

Abstract
The ability of Streptococcus pyogenes to infect different niches within its human host most likely relies on its ability to utilize alternative carbon sources. In examining this question, we discovered that all sequenced S. pyogenes strains possess the genes for the malic enzyme (ME) pathway, which allows malate to be used as a supplemental carbon source for growth. ME is comprised of four genes in two adjacent operons, with the regulatory two-component MaeKR required for expression of genes encoding a malate permease (maeP) and malic enzyme (maeE). Analysis of transcription indicated that expression of maeP and maeE is induced by both malate and low pH, and induction in response to both cues is dependent on the MaeK sensor kinase. Furthermore, both maePE and maeKR are repressed by glucose, which occurs via a CcpA-independent mechanism. Additionally, malate utilization requires the PTS transporter EI enzyme (PtsI), as a PtsI– mutant fails to express the ME genes and is unable to utilize malate. Virulence of selected ME mutants was assessed in a murine model of soft tissue infection. MaeP–, MaeK–, and MaeR– mutants were attenuated for virulence, whereas a MaeE– mutant showed enhanced virulence compared to that of the wild type. Taken together, these data show that ME contributes to S. pyogenes' carbon source repertory, that malate utilization is a highly regulated process, and that a single regulator controls ME expression in response to diverse signals. Furthermore, malate uptake and utilization contribute to the adaptive pH response, and ME can influence the outcome of infection.
INTRODUCTION
Although it has a relatively small genome (approximately 1.8 Mbp), the pathogenic Gram-positive bacterium Streptococcus pyogenes has a remarkable ability to adapt to a variety of human tissues. This trait allows it to cause numerous diseases, ranging from superficial and self-limiting infections in soft tissues like the skin (impetigo) and pharynx (pharyngitis) to more problematic infections at a number of diverse anatomical sites (1). Understanding the complex regulatory interactions that allow it to adapt to these diverse environments provides a unique opportunity to gain insight into how a pathogen can efficiently employ a relatively limited genetic repertory to maximize its ability to cause disease.
An important question is how S. pyogenes uses its limited metabolic potential to grow efficiently in diverse tissues. Considerable evidence has accrued to suggest that the patterns by which S. pyogenes exploits available growth substrates are intimately associated with both temporal and compartment-specific patterns of virulence gene expression (2–4). As a lactic acid bacterium, S. pyogenes relies exclusively on fermentation via the homolactic and mixed-acid pathways to generate energy (5–7). However, the specific carbon sources it preferentially utilizes in different tissues, the temporal patterns with which these are consumed, and how these patterns impact regulation of virulence gene expression are not well understood.
One approach to gain insight into conditions encountered during infection has involved comparison of the S. pyogenes transcriptome between organisms recovered from various models of infection to organisms cultured under different in vitro conditions. In general, these studies have revealed that at the latter time points of infection, patterns of gene expression most closely resemble those observed in vitro in environments of low pH (pH 6.0 to 6.5) and low concentrations of glucose (8–10). These two conditions are likely related, as the fermentation of glucose by lactic acid bacteria results in the highest rates of production of acidic end products, including lactate, acetate, and formate (6, 7). This suggests that S. pyogenes' choice of carbon source results in a significant remodeling of its local tissue environment. It also indicates that over the course of infection, it must both adapt to its self-inflicted acid stress as well as exploit alternative carbon sources. In this regard, transcriptome profiling revealed that one of the most highly differentially activated gene clusters under conditions of acid stress and glucose starvation and in murine soft tissue encodes a putative operon of two genes predicted to function in the catabolism of malate (10), annotated as the malic permease (maeP) and the malic enzyme (maeE) (Fig. 1A).
FIG 1.

The malic enzyme (ME) pathway in S. pyogenes. (A) The arrangement of the open reading frames that comprise the ME locus of S. pyogenes is shown by large arrows. Gene names are shown below, and the genomic loci listed within the open reading frames are based on the genome of S. pyogenes HSC5 (23). Known (black font) and predicted (gray font) regulatory elements of the intergenic region of S. pyogenes and Enterococcus faecalis JH2-2 (Ef JH2-2 [17]) and Lactobacillus casei BL23 (Lc BL23 [15]) are shown below. Arrows indicate sites in DNA bound by MaeR, while sites bound by CcpA are boxes labeled “cre” (catabolite regulatory element). Numbers below indicate intergenic distances in numbers of base pairs. (B) Schematic of the ME pathway. The subcellular localization, function, and reactions catalyzed by the various components of the ME pathway that are listed in the figure are shown.
The dicarboxylic organic acid malate is found in abundance in tissue and in the environment, so it is not surprising that numerous malate degradation pathways have been identified among both prokaryotic and eukaryotic organisms (11–17). In lactic acid bacteria, two distinct pathways have been identified that make very different contributions to physiology. The most common of these is malolactic fermentation (MLF), which allows for the conversion of malate into lactate through the function of the malolactic enzyme (MLE). Typically, MLF does not contribute to growth yields but does play an important role in maintenance of ATP pools during starvation and in protection from acid killing (18–21). Since malate is a stronger acid than lactate, its decarboxylation by MLF results in alkalinization of the cytoplasm, and the resulting pH gradient drives the malate/lactate antiporter coupled to ATP synthesis (7, 18–21).
Less commonly found is an alternative degradation pathway that converts malate to pyruvate and carbon dioxide (18) that is known as the malic enzyme (ME) pathway (Fig. 1B). A unique feature of ME is that, unlike MLF, it enables cells to utilize malate as a carbon source for growth (16, 18). However, while the MLF system has been extensively studied, the regulation and physiological significance of the ME pathway are not as well understood. Studies in several lactic acid species, including Enterococcus faecalis, Streptococcus bovis, and Lactobacillus casei (13, 15, 17, 22), have indicated that ME requires 4 genes organized into two adjacent operons (Fig. 1A). These include the maePE operon that encodes the transmembrane permease (maeP) and cytosolic malic enzyme (maeE). Expression of these genes is dependent on the adjacent two-component system (TCS), which includes a sensor histidine kinase (maeK) and response regulator (maeR) (15, 17, 18). This similar organization is observed in the S. pyogenes chromosome (Fig. 1A), and as noted above, maePE is upregulated by acid stress and infection in S. pyogenes. In addition, examination of the S. pyogenes profiling data shows that both the maePE operon and the adjacent TCS had similar patterns of regulation, suggesting that these two systems function together (10).
