in the range of 0 to 20 mM for 48 h, and cytotoxicity was measured by an MTS assay. cFP did not show a significant level of cytotoxicity in the range of 0 to 10 mM. However, cFP was cytotoxic to J774A.1 cells at concentrations of >10 mM; therefore, <5 mM cFP was used in subsequent experiments (Fig. 1B).
cFP suppresses the production of proinflammatory cytokines from LPS-stimulated J774A.1 cells.
To investigate whether microbe-released cFP can influence host immune responses, resting J774A.1 cells were treated with cFP at a concentration of 0 to 5 mM for 48 h, and the secretion of innate cytokines was measured by ELISA of culture supernatants. cFP did not induce the production of TNF-α, IL-6, or IL-1β in resting macrophages, based on the negative and positive controls (see Fig. S1 in the supplemental material).
The inhibitory effect of cFP on cytokine production in resting cells could not be determined because the basal expression levels of the cytokines were too low. Therefore, J774A.1 cells were stimulated with LPS, and the effect cFP on the release of proinflammatory cytokines was examined by ELISA. LPS substantially induced the production of the proinflammatory cytokines TNF-α and IL-6. Interestingly, cFP inhibited LPS-induced expression of TNF-α and IL-6 in a dose-dependent manner (Fig. 2A). Maximal inhibition of cytokine release was observed at a cFP concentration of 4 mM, at which cell viability was not affected (Fig. 1B). The effect of cFP on the expression of proinflammatory cytokines at the transcriptional level was also examined. Without the addition of cFP, LPS-induced mRNA expression of Tnfa and Il6 peaked at 3 h of stimulation and decreased thereafter. Consistent with its effects on cytokine protein production, cFP strongly inhibited the LPS-induced expression of Tnfa and Il6 mRNAs (Fig. 2B).
FIG 2.

cFP reduces proinflammatory cytokine levels in mouse macrophages stimulated with LPS. (A) J774A.1 cells were stimulated with LPS (100 ng/ml) for 48 h in the presence of the indicated concentrations of cFP. The levels of TNF-α and IL-6 in the culture supernatants were measured by ELISA. Data represent means ± SD (n = 4). Statistical differences between groups were analyzed by using Student's t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (B) J774A.1 cells were stimulated with LPS (100 ng/ml) for the indicated numbers of hours in the presence or absence of 4 mM cFP. Relative mRNA levels of Tnf and Il6 were measured by qRT-PCR and normalized to the Hprt mRNA level. Data represent means ± SD (n = 3). Statistical analyses were performed by using Student's t test (***, P < 0.001).
cFP suppresses the production of antimicrobial agents in a macrophage cell line.
The effect of cFP on the production of antimicrobial chemicals generated by mammalian cells, including nitric oxide (NO) and ROS, was next examined. First, the effect of cFP on NO production in LPS-stimulated J774A.1 cells was quantitatively measured by using a Griess assay. cFP reduced NO production by LPS-stimulated J774A.1 cells in a dose-dependent manner (Fig. 3A). cFP also reduced the mRNA expression of the Inos gene, which encodes inducible nitric oxide synthase (iNOS), an enzyme that catalyzes the production of NO (Fig. 3B) (27).
FIG 3.

Effects of cFP on production of nitric oxide (NO) and reactive oxygen species (ROS) in J774A.1 cells. (A) J774A.1 cells were stimulated with LPS (100 ng/ml) for 24 h in the presence of the indicated concentrations of cFP, and the concentrations of nitrite were detected by a Griess reagent assay. Data represent means ± SD (n = 4). Statistical differences between groups were analyzed by using Student's t test (*, P < 0.05; **, P < 0.01; NS, not significant). (B) The mRNA level of Inos was measured by qRT-PCR after the indicated incubation times and normalized to the Hprt mRNA level. Data represent means ± SD (n = 3). Statistical analyses were performed by using Student's t test (***, P < 0.001). (C) J774A.1 cells were stimulated with 100 ng/ml LPS in the presence or absence of 1 mM or 4 mM cFP for 2 h. ROS generation was detected by DCFH-DA fluorescence.
Next, the effect of cFP on ROS production in LPS-stimulated J774A.1 cells was examined. To detect intracellular ROS levels, cells were labeled with DCFH-DA, which is a cell-permeable chemical that is rapidly oxidized to a highly fluorescent form by ROS. As shown in Fig. 3C, cFP inhibited ROS production in a dose-dependent manner in LPS-stimulated J774A.1 cells.
cFP suppresses the production of innate cytokines and nitric oxide in primary macrophages.
The above-described experiments were performed by using the J774A.1 cell line, which is a transformed monocyte/macrophage cell line. To confirm that these results can be repeated in more physiologically relevant primary cells, bone marrow-derived macrophages (BMDMs) were used. Bone marrow was isolated from C57BL/6 mice and differentiated into macrophages ex vivo in a medium supplemented with L929 cell-conditioned medium as a source of macrophage colony-stimulating factor (M-CSF). The role of cFP in the production of proinflammatory cytokines and NO and the expression of Inos was examined by using LPS-stimulated BMDMs. Consistent with the J774A.1 cell data, cFP inhibited the production of the proinflammatory cytokines TNF-α and IL-6 at both the protein and mRNA levels (Fig. 4A) and also inhibited the production of NO and the expression of Inos mRNA (Fig. 4B) in LPS-stimulated BMDMs. These results suggest that cFP also exerts its inhibitory effects in more physiologically relevant primary macrophages.
FIG 4.

cFP suppresses production of innate cytokines and nitric oxide in primary macrophages. (A) BMDMs were stimulated with LPS (100 ng/ml) for the indicated times in the presence or absence of 4 mM cFP. (Top) The levels of TNF-α and IL-6 in the culture supernatants were measured by an ELISA. Data represent means ± SD (n = 3). (Bottom) BMDMs were stimulated with LPS (100 ng/ml) for 3 h in the presence or absence of 4 mM cFP. The mRNA levels of Tnf and Il6 were measured by qRT-PCR and normalized to the Hprt mRNA level. Statistical differences between groups were analyzed by using Student's t test (*, P < 0.05). (B) BMDMs were stimulated with LPS (100 ng/ml) for the indicated times in the presence or absence of 4 mM cFP. The concentrations of nitrite were detected by a Griess reagent assay (left), and the mRNA level of Inos was measured by qRT-PCR (right). Data represent means ± SD (n = 3). Statistical analyses between groups were performed by using Student's t test (*, P < 0.05).
cFP inhibits TNF-α production in J774A.1 cells at an early time point.
Understanding the kinetics of innate cytokine production and its inhibition is important for innate immunity, because the innate immune system provides the first line of defense for the body and prevents invading microbes from proliferating and colonizing. The effect of cFP on TNF-α production by LPS-stimulated J774A.1 cells was therefore measured at various time points by intracellular cytokine staining. LPS rapidly induced TNF-α production, starting at 1 h and peaking at 3 h (Fig. 5). cFP at a final concentration of 1 mM did not exert a significant effect on TNF-α production. However, cFP at 4 mM rapidly blocked the production of TNF-α at as early as 1 h (Fig. 5), suggesting that cFP acts at a relatively early time point.
FIG 5.

cFP delays TNF-α expression induced by LPS. (A) J774A.1 cells were treated with 100 ng/ml of LPS in the presence or absence of cFP for the indicated times. TNF-α-positive cells were stained by intracellular cytokine staining and analyzed by fluorescence-activated cell sorter analysis. Each experiment was performed in triplicate and repeated at least three times, which yielded similar results. (B) Graphical representation of the data shown in panel A. Data represent means ± SD (n = 3). Statistical analyses between cFP-treated and untreated groups were performed by using Student's t test (*, P < 0.05; **, P < 0.01).
In all the experiments in this study, cFP was added to the culture medium together with LPS to examine the effects of cFP on cultured cells. We therefore investigated the possibility that cFP binds directly to LPS. If this occurs, cFP would reduce the effective dose of LPS available for cell activation and thereby limit LPS-induced cytokine and NO production, consistent with our previous results. To test this possibility, the ability of cFP to physically bind to LPS was examined by ITC. We found that cFP did not specifically bind to LPS (see Fig. S2 in the supplemental material), which indicates that the inhibitory effect of cFP was not due to a sequestration of cFP by LPS.
cFP does not affect phagocytic activity or TLR internalization.