Interestingly, while other ME operons are activated by malate (15, 17, 18) and repressed by glucose (15, 17), regulation by pH has been described only for Lactococcus casei (18). Whether this system is regulated by pH in other bacterial species that contain a functional ME pathway and the physiological role of this regulation are not known. Rather, pH regulation is more commonly associated with the MLF pathway, where it is associated with acid resistance (20, 21). Examination of the S. pyogenes genome has not revealed the presence of MLF genes (23), so the significance of pH regulation of the ME pathway and whether it compensates for MLF in acid tolerance are not clear. In this study, we examined the contribution of malate catabolism and its unique pattern of regulation to S. pyogenes physiology and virulence. This analysis revealed that S. pyogenes has a functional ME pathway, that catabolism of malate contributes to growth, and that its regulation shares some similarities with other lactic acid bacteria but also has several unique features. Finally, we show that the presence or absence of ME genes can influence virulence in a murine model of soft tissue infection.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
The Escherichia coli strain TOP10 (Invitrogen) was used for cloning using standard molecular biology techniques. The Streptococcus pyogenes strain HSC5 (23) and mutant derivatives were utilized in this study. Strains were grown in Todd-Hewitt broth with 0.2% yeast extract (THY; Difco) or C medium (24). C medium was adjusted to pH 7.5 as described previously (24). Routine growth conditions utilized sealed culture tubes incubated at 37°C under static conditions. Streptococcal strains grown on solid medium containing 1.4% Bacto agar (Difco) were cultured in a sealed jar with a commercial gas generator (GasPak catalogue no. 70304; BBL). For experiments utilizing malate supplementation, filter-sterilized 5% (wt/vol) stock solution buffered to pH 7.0 with NaOH (Sigma) was used to add malate (Sigma) to a final concentration of 0.5% to the medium. For experiments utilizing glucose or maltose supplementation, filter-sterilized 20% (wt/vol) stock solution was used to add glucose or maltose (Sigma) to a final concentration of 0.2% to the medium. For experiments utilizing buffered medium, 1 M stock solutions of HEPES (pH 7.5) or morpholineethanesulfonic acid (MES) (pH 6.0) (both obtained from Sigma) were added to a final concentration of 0.1 M to the medium. All media used were sterilized in an autoclave prior to supplementation. When appropriate, antibiotics were added at the following concentrations: erythromycin at 1 μg/ml, kanamycin at 250 μg/ml, and chloramphenicol at 3 μg/ml.
Construction of mutants.
All references to genomic loci are based on the genome of HSC5 (23). In-frame deletion mutations in the genes encoding MaeP (L897_04180), MaeE (L897_04185), MaeR (L897_04170), and MaeK (L897_04175), as well as the modified allele for HPr (L897_05590) (see Table S1 in the supplemental material), were generated using allelic replacement and the PCR primers listed in Table S2 in the supplemental material. The deletion alleles were transferred to the HSC5 chromosome by using the allelic replacement vector pGCP213 (25) as described previously (26) and listed in Table S3 in the supplemental material. Each deletion allele was obtained through overlap extension PCR (27) using the primers listed in Table S2. All molecular constructs and chromosomal structures of all mutants were verified using PCR and DNA sequencing (Genewiz, South Plainfield, NJ) by using oligonucleotide primers (IDT, Coralville, IA) of the appropriate sequences.
Complementation of ME mutants.
To complement the maeE in-frame deletions, DNA fragments containing maeE from HSC5 in the absence of its promoter were amplified using the primers listed in Table S2 in the supplemental material and inserted under the control of the rofA promoter in pABG5 as previously described (28). The resulting plasmid, pEP66, was then used for ectopic expression of MaeE (see Table S3). For complementation of the maeK in-frame deletion, a reversion strategy was used to restore the wild-type (WT) locus in the MaeK– mutant background. A DNA fragment containing the maeK open reading frame and flanking regions was amplified from HSC5 using primers listed in Table S2 and inserted into the plasmid pCRK (29). The resulting plasmid, pEP74, was then used to create the strain MaeKr as described previously (30) (see Table S1).
Metabolic assays.
Malate, lactate, and formate concentrations were measured using commercially available kits (Sigma). Briefly, cultures of each individual strain tested were grown overnight in C medium with the appropriate supplement added (see the text). Cultures were then subjected to centrifugation, filtered through 0.22-μm-pore-size filters (Millipore), and then assessed per the manufacturer's protocol. Data shown are the means and standard deviations from duplicate determination of three separate biological samples prepared from at least 2 independent experiments.
Isolation of RNA and transcript analysis.
Transcript abundance of selected genes was analyzed as previously described (31). Briefly, overnight cultures were diluted 1:25 into fresh C medium with the appropriate supplement added (see the text) and harvested at mid-log phase (optical density at 600 nm [OD600] of 0.2). Total RNA was isolated using Qiagen RNeasy minikit per the manufacturer's protocol. RNA was subjected to reverse transcription (RT) using iScript (Bio-Rad) per the manufacturer's protocol. RT-PCR analysis of cDNA samples were performed using iQ SYBR green supermix (Bio-Rad) and the primers listed in Table S2. Relative transcript abundance was determined using the ΔΔCT method using recA transcript as a standard and are presented in comparison to unmodified C medium or in comparison to the wild type. The data shown are the means and the standard deviations from triplicate determinations of at least two separate biological samples prepared from at least two independent experiments.
Infection of mice.
As previously described (32, 33), 5- to 6-week-old female SKH1 hairless mice (Charles River Labs) were injected subcutaneously with approximately 107 CFU of the S. pyogenes strains indicated in the text. Following infection, the resulting ulcers formed were documented over a period of several days by digital photography, and lesion areas were measured as previously described (32). Data presented are pooled from at least two independent experiments with at least 10 mice per experimental group.
Growth rate calculations.
Indicated bacterial strains were back-diluted 1:50 into 1 ml of fresh C medium (unmodified or altered as indicated in the text), and their growth was monitored at 37°C using a Tecan Infinite M200 Pro plate reader. During growth, the plate was shaken every 10 min for 30 s, followed by a 5-s wait period and measurement of the OD600. Data were normalized relative to uninoculated medium, and growth rates were calculated as described previously (34). Growth rates are reported as doubling time (t1/2) and were determined from a series of 7 time points collected over a 60-min period that defines the peak rate of growth, which typically occurred prior to the culture reaching 15 to 30% of the max OD600. Growth yields were calculated from the maximum OD600 reached by the culture and are expressed as a percentage relative to that of the wild-type strain under identical conditions. The average doubling time and percent growth yield were calculated from each replicate from at least three independent experiments.