To explore the effect of cFP on macrophage functions, the effect of cFP on phagocytosis, a major mechanism through which macrophages clear pathogens, was examined. Phagocytosis by J774A.1 cells was measured by using a pHrodo Green E. coli BioParticles conjugate. Treatment with 4 mM cFP did not have a significant effect on the phagocytic activity of J774A.1 cells (see Fig. S3 in the supplemental material).
To study the molecular mechanisms of the cFP-mediated inhibition of the production of proinflammatory cytokines and antimicrobial agents, we first examined whether cFP affects TLR4 endocytosis, because LPS triggers the inflammatory signaling pathway by inducing TLR4 endocytosis in macrophages (28). Even after 60 min, cFP did not have a significant effect on TLR4 internalization induced by LPS (see Fig. S4 in the supplemental material). Taken together, these results suggest that cFP has no discernible effect on phagocytosis or TLR4 internalization.
cFP affects the NF-κB signaling pathway.
The above-described results led us to examine whether cFP exerts its effect by influencing a downstream signaling pathway associated with TLR4 stimulation. The central downstream pathway of TLR4 is the IKK/NF-κB pathway. Upon stimulation of TLR4, IKK becomes autophosphorylated and subsequently phosphorylates IκB, which, in its unphosphorylated form, binds to NF-κB and inhibits its nuclear translocation. Phosphorylated IκB (p-IκB) is ubiquitinated and degraded by a proteasome. The released NF-κB translocates into the nucleus and induces the expression of its target genes (14, 29). IκB levels recover as the signal wanes since IκB is continuously resynthesized; thus, the degradation of IκB reflects the active transduction of a stimulatory signal (14, 29). The role of cFP in these signaling events was examined by using LPS-stimulated J774A.1 cells. LPS led to IKK phosphorylation and a transient degradation of IκBα, as previously reported (30). Interestingly, cFP inhibited IKK phosphorylation and reduced and delayed the degradation of IκBα in whole-cell lysates from LPS-stimulated J774A.1 cells (Fig. 6A and B). cFP also reduced the accumulation of NF-κB p65 in the nuclear extract compared to controls without cFP treatment (Fig. 6C and D).
FIG 6.

cFP impairs NF-κB signaling in macrophages. (A) Phosphorylation of IκB and IKK. Phosphorylated IKK and IκBα were detected in whole-cell extracts by Western blotting. β-Actin was used as the reference protein. Data are representative of three independent experiments that yielded similar results. (B) Quantitation of phosphorylated IKK, IκBα, and β-actin bands was performed by using ImageJ software (NCBI freeware). (C) Nuclear translocation of p65. J774A.1 cells were treated with LPS in the presence or absence of 4 mM cFP for various amounts of time. The cells were then lysed, nuclear extracts were prepared, and the p65 protein was detected in the nuclear extracts by Western blotting. Lamin B1 was used as a loading control for nuclear protein. (D) Quantitation of p65 and lamin B bands was performed by using ImageJ software. (E) NF-κB target gene expression. J774A.1 cells were treated with LPS in the presence or absence of 4 mM cFP for 3 h. The mRNA levels of the indicated genes were measured by qRT-PCR. Statistical differences between groups were analyzed by using Student's t test (*, P < 0.05). (F) NF-κB promoter activity. HEK293T cells were transfected with a pNF-κB luciferase vector (containing NF-κB binding sites) and a Renilla luciferase reporter plasmid by using polyethyleneimine. Cells were allowed to rest for 24 h and then stimulated with 100 ng/ml LPS in the presence or absence of 4 mM cFP for the indicated times. Luciferase activity in the cell extract was measured. Statistical differences between groups were analyzed by using Student's t test (*, P < 0.05).
To examine whether cFP inhibits NF-κB-mediated gene expression, the expression of NF-κB target genes in LPS-stimulated J774A.1 cells was analyzed. cFP greatly reduced the expression of the NF-κB target genes Rantes, Ip10, and Mcp1 in these cells (Fig. 6C). Taken together, these results suggest that cFP inhibits the IKK/NF-κB signaling pathway in LPS-stimulated macrophages.
A transient-reporter assay was used to confirm the effect of cFP on NF-κB-mediated gene expression. HEK293T cells were transfected with a reporter construct containing NF-κB binding sites in its promoter. The cells were then stimulated with TNF-α in the presence or absence of 4 mM cFP for the indicated times, and reporter activity was measured. Stimulation with TNF-α triggered luciferase activity, indicating that TNF-α induced NF-κB binding to its promoter (Fig. 6D). cFP significantly reduced TNF-α-induced luciferase activity, indicating that cFP inhibits NF-κB-mediated gene expression (Fig. 6D). Since TNF-α rather than LPS was used in this experiment, this result further supports the notion that the inhibitory activity of cFP is not limited to LPS stimulation but is applicable to other NF-κB-activating signals.
Isolation of a mutant of MO6-24/O with a defect in cFP production.
Since cFP was found to inhibit the production of proinflammatory cytokines and antimicrobial agents, we hypothesized that cFP gives bacteria a survival advantage in the host environment during infection. To test this hypothesis, we first isolated a derivative of V. vulnificus that has a defect in cFP production. For this, we utilized the fact that E. coli strain LE392 does not produce detectable levels of cFP, as assessed by HPLC (data not shown). A genomic library of V. vulnificus MO6-24/O was constructed in LE392 cells, and each clone of the library was examined for the ability to produce cFP by using the bioindicator MT102(pSB403). About 25,000 genomic library clones of V. vulnificus were assessed for the production of cFP by using MT102(pSB403), and two clones (pCP-1151 and pCP-1440) that activated the bioindicator to the level of wild-type V. vulnificus MO6-24/O were isolated (see Fig. S5A in the supplemental material). This suggests that these two clones carry genes that allow cFP production by LE392, which does not naturally produce cFP. cFP production in the two clones, measured by using HPLC, confirmed the result (see Fig. S5B in the supplemental material). LE392 harboring the control vector pCP13/B gave a signal comparable to that in uninoculated LB medium (see Fig. S5B in the supplemental material).
DNA nucleotide sequencing showed that the inserts in pCP-1151 and pCP-1440 contain an identical region of chromosome II of V. vulnificus MO6-24/O, and pCP-1151 was chosen for further analysis. The clone pCP-1151 contains an ∼23-kb genomic DNA fragment of V. vulnificus, from VVMO6_03006 to VVMO6_03031 (see Fig. S6A in the supplemental material). To identify the gene that is responsible for conferring cFP production to the pCP-1151 clone, random transposon insertion mutants were made, and each mutant was tested for the ability to induce the production of cFP in LE392 cells. A transposon insertion in the VVMO6_03017 gene caused a failure to activate the bioindicator, while transposon insertions in other genes in the clone did not affect the activation of the bioindicator (see Fig. S6B in the supplemental material).
The deduced amino acid sequence of VVMO6_03017 suggests that the protein is a membrane protein that does not show homology to any known genes. A deletion of this gene abolished cFP production in V. vulnificus MO6-24/O cells, and the introduction of the clone in trans into the mutant restored cFP production (see Fig. S7 in the supplemental material). Although the biological function of VVMO6_03017 in association with cFP biosynthesis is not known, this result clearly showed that this gene is required for the production of cFP in V. vulnificus MO6-24/O cells, and the gene was therefore named llcA (low level of cFP producer A). The growth rate of the ΔllcA mutant did not differ from that of wild-type strain MO6-24/O (see Fig. S8 in the supplemental material).
cFP enhances bacterial survival in animal hosts.
To test the hypothesis that cFP production by bacteria results in a survival advantage during infection, ICR mice were infected with WT (MO6-24/O) or mutant (ΔllcA) V. vulnificus by subcutaneous injection, and the virulence of the bacteria was determined by measuring the survival rate of the infected mice. Mice infected with 5 × 104 CFU of WT V. vulnificus had 0% viability after 72 h of infection. However, mice infected with the ΔllcA mutant had an increased viability of 50% (Fig. 7A). When mice were infected with higher CFU (1 × 105 or 1 × 106) of V. vulnificus, the difference in virulence between the WT and mutant waned (Fig. 7B and C), suggesting that the effect of cFP on virulence is modest. These results support the hypothesis that, at least modestly, cFP is involved in virulence in the host environment during infection.