Statistical analyses.
Differences between mean values obtained for wild-type and mutant strains in in vitro assays were tested for significance using the Student t test. For infection of mice, differences in lesion area between the wild type and individual mutants were tested for significance using the Mann-Whitney U test. Computation of test statistics utilized Instat (version 3.1) and Prism (version 6.0) from the GraphPad software (San Diego, CA). For all tests, the null hypothesis was rejected for P values of <0.05.
RESULTS
ME is necessary for S. pyogenes malate-enhanced growth.
It is unclear why the ME pathway in S. pyogenes is regulated by pH, as MLF and not ME is typically associated with acid stress resistance in other lactic acid species (20, 21). However, S. pyogenes lacks the genes necessary for MLF (23), so the contribution of the ME pathway to streptococcal physiology was investigated. The malic enzyme uses NAD+ to oxidize malate to produce CO2, NADH, and pyruvate (Fig. 1B). Since pyruvate can be further metabolized to produce ATP, the signature function of ME is to allow cells to utilize malate as a carbon source for growth (16). To test this growth phenotype, S. pyogenes HSC5 was cultured overnight in a carbohydrate-reduced medium (C medium) in the presence or absence of 0.5% malate. Although cultures had comparable growth rates in both conditions (t1/2 = 56 min or 63 min, respectively), 0.5% malate enhanced growth yields by approximately 50% (Fig. 2A). Additionally, pH measurements of cell-free supernatants taken throughout growth indicate that malate utilization does not alter the pH of the medium compared to that of unmodified C medium (see Fig. S2A in the supplemental material). This is due to the fact that, unlike when grown in medium supplemented with glucose, when grown on malate, the bacteria utilize mixed-acid fermentation, producing large amounts of formate, which has a much higher pKa than the lactate commonly produced (see Fig. S2B).
FIG 2.

ME mutants are deficient in malate catabolism. (A) WT bacteria were tested for malate utilization by measuring growth over the course of 16 h of cultures grown in either unmodified C medium or C medium supplemented with 0.5% malate. Data are presented as means and standard deviations from 3 independent experiments. (B and D) WT and ME mutants were grown in unmodified C medium or C medium supplemented with 0.5% malate. Following 16 h of incubation, growth yields were measured by OD600. Data are presented as the means and standard deviations from 3 independent experiments. (C and E) Malate concentrations from cell-free culture supernatants from overnight cultures of WT or ME mutants grown in C medium supplemented with 0.5% malate were measured (see Materials and Methods). Data are presented as percent remaining (compared to initial concentration) and are presented as means and standard deviations from 3 biological samples analyzed in duplicate. Asterisks indicate significant differences (***, P < 0.001) compared to WT in C medium.
In-frame deletion mutants in maeP (malate permease), maeE (malic enzyme), maeK (malate sensor kinase), and maeR (malate response regulator) were constructed and were found to have growth characteristics identical to those of the wild type in unmodified C medium. However, all mutants failed to show an increased growth yield in the presence of malate (Fig. 2B). Malate concentrations were measured from cell-free supernatants of overnight cultures grown in 0.5% malate to determine malate consumption by the wild type and the four ME mutants. WT cultures exhibited an approximately 80% reduction from the initial concentration of 37.3 mM, while malate concentrations were unchanged by growth of the mutants (Fig. 2C). With the exception of maeE, it was not possible to express the ME genes from a plasmid for complementation. As an alternative, allelic replacement was used to restore the full-length maeK gene in a ΔMaeK mutant background to make the reversion strain MaeKr. In this way, we were able to complement at least one gene from each operon. Complementation of maeE and reversion of maeK restored both enhanced growth yields in the presence of malate (Fig. 2D) and consumption of malate (Fig. 2E).
Expression of ME genes is dependent on malate and requires MaeK.
In other lactic acid bacteria, expression of ME requires both the presence of malate and the ME TCS (15, 17, 18). To determine if this common regulatory mechanism is also utilized in S. pyogenes, an analysis of transcript levels of ME genes using real-time RT-PCR was performed. The results indicated that during the exponential phase of growth (OD600 = 0.2), maeP and maeE were highly upregulated in the presence of malate by 100- and 200-fold, respectively (Fig. 3A). This response was dependent on MaeK, as transcript levels were equivalent in the presence or absence of malate in an MaeK− mutant. Restoration of the protein in an MaeKr reversion strain also restored malate-induced transcription (3A). Transcription of maeK and maeR was also increased in the presence of malate in WT cells, with both genes showing an approximately 3-fold increase compared to that in the unmodified medium (Fig. 3B).
FIG 3.
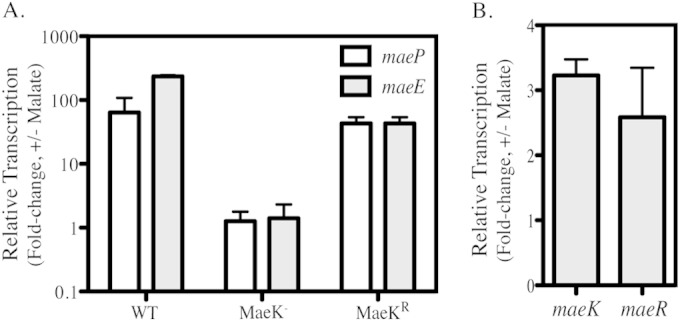
MaeK regulates malate-dependent expression of maePE. (A) WT, MaeK–, and MaeKr strains were grown in C medium supplemented with 0.5% malate until exponential phase (OD600 of 0.2). Total RNA was isolated and used for real-time RT-PCR analysis of maeP and maeE transcripts. (B) WT bacteria were grown in C medium supplemented with 0.5% malate as described before. Total RNA was isolated and used for real-time RT-PCR analysis of maeK and maeR transcripts. Data presented for all genes are the ratios of transcript abundance in modified medium to that of unmodified C medium and represent the means and standard deviations from 3 biological samples, each analyzed in triplicate.
Glucose regulation of ME is CcpA independent.
Malate catabolism in other lactic acid bacteria is repressed by glucose, indicating this pathway is regulated through a mechanism of carbon catabolite repression (CCR) (15, 17). CCR allows the bacteria to metabolize preferable carbon sources in the environment, usually through transcriptional repression of genes involved in the processing of alternative, and less favorable, carbon sources (reviewed in references 35–37). A key transcriptional regulator of global CCR in Gram-positive bacteria is CcpA (35), which has been shown to regulate ME in response to glucose in both Lactobacillus casei and Enterococcus faecalis (15, 17). To test for CCR regulation of the ME pathway in S. pyogenes, transcription of the four ME genes was analyzed in the absence or presence of glucose (0.2%) by real-time RT-PCR. Results showed a significant repression of 4- to 6-fold (log2 scale) for all four ME genes (Fig. 4), consistent with observations in other lactic acid species (15, 17). However, in contrast to these other species, repression occurred independently of CcpA, as the addition of glucose still repressed expression of all ME genes in a CcpA– mutant (Fig. 4) (38). Repression does have the characteristics of CCR, as glucose was repressive even in the presence of malate (Fig. 4), indicating that S. pyogenes has adopted a CcpA-independent CCR mechanism for regulation of malate catabolism.
FIG 4.
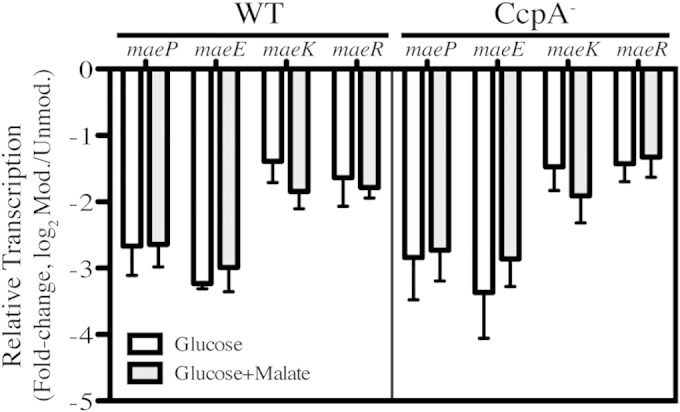
Carbon catabolite repression of ME genes is CcpA independent. WT and CcpA– bacteria were grown in C medium supplemented with 0.2% glucose until exponential phase (OD600 of 0.2). Total RNA was isolated and used for real-time RT-PCR analysis of the individual mae transcripts. Data are presented as the ratios of transcript abundance in modified medium to that in unmodified C medium and represent the means and standard deviations derived from 4 biological samples, each analyzed in triplicate.
Malate catabolism is regulated by PTS-mediated phosphorylation.
An alternative mechanism of CCR in bacteria is known as induction prevention, which is dependent on the sugar phosphotransferase (PTS) system and the phosphorylation state of a conserved histidine residue of the phosphocarrier protein HPr (36), which in the case of S. pyogenes is His15 (39). If the ME loci are controlled by a mechanism similar to induction prevention, then cells unable to produce phosphorylated His-HPr (P∼His-HPr) should be unable to utilize malate. To test this hypothesis, two mutants were constructed. The first is an allelic exchange mutant with a swap of a chloramphenicol cassette with ptsI, which encodes EI, the enzyme responsible for phosphorylation of the His15 site within the HPr protein. The second mutant contains a single amino acid substitution in HPr, replacing His15 with alanine, which has been shown to maintain HPr capability to be phosphorylated at Ser46 and is functional for sugar transport but lacking in the ability to participate in regulation (HPrH15A) (36, 40, 41).
When grown in the presence of malate, both the PtsI– mutant and the HPrH15A mutant have a significant growth defect compared to WT growth, resulting in a reduced growth rate and lower final culture density (Fig. 5A and B). To verify that this growth defect was specific for malate utilization and not a general defect under all conditions, strains were also grown in unmodified C medium, as well as in C medium supplemented with 0.2% maltose (a non-PTS sugar) (34). Comparisons of final yield from overnight cultures demonstrate that growth of both the PtsI– and HPrH15A mutants is similar to the WT in unmodified medium (Fig. 5B). In addition, upon supplementation of maltose, all three strains showed an identical increase in growth (Fig. 5B). Thus, mutations that block formation of P∼His-HPr are deficient in malate utilization but are still able to utilize non-PTS carbon sources.
FIG 5.

Carbon catabolite repression of ME genes controlled by P∼His-HPr. WT, PtsI–, and HPrH15A strains were grown in unmodified C medium or C medium plus 0.5% malate. (A) Growth of the WT and PTS mutants in malate-supplemented medium was measured by OD600 over the course of 16 h. Data presented are from a representative experiment. (B) The WT and PTS mutants were grown in unmodified C medium or C medium supplemented with 0.5% malate or 0.2% maltose. Following 16 h of incubation, growth yields were measured by OD600. Data are presented as the means and standard deviations from 3 independent experiments. Asterisks indicate significant differences (**, P < 0.01) compared to the WT under each condition. (C) WT and HPrH15A strains were grown in C medium supplemented with 0.5% malate until exponential phase (OD600 of 0.2). Total RNA was isolated and used for real-time RT-PCR analysis of transcript abundance of the individual mae transcripts. Data are presented as ratios of transcript abundance of HPrH15A to that of WT and represent the means and standard deviations derived from 3 biological samples, analyzed in triplicate.
Expression of the ME genes was then examined in the presence of malate, and it was discovered that compared to the WT, the HPrH15A mutant had a substantial reduction in transcript levels for all four ME genes (Fig. 5C). Taken together, these data demonstrate that the ME pathway in S. pyogenes is repressed by glucose through a mechanism similar to induction prevention.
pH regulation of ME is independent of malate but dependent on maeK.