FIG 7.

cFP enhances virulence of V. vulnificus in mice. Four- to five-week-old ICR mice were injected with V. vulnificus strain MO6-24/O (WT strain), the ΔllcA mutant, or PBS (n = 20). Bacteria (5 × 104, 1 × 105, or 1 × 106 CFU) were injected subcutaneously in 100 μl of PBS, and the numbers of surviving mice are shown. Statistical differences between WT- and mutant-infected groups were analyzed by Pearson's chi-squared test (**, P < 0.01; n.s., not significant).
Next, we examined whether the llcA gene complements the virulence of the WT in a mouse infection experiment. For this, the WT or the ΔllcA mutant was introduced with either a control plasmid (pRK415) or plasmid pRK-llcA. ICR mice were then infected with WT(pRK415), ΔllcA(pRK415), and ΔllcA(pRK-llcA) by subcutaneous injection, and the virulence of the bacteria was determined by measuring the survival rate of the infected mice. These experimental conditions are slightly different from those used in the previous mouse experiment. V. vulnificus conjugated with either the control plasmid (pRK415) or plasmid pRK-llcA has to be maintained in the presence of tetracycline in the mouse. Therefore, tetracycline was injected into a mouse together with the bacteria, which selects bacteria that contain the plasmid. Because of the low copy number and the instability of the plasmid, it is expected that the number of live V. vulnificus cells under these conditions is not as high as that in the absence of the antibiotics. In these experimental settings, 1 × 106 or 2 × 106 CFU of the ΔllcA mutant complemented with the llcA gene [ΔllcA(pRK415)] almost completely restored virulence to WT levels (Fig. 8), demonstrating that LlcA is responsible for virulence.
FIG 8.

LlcA is responsible for virulence of V. vulnificus in mice. Four- to five-week-old ICR mice were injected with the V. vulnificus WT(pRK415) strain, the ΔllcA(pRK415) mutant, and the ΔllcA(pRK-llcA) complementation strain or PBS (A, n = 5; B, n = 10; C, n = 5). Bacteria (2 × 106, 1 × 106, or 1 × 105 CFU) were injected subcutaneously in 100 μl of PBS containing 2 μg/ml tetracycline, and the numbers of surviving mice are shown. Statistical differences between the ΔllcA(pRK415)- and WT(pRK415)- or ΔllcA(pRK-llcA)-treated groups were analyzed by Pearson's chi-squared test (**, P < 0.01).
DISCUSSION
In this study, we investigated the effects of cFP produced by the lethal human pathogen V. vulnificus on the mammalian innate immune system using the J774A.1 monocyte/macrophage cell line and BMDMs. cFP suppressed the LPS-induced production of proinflammatory cytokines and antimicrobial agents in these cells by interfering with the NF-κB signaling pathway. cFP provided the bacteria with a survival advantage in the host environment.
Since the innate immune system acts immediately upon pathogen invasion, suppression of this system is a well-known strategy that many pathogenic microorganisms employ for their survival and propagation during infection of the host body. In particular, a variety of pathogens inhibit the NF-κB signaling pathway to evade the innate immune system of the host (31). Bacteria can inhibit the NF-κB pathway at the level of TLR signaling, the IKK complex, or NF-κB-dependent transcription (31). Although it is yet to be determined which level of the NF-κB signaling pathway is targeted by cFP, our current study provides some clues about the target molecule. For instance, cFP did not affect TLR4 internalization, but it did inhibit IKK phosphorylation and its downstream signaling. These results suggest that the target molecule lies between TLR internalization and IKK phosphorylation in the signaling pathway. The exact molecular target of cFP has yet to be determined.
Our study revealed that cFP acts at an early time point. cFP-mediated inhibition of the NF-κB signaling pathway occurred by as early as 5 min after stimulation, and a reduction of TNF-α and IL-6 expression occurred by as early as 1 to 3 h poststimulation. This early-acting property of cFP may be related to the capacity of V. vulnificus to evade the innate immune system upon initial infection, swiftly inducing symptoms with potentially lethal results in the host (3).