Prior transcriptional profiling revealed that maeP and maeE are among the genes most highly regulated by pH in S. pyogenes (10). Additionally, growth of WT cells in acidified media was enhanced with the addition of malate, demonstrating that malate catabolism can occur in a low-pH environment (see Fig. S3 in the supplemental material). To further characterize the role of environmental pH on the ME pathway, WT S. pyogenes was grown in C medium buffered to either low (pH 6.0) or neutral (pH 7.5) pH, and transcription of the ME genes was analyzed by real-time RT-PCR. Compared to unbuffered medium and in the absence of the addition of malate, growth at low pH, but not neutral pH, enhanced abundance of the maeP and maeE transcripts by approximately 10- and 20-fold, respectively (Fig. 6A). Neither low nor high pH environments altered expression of maeK or maeR compared with that in unmodified medium (Fig. 6B). However, MaeK itself was required for the enhanced expression of maeP and maeE, as the abundance of these transcripts did not increase in the MaeK– mutant during growth at low pH, but regulation was restored in the MaeKr strain (Fig. 6C). Finally, to address the hierarchy of stimuli between malate and pH, quantitative RT-PCR was performed on cells in the presence of both high malate (0.5%) concentrations and neutral pH and compared to malate alone. Results show that, for both maeP and maeE, transcription is dramatically increased in the presence of 0.5% malate and that this enhanced expression is unaffected by the pH of the medium (Fig. 6D). Overall, these data show that mae gene expression is regulated by environmental pH and that this regulation is mediated through MaeK and is independent of malate regulation.
FIG 6.

pH regulation of ME is malate independent but requires MaeK. (A and B) WT bacteria were grown in C medium buffered to pH 6.0 or pH 7.5 until exponential phase (OD600 of 0.2). Total RNA was isolated and used for real-time RT-PCR analysis of the individual mae transcripts. Data are presented as the ratios of transcript abundance in buffered medium to that in unmodified C medium and represent the means and standard deviations derived from 3 biological samples, each analyzed in triplicate. (C) WT, MaeK–, and MaeKr strains were grown in C medium (pH 6.0), and total RNA was isolated as described before and used for real-time RT-PCR analysis of maeP and maeE transcripts. Data are presented as ratios of transcript abundance in buffered medium to that in unmodified C medium and represent the means and standard deviations derived from 3 biological samples, each analyzed in triplicate. (D) WT bacteria were grown in C medium plus 0.5% malate or C medium plus 0.5% malate buffered to pH 7.5, and total RNA was isolated as described before and used for real-time RT-PCR analysis of maeP and maeE transcripts. Data are presented as ratios of transcript abundance in modified medium to that in unmodified C medium and represent the means and standard deviations derived from 3 biological samples, each analyzed in triplicate.
Loss of MaeE causes enhanced virulence in vivo.
The in vitro experiments presented in this work were done with bacterial cultures grown in C medium, which is characterized as having low carbohydrate concentrations and high salt and peptide levels (10). It has been demonstrated previously that these conditions are highly analogous to the in vivo milieu of carbon sources within the murine soft tissue environment (10). Additionally, malate, being one of the intermediate products of the citric acid cycle, is abundant in host tissue (42). Therefore, the fact that S. pyogenes is able to utilize malate in vitro when added to C medium lends strong support that it can also be utilized in vivo during host tissue infections.
Thus, it was of interest to determine if malate utilization and the ME pathway play a role in virulence. To assess this, a murine soft tissue model was used. Briefly, approximately 107 bacteria were injected subcutaneously into the flank of immunocompetent hairless mice. Infection of WT S. pyogenes HSC5 produces a localized necrotic lesion and formation of an eschar within 24 h but does not cause a systemic infection (33). Lesion size increases over time, peaking in size at day 3 postinfection (32). Measurement of the lesion area over time is therefore used as a marker for virulence in this model. For this analysis, mice were infected with the WT, MaeP–, MaeE–, MaeK–, or MaeR– strains, and lesion areas were compared 3 days postinfection. Mice infected with the MaeK–, MaeR–, and MaeP– strains all formed lesions that were significantly smaller than those in mice infected with the WT (Fig. 7). Conversely, mice infected with the MaeE– strain formed lesions that were significantly larger than those in mice infected with the WT (Fig. 7). These results demonstrate that malate catabolism is an important factor during a soft tissue infection and that the loss of individual ME genes can have differential effects on the outcome of an infection.
FIG 7.
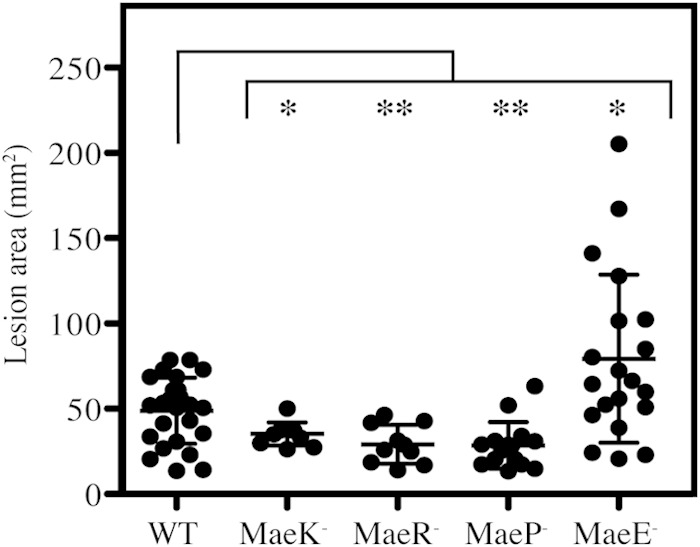
Loss of MaeE causes enhanced virulence in vivo. Hairless SKH1 mice were infected subcutaneously with WT or individual ME mutant strains, and the resulting lesions formed at day 3 postinfection were measured. Each symbol plotted represents the value derived from an individual animal. Data shown are pooled from at least 2 independent experiments, with the mean and standard deviation indicated. Differences between groups were tested for significance using the Mann-Whitney U test (*, P < 0.05; **, P < 0.01).
DISCUSSION
In this study, we have shown that the ME genes of S. pyogenes allow the bacterium to use malate as a carbon source for growth. Additionally, we have shown that this pathway is subjected to regulation by both positive and negative signals, including glucose, malate, and pH. The former of these is via PTS-mediated phosphorylation, while the latter two signals are recognized by the MaeKR regulatory system. Finally, these data show that loss of any individual mae gene can alter the outcome of a soft tissue infection in mice, suggesting that the abilities to transport and utilize malate are both key processes in pathogenesis.
Regulation of ME gene expression in S. pyogenes was found to share features in common with other bacterial species (15, 17, 18). Most notably is that they are induced by malate and that induction requires the MaeKR TCS. A prior analysis of the maePE and maeKR promoter regions in L. casei identified the DNA-binding site for MaeR as a series of direct repeats, and a similar site is shared among other lactic acid bacteria, including S. pyogenes (see Fig. S1 in the supplemental material) (15, 17). Another common feature to ME pathway regulation is that all species repress ME gene expression in the presence of glucose, a regulatory mechanism known as carbon catabolite repression (CCR) (36, 37, 43). However, in S. pyogenes, glucose-mediated regulation of the ME loci functions independently of the major carbon catabolite protein CcpA. In support of this finding is the fact that, unlike the other characterized lactic acid bacteria, the promoter region in S. pyogenes lacks any identifiable cre sites (Fig. 1; see also Fig. S1) (15, 35, 43, 44).