Our luciferase activity analysis showed that cFP has similar NF-κB-suppressive activity in LPS-stimulated macrophages and in TNF-α-stimulated HEK293T cells, suggesting that the inhibition of NF-κB activity is not a cell type-restricted phenomenon. In addition, cFP did not affect either phagocytic activity or TLR4 endocytosis in a monocyte/macrophage cell line. Thus, it seems that cFP does not affect receptor-mediated signaling of host cells, suggesting that the cFP-mediated effect is highly specific to NF-κB signaling rather than influencing the broad activities involved in cellular function.
Quorum-sensing molecules regulate biofilm formation as well as toxin and virulence factor production (32, 33). Recent studies showed that quorum-sensing molecules secreted from bacteria also affect host immune systems (32, 34–36). In particular, N-(3-oxo-dodecanoyl)homoserine lactone (3-oxo-C12-HSL), a member of the N-acyl-homoserine lactone (AHL) family that is produced by Pseudomonas aeruginosa, inhibits lymphocyte proliferation and TNF-α production by LPS-stimulated macrophages (37). In addition, 3-oxo-C12-HSL represses genes encoding inflammatory regulators by impairing the regulation of NF-κB functions (35). Pretreatment with the quorum-sensing small volatile aromatic molecule 2-amino-acetophenon (2-AA) limits the inflammatory response of the host by reducing proinflammatory cytokine production (36). Recently, we showed that 4-hydroxy-2-heptylquinoline (HHQ) and 2-heptyl-3,4-dihydroxyquinoline (Pseudomonas quinolone signal [PQS]), members of the 4-hydroxy-2-alkylquinoline (HAQ) family, also have an inhibitory function in innate immunity (34, 38). cFP acts as a quorum-sensing molecule in V. vulnificus as well as in other pathogenic Vibrio species, including V. cholerae, by affecting the expression of the ToxR-dependent gene ompU, which is important for pathogenicity (7). Our current study is another example of the regulation of the host immune system by quorum-sensing signaling molecules. In addition to cFP, other cyclic dipeptides isolated from various microorganisms also play physiological roles in higher organisms (5, 13, 39–41). Future studies will undoubtedly reveal roles for other cyclic dipeptide molecules in microbe-host interactions.
We performed BLAST searches for the llcA gene. The gene encodes a hypothetical membrane protein found in the genus Vibrio, but the protein does not have any homology with any known protein except for a few proteins annotated as “arginine/ornithine antiporters” (ArcD) in a few Vibrio species, such as V. maritimus, V. ponticus, and V. variabilis. However, these ArcD proteins have different sizes than and no significant homology with the ArcD proteins of V. cholerae and other bacteria. These results suggest that the annotation may not be solid. Therefore, we did not follow this nomenclature in our study. We also performed a protein domain analysis on the protein, but we could not find any known functional domain in the protein. However, based on the fact that it is a membrane protein, we speculate that it might be involved in cFP transport across the cell membrane.
In conclusion, our results provide evidence that cFP has immune-suppressive effects that enhance the pathogen's ability to infect and survive in a host environment. Our study sheds light on the role of cFP in host-pathogen interactions and aids in the development of potential therapeutic strategies for diseases related to V. vulnificus. Further studies are needed to elucidate the exact molecular target of cFP in the NF-κB pathway.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Research Foundation of Korea (NRF) grants funded by the South Korean government (2014-023419 to G.R.L. and NRF-2011-0018115 to K.-S.K.), Ministry of Science, ICT & Future Planning, Republic of Korea.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02878-14.