Instead, an alternative method of CCR, induction prevention, is likely regulating ME genes in S. pyogenes. Evidence to support this idea includes the fact that multiple genetic strains that are unable to form P∼His-HPr (either through loss of the EI enzyme or direct mutation of HPr) are likewise deficient in ME transcription and malate utilization. This method of regulation, therefore, enables the cell to activate ME genes only in the presence of a high concentration of P∼His-HPr. During normal growth utilizing preferred carbohydrates, levels of intracellular P∼His-HPr would likely be low (36, 45). This is due to either rapid accumulation of P∼Ser-HPr or transfer of the phosphate group on P∼His-HPr to downstream PTS transporters to allow uptake of PTS sugars. In this way, the bacterium is able to preferentially utilize a number of available PTS sugars before turning on the alternative ME pathway.
In order for this form of regulation to control ME expression, it requires a transfer of the phosphate from P∼His-HPr to an ME regulatory protein. Phosphorylation of non-PTS protein by P∼His-HPr is known to occur in a variety of species (for a review, see reference 36) and often involves a PTS regulatory domain (PRD)-containing protein. Currently, the only non-PTS protein shown to act as a phosphate acceptor from P∼His-HPr in S. pyogenes is the transcriptional regulator Mga (46). This protein has been characterized as containing several unique, but related, PRD domains (PRD_Mga), and previous work has shown that P∼His-HPr is able to phosphorylate specific residues within these domains in vitro (46, 47). Although Mga has been shown to regulate a large number of target genes (3, 4, 47, 48), the ME cluster has not yet been identified as part of its regulon. Alternatively, there are two additional transcription regulators in S. pyogenes HSC5 predicted to include a PRD_Mga domain (E. Paluscio and M. G. Caparon, unpublished data), both within the RofA family of regulators (46, 49–51). It remains possible that one of these proteins may be necessary for ME gene expression. One likely mechanism for this regulation would be that the phosphate from the P∼His-HPr gets transferred to one of the PRD transcriptional regulators, which then allows this protein to bind to the promoter region of maeKR and induce its expression. Further evidence to support this hypothesis is the presence of several putative regulatory elements within the mae promoter region of S. pyogenes (see Fig. S1), which are absent from the promoters of the other lactic acid bacteria. These sequences may serve as binding sites for one of the PRD-containing regulatory proteins mentioned above.
This work also demonstrated that the maePE operon is regulated by a pH-dependent mechanism, whereby acidic pH induces transcription of these genes and neutral pH is inhibitory. In S. pyogenes, it appears that, in addition, the MaeKR TCS was necessary for this regulation, as the loss of MaeK prevents the pH-dependent expression of maePE seen in wild-type cells. Interestingly, this work is the first to identify a signal other than malate that is recognized by the MaeKR regulatory system. Though uncommon, MaeK is not the first transcriptional regulator identified that is able to respond to multiple extracellular signals. In Escherichia coli, the cad operon is regulated by CadC, which recognizes both acidic pH and lysine to induce transcription of the cad genes (52, 53). In Streptococcus mutans, the AguR protein, which controls the expression of the agmatine deiminase system (AgDS), recognizes acidic pH and agmatine (54, 55). An important distinction between CadC, AguR, and MaeK is that the former two proteins require both signals to be present in order to allow for activation and transcription of their target genes. This work has shown, however, that MaeK functions in the presence of either signal and does not require both for transcriptional activation. Nonetheless, given that all three proteins respond to multiple signals, one of which is low pH, they may all share some similar mechanisms of activation. It is hypothesized that for both CadC and AguR, the acidic pH environment induces a conformational change in the protein, and this change then allows for binding of the substrate (lysine and agmatine, respectively) (53, 54). Likewise, in the presence of acidic pH, MaeK may undergo a conformational change that induces activation of the protein. However, unlike CadC and AguR, the MaeK protein likely has a separate malate sensor domain that can bind malate in the presence or absence of acidic pH. This mechanism would predict that MaeK has two distinct regions required for signal recognition and that either can control the activity of the protein.
The identification of a pH-dependent response for ME expression is of particular interest due to the metabolism of the bacterium. S. pyogenes is a member of the lactic acid bacteria, a group that relies on a mix of homolactic and mixed-acid fermentation as a means of generating energy in the cell (6, 56). Over time, in the presence of rapidly metabolized carbohydrates such as glucose, high concentrations of organic acid end products will accumulate (see Fig. S2 in the supplemental material) (5, 6, 56, 57). In this way, there is a direct link between carbohydrate availability and pH, with depletion of glucose leading to a corresponding reduction of the surrounding pH. In this respect, low pH could function as an early warning signal for changes in carbohydrate availability. Thus, low pH may function as an inducer of expression for multiple alternative catabolic operons and is likely not exclusive to malate catabolism alone. Additionally, the MaeKR TCS may be necessary for controlling this pH-adaptive response for these other catabolic operons.
Taken together, this work demonstrates that under conditions of low glucose or acidic pH, malic acid catabolic genes are highly expressed. Given that these signals are among those that S. pyogenes encounters at specific points in a soft tissue infection (8–10, 57), the question of whether this alternative metabolic pathway was important for virulence was of particular interest. Although all of the ME mutants have similar phenotypes in vitro, a loss of growth on malate, the mechanism to cause this deficiency is different for each strain. All three attenuated strains are unable to transport extracellular malate into the cell (either through deletion of the malate transporter gene or loss of maeP expression in an MaeK– or MaeR– mutant). Alternatively, an MaeE– mutant is able to transport malate into the cell but cannot convert this molecule into pyruvate. This differentiation may, in part, explain the in vivo phenotypes observed.
In this case, it remains possible that malate may serve a secondary, as-yet-unknown benefit to the bacterial cell independent of increased pyruvate concentrations. Thus, the ability of an MaeE– mutant to allow uptake of malate may allow for this unused malate to be shuttled into an alternative pathway, ultimately serving to benefit S. pyogenes during infection. Alternatively, MaeP–, MaeK–, and MaeR– mutants would be depleted of any internal malate accumulation, and this loss would ultimately decrease fitness for the cells compared to a WT or an MaeE– mutant. Another possibility is that the accumulation of intracellular malate in the MaeE– mutant may cause the misregulation of virulence factor expression, leading to enhanced virulence. In preliminary studies, we have found that while expression of the SpeB cysteine protease does not differ between WT and the MaeE– and MaeP– mutants (see Fig. S4A in the supplemental material), the addition of malate alters the temporal pattern of SpeB expression in both mutants compared to that in the WT (see Fig. S4B). Since this alteration in SpeB expression is similar between the two mutants, it cannot explain the enhanced virulence of the MaeE– mutant. However, it does support the possibility that alterations to malate metabolism can result in changes in patterns of virulence factor expression. Further analyses of virulence factor expression will be required in order to determine if specific factors are specifically misregulated in the MaeE– mutant and whether these factor are responsible for hypervirulence. This work does, however, provide novel insights into the unique regulatory mechanisms utilized by S. pyogenes for malate degradation, as well as demonstrate for the first time the importance of this alternative metabolic pathway on influencing pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Y. Le Breton and K. McIver for providing the plasmid pCRK.
This work was supported by Public Health Service Grant AI070759 from the National Institutes of Health. The work cited in this publication was performed in a facility supported by NCRR grant C06 RR015502.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02814-14.
REFERENCES
- 1.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Churchward G. 2007. The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol Microbiol 64:34–41. doi: 10.1111/j.1365-2958.2007.05649.x. [DOI] [PubMed] [Google Scholar]
- 3.Hondorp ER, McIver KS. 2007. The Mga virulence regulon: infection where the grass is greener. Mol Microbiol 66:1056–1065. doi: 10.1111/j.1365-2958.2007.06006.x. [DOI] [PubMed] [Google Scholar]
- 4.Kreikemeyer B, McIver KS, Podbielski A. 2003. Virulence factor regulation and regulatory networks in Streptococcus pyogenes and their impact on pathogen-host interactions. Trends Microbiol 11:224–232. doi: 10.1016/S0966-842X(03)00098-2. [DOI] [PubMed] [Google Scholar]
- 5.Martinussen J, Solem C, Holm AK, Jensen PR. 2012. Engineering strategies aimed at control of acidification rate of lactic acid bacteria. Curr Opin Biotechnol 24:124–129. doi: 10.1016/j.copbio.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Neijssel OM, Snoep JL, Teixeira de Mattos MJ. 1997. Regulation of energy source metabolism in streptococci. Soc Appl Bacteriol Symp Ser 26:12S–19S. [PubMed] [Google Scholar]
- 7.Zaunmuller T, Eichert M, Richter H, Unden G. 2006. Variations in the energy metabolism of biotechnologically relevant heterofermentative lactic acid bacteria during growth on sugars and organic acids. Appl Microbiol Biotechnol 72:421–429. doi: 10.1007/s00253-006-0514-3. [DOI] [PubMed] [Google Scholar]
- 8.Graham MR, Virtaneva K, Porcella SF, Barry WT, Gowen BB, Johnson CR, Wright FA, Musser JM. 2005. Group A Streptococcus transcriptome dynamics during growth in human blood reveals bacterial adaptive and survival strategies. Am J Pathol 166:455–465. doi: 10.1016/S0002-9440(10)62268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham MR, Virtaneva K, Porcella SF, Gardner DJ, Long RD, Welty DM, Barry WT, Johnson CA, Parkins LD, Wright FA, Musser JM. 2006. Analysis of the transcriptome of group A Streptococcus in mouse soft tissue infection. Am J Pathol 169:927–942. doi: 10.2353/ajpath.2006.060112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loughman JA, Caparon M. 2006. Regulation of SpeB in Streptococcus pyogenes by pH and NaCl: a model for in vivo gene expression. J Bacteriol 188:399–408. doi: 10.1128/JB.188.2.399-408.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Augagneur Y, Ritt JF, Linares DM, Remize F, Tourdot-Marechal R, Garmyn D, Guzzo J. 2007. Dual effect of organic acids as a function of external pH in Oenococcus oeni. Arch Microbiol 188:147–157. doi: 10.1007/s00203-007-0230-0. [DOI] [PubMed] [Google Scholar]
- 12.Etienne A, Genard M, Lobit P, Mbeguie AMD, Bugaud C. 2013. What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J Exp Bot 64:1451–1469. doi: 10.1093/jxb/ert035. [DOI] [PubMed] [Google Scholar]
- 13.Kawai S, Suzuki H, Yamamoto K, Inui M, Yukawa H, Kumagai H. 1996. Purification and characterization of a malic enzyme from the ruminal bacterium Streptococcus bovis ATCC 15352 and cloning and sequencing of its gene. Appl Environ Microbiol 62:2692–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleijn RJ, Buescher JM, Le Chat L, Jules M, Aymerich S, Sauer U. 2009. Metabolic fluxes during strong carbon catabolite repression by malate in Bacillus subtilis. J Biol Chem 285:1587–1596. doi: 10.1074/jbc.M109.061747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landete JM, Garcia-Haro L, Blasco A, Manzanares P, Berbegal C, Monedero V, Zuniga M. 2010. Requirement of the Lactobacillus casei MaeKR two-component system for l-malic acid utilization via a malic enzyme pathway. Appl Environ Microbiol 76:84–95. doi: 10.1128/AEM.02145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.London J, Meyer EY. 1970. Malate utilization by a group D Streptococcus: regulation of malic enzyme synthesis by an inducible malate permease. J Bacteriol 102:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mortera P, Espariz M, Suarez C, Repizo G, Deutscher J, Alarcon S, Blancato V, Magni C. 2012. Fine-tuned transcriptional regulation of malate operons in Enterococcus faecalis. Appl Environ Microbiol 78:1936–1945. doi: 10.1128/AEM.07280-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landete JM, Ferrer S, Monedero V, Zuniga M. 2013. Malic enzyme and malolactic enzyme pathways are functionally linked but independently regulated in Lactobacillus casei BL23. Appl Environ Microbiol 79:5509–5518. doi: 10.1128/AEM.01177-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemme A, Sztajer H, Wagner-Dobler I. 2010. Characterization of mleR, a positive regulator of malolactic fermentation and part of the acid tolerance response in Streptococcus mutans. BMC Microbiol 10:58. doi: 10.1186/1471-2180-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheng J, Baldeck JD, Nguyen PT, Quivey RG Jr, Marquis RE. 2010. Alkali production associated with malolactic fermentation by oral streptococci and protection against acid, oxidative, or starvation damage. Can J Microbiol 56:539–547. doi: 10.1139/W10-039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheng J, Marquis RE. 2007. Malolactic fermentation by Streptococcus mutans. FEMS Microbiol Lett 272:196–201. doi: 10.1111/j.1574-6968.2007.00744.x. [DOI] [PubMed] [Google Scholar]