REFERENCES
- 1.Gulig PA, Bourdage KL, Starks AM. 2005. Molecular pathogenesis of Vibrio vulnificus. J Microbiol 43:118–131. [PubMed] [Google Scholar]
- 2.Jones JM, Warchol ME. 2009. Expression of the Gata3 transcription factor in the acoustic ganglion of the developing avian inner ear. J Comp Neurol 516:507–518. doi: 10.1002/cne.22128. [DOI] [PubMed] [Google Scholar]
- 3.Horseman MA, Surani S. 2011. A comprehensive review of Vibrio vulnificus: an important cause of severe sepsis and skin and soft-tissue infection. Int J Infect Dis 15:e157–e166. doi: 10.1016/j.ijid.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Prasad C. 1995. Bioactive cyclic dipeptides. Peptides 16:151–164. doi: 10.1016/0196-9781(94)00017-Z. [DOI] [PubMed] [Google Scholar]
- 5.Holden MT, Ram Chhabra S, de Nys R, Stead P, Bainton NJ, Hill PJ, Manefield M, Kumar N, Labatte M, England D, Rice S, Givskov M, Salmond GP, Stewart GS, Bycroft BW, Kjelleberg S, Williams P. 1999. Quorum-sensing cross talk: isolation and chemical characterization of cyclic dipeptides from Pseudomonas aeruginosa and other gram-negative bacteria. Mol Microbiol 33:1254–1266. [DOI] [PubMed] [Google Scholar]
- 6.Bellezza I, Peirce MJ, Minelli A. 2014. Cyclic dipeptides: from bugs to brain. Trends Mol Med 20:551–558. doi: 10.1016/j.molmed.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Park DK, Lee KE, Baek CH, Kim IH, Kwon JH, Lee WK, Lee KH, Kim BS, Choi SH, Kim KS. 2006. Cyclo(Phe-Pro) modulates the expression of ompU in Vibrio spp. J Bacteriol 188:2214–2221. doi: 10.1128/JB.188.6.2214-2221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strom K, Sjogren J, Broberg A, Schnurer J. 2002. Lactobacillus plantarum MiLAB 393 produces the antifungal cyclic dipeptides cyclo(L-Phe-L-Pro) and cyclo(L-Phe-trans-4-OH-L-Pro) and 3-phenyllactic acid. Appl Environ Microbiol 68:4322–4327. doi: 10.1128/AEM.68.9.4322-4327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne PJ, Hunt AL, Rostoll K, Van Der Walt JJ, Graz CJM. 1998. The biological activity of selected cyclic dipeptides. J Pharm Pharmacol 50:1331–1337. doi: 10.1111/j.2042-7158.1998.tb03355.x. [DOI] [PubMed] [Google Scholar]
- 10.Brauns SC, Milne P, Naude R, Van de Venter M. 2004. Selected cyclic dipeptides inhibit cancer cell growth and induce apoptosis in HT-29 colon cancer cells. Anticancer Res 24:1713–1719. [PubMed] [Google Scholar]
- 11.Brauns SC, Dealtry G, Milne P, Naude R, Van de Venter M. 2005. Caspase-3 activation and induction of PARP cleavage by cyclic dipeptide cyclo(Phe-Pro) in HT-29 cells. Anticancer Res 25:4197–4202. [PubMed] [Google Scholar]
- 12.Lee KH, Rhee KH. 2008. Radioprotective effect of cyclo(L-phenylalanyl-L-prolyl) on irradiated rat lung. J Microbiol Biotechnol 18:369–376. [PubMed] [Google Scholar]
- 13.Ortiz-Castro R, Diaz-Perez C, Martinez-Trujillo M, del Rio RE, Campos-Garcia J, Lopez-Bucio J. 2011. Transkingdom signaling based on bacterial cyclodipeptides with auxin activity in plants. Proc Natl Acad Sci U S A 108:7253–7258. doi: 10.1073/pnas.1006740108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawai T, Akira S. 2007. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Newton K, Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4:a006049. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJJ. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 17.Hayden MS, Ghosh S. 2004. Signaling to NF-kappaB. Genes Dev 18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 18.Weischenfeldt J, Porse B. 2008. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc 2008:pdb.prot5080. doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 19.Velazquez-Campoy A, Freire E. 2005. ITC in the post-genomic era…? Priceless Biophys Chem 115:115–124. doi: 10.1016/j.bpc.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 20.Reddy GP, Hayat U, Abeygunawardana C, Fox C, Wright AC, Maneval DR Jr, Bush CA, Morris JG Jr. 1992. Purification and determination of the structure of capsular polysaccharide of Vibrio vulnificus M06-24. J Bacteriol 174:2620–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darzins A, Chakrabarty AM. 1984. Cloning of genes controlling alginate biosynthesis from a mucoid cystic fibrosis isolate of Pseudomonas aeruginosa. J Bacteriol 159:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Lorenzo V, Herrero M, Jakubzik U, Timmis KN. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol 172:6568–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winson MK, Swift S, Fish L, Throup JP, Jorgensen F, Chhabra SR, Bycroft BW, Williams P, Stewart GS. 1998. Construction and analysis of luxCDABE-based plasmid sensors for investigating N-acyl homoserine lactone-mediated quorum sensing. FEMS Microbiol Lett 163:185–192. doi: 10.1111/j.1574-6968.1998.tb13044.x. [DOI] [PubMed] [Google Scholar]
- 24.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 25.Milton DL, O'Toole R, Horstedt P, Wolf-Watz H. 1996. Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol 178:1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keen NT, Tamaki S, Kobayashi D, Trollinger D. 1988. Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70:191–197. doi: 10.1016/0378-1119(88)90117-5. [DOI] [PubMed] [Google Scholar]
- 27.Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding AH, Troso T, Nathan C. 1992. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 256:225–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 28.Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC. 2011. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulze-Luehrmann J, Ghosh S. 2006. Antigen-receptor signaling to nuclear factor kappa B. Immunity 25:701–715. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Kubo-Murai M, Hazeki K, Nigorikawa K, Omoto T, Inoue N, Hazeki O. 2008. IRAK-4-dependent degradation of IRAK-1 is a negative feedback signal for TLR-mediated NF-kappaB activation. J Biochem 143:295–302. doi: 10.1093/jb/mvm234. [DOI] [PubMed] [Google Scholar]
- 31.Le Negrate G. 2012. Subversion of innate immune responses by bacterial hindrance of NF-kappaB pathway. Cell Microbiol 14:155–167. doi: 10.1111/j.1462-5822.2011.01719.x. [DOI] [PubMed] [Google Scholar]
- 32.Freestone P. 2013. Communication between bacteria and their hosts. Scientifica 2013:361073. doi: 10.1155/2013/361073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez JE, Keshavan ND. 2006. Messing with bacterial quorum sensing. Microbiol Mol Biol Rev 70:859–875. doi: 10.1128/MMBR.00002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim K, Kim YU, Koh BH, Hwang SS, Kim SH, Lepine F, Cho YH, Lee GR. 2010. HHQ and PQS, two Pseudomonas aeruginosa quorum-sensing molecules, down-regulate the innate immune responses through the nuclear factor-kappaB pathway. Immunology 129:578–588. doi: 10.1111/j.1365-2567.2009.03160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, Lehmann M, Meijler MM, Janda KD, Ulevitch RJ. 2008. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science 321:259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- 36.Bandyopadhaya A, Kesarwani M, Que YA, He J, Padfield K, Tompkins R, Rahme LG. 2012. The quorum sensing volatile molecule 2-amino acetophenon modulates host immune responses in a manner that promotes life with unwanted guests. PLoS Pathog 8:e1003024. doi: 10.1371/journal.ppat.1003024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Telford G, Wheeler D, Williams P, Tomkins PT, Appleby P, Sewell H, Stewart GS, Bycroft BW, Pritchard DI. 1998. The Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-L-homoserine lactone has immunomodulatory activity. Infect Immun 66:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim K, Kim SH, Lepine F, Cho YH, Lee GR. 2010. Global gene expression analysis on the target genes of PQS and HHQ in J774A.1 monocyte/macrophage cells. Microb Pathog 49:174–180. doi: 10.1016/j.micpath.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 39.de Carvalho MP, Abraham WR. 2012. Antimicrobial and biofilm inhibiting diketopiperazines. Curr Med Chem 19:3564–3577. doi: 10.2174/092986712801323243. [DOI] [PubMed] [Google Scholar]
- 40.Cornacchia C, Cacciatore I, Baldassarre L, Mollica A, Feliciani F, Pinnen F. 2012. 2,5-Diketopiperazines as neuroprotective agents. Mini Rev Med Chem 12:2–12. doi: 10.2174/138955712798868959. [DOI] [PubMed] [Google Scholar]
- 41.Martins MB, Carvalho I. 2007. Diketopiperazines: biological activity and synthesis. Tetrahedron 63:9923–9932. doi: 10.1016/j.tet.2007.04.105. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.