- 22.Kawai S, Suzuki H, Yamamoto K, Kumagai H. 1997. Characterization of the l-malate permease gene (maeP) of Streptococcus bovis ATCC 15352. J Bacteriol 179:4056–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Port GC, Paluscio E, Caparon MG. 2013. Complete genome sequence of emm type 14 Streptococcus pyogenes strain HSC5. Genome Announc 1(4):e00612-13. doi: 10.1128/genomeA.00612-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyon WR, Gibson CM, Caparon MG. 1998. A role for trigger factor and an rgg-like regulator in the transcription, secretion and processing of the cysteine proteinase of Streptococcus pyogenes. EMBO J 17:6263–6275. doi: 10.1093/emboj/17.21.6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen HV, Guiton PS, Kline KA, Port GC, Pinkner JS, Neiers F, Normark S, Henriques-Normark B, Caparon MG, Hultgren SJ. 2012. The metal ion-dependent adhesion site motif of the Enterococcus faecalis EbpA pilin mediates pilus function in catheter-associated urinary tract infection. mBio 3(4):e00177-12. doi: 10.1128/mBio.00177-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruiz N, Wang B, Pentland A, Caparon M. 1998. Streptolysin O and adherence synergistically modulate proinflammatory responses of keratinocytes to group A streptococci. Mol Microbiol 27:337–346. doi: 10.1046/j.1365-2958.1998.00681.x. [DOI] [PubMed] [Google Scholar]
- 27.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535. [PubMed] [Google Scholar]
- 28.Meehl MA, Pinkner JS, Anderson PJ, Hultgren SJ, Caparon MG. 2005. A novel endogenous inhibitor of the secreted streptococcal NAD-glycohydrolase. PLoS Pathog 1:e35. doi: 10.1371/journal.ppat.0010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Breton Y, Mistry P, Valdes KM, Quigley J, Kumar N, Tettelin H, McIver KS. 2013. Genome-wide identification of genes required for fitness of group A Streptococcus in human blood. Infect Immun 81:862–875. doi: 10.1128/IAI.00837-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watson ME Jr, Nielsen HV, Hultgren SJ, Caparon MG. 2013. Murine vaginal colonization model for investigating asymptomatic mucosal carriage of Streptococcus pyogenes. Infect Immun 81:1606–1617. doi: 10.1128/IAI.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho KH, Caparon MG. 2008. tRNA modification by GidA/MnmE is necessary for Streptococcus pyogenes virulence: a new strategy to make live attenuated strains. Infect Immun 76:3176–3186. doi: 10.1128/IAI.01721-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brenot A, King KY, Janowiak B, Griffith O, Caparon MG. 2004. Contribution of glutathione peroxidase to the virulence of Streptococcus pyogenes. Infect Immun 72:408–413. doi: 10.1128/IAI.72.1.408-413.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bunce C, Wheeler L, Reed G, Musser J, Barg N. 1992. Murine model of cutaneous infection with Gram-positive cocci. Infect Immun 60:2636–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Port GC, Vega LA, Nylander AB, Caparon MG. 2014. Streptococcus pyogenes polymyxin B-resistant mutants display enhanced ExPortal integrity. J Bacteriol 196:2563–2577. doi: 10.1128/JB.01596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 36.Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70:939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruckner R, Titgemeyer F. 2002. Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol Lett 209:141–148. doi: 10.1016/S0378-1097(02)00559-1. [DOI] [PubMed] [Google Scholar]
- 38.Kietzman CC, Caparon MG. 2009. CcpA and LacD.1 affect temporal regulation of Streptococcus pyogenes virulence genes. Infect Immun 78:241–252. doi: 10.1128/IAI.00746-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gera K, Le T, Jamin R, Eichenbaum Z, McIver KS. 2014. The PEP phosphotransferase system (PTS) in the group A Streptococcus acts to reduce SLS activity and lesion severity during soft tissue infection. Infect Immun 84:1192–1204. doi: 10.1128/IAI.01271-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antunes A, Martin-Verstraete I, Dupuy B. 2011. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79:882–899. doi: 10.1111/j.1365-2958.2010.07495.x. [DOI] [PubMed] [Google Scholar]
- 41.Deutscher J, Herro R, Bourand A, Mijakovic I, Poncet S. 2005. P-Ser-HPr—a link between carbon metabolism and the virulence of some pathogenic bacteria. Biochim Biophys Acta 1754:118–125. doi: 10.1016/j.bbapap.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 42.Heart E, Cline GW, Collis LP, Pongratz RL, Gray JP, Smith PJ. 2009. Role for malic enzyme, pyruvate carboxylation, and mitochondrial malate import in glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 296:E1354–E1362. doi: 10.1152/ajpendo.90836.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deutscher J. 2008. The mechanisms of carbon catabolite repression in bacteria. Curr Opin Microbiol 11:87–93. doi: 10.1016/j.mib.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Fujita Y. 2009. Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci Biotechnol Biochem 73:245–259. doi: 10.1271/bbb.80479. [DOI] [PubMed] [Google Scholar]
- 45.Monedero V, Maze A, Boel G, Zuniga M, Beaufils S, Hartke A, Deutscher J. 2007. The phosphotransferase system of Lactobacillus casei: regulation of carbon metabolism and connection to cold shock response. J Mol Microbiol Biotechnol 12:20–32. doi: 10.1159/000096456. [DOI] [PubMed] [Google Scholar]
- 46.Hondorp ER, Hou SC, Hause LL, Gera K, Lee CE, McIver KS. 2013. PTS phosphorylation of Mga modulates regulon expression and virulence in the group A Streptococcus. Mol Microbiol 88:1176–1193. doi: 10.1111/mmi.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hondorp ER, Hou SC, Hempstead AD, Hause LL, Beckett DM, McIver KS. 2012. Characterization of the group A Streptococcus Mga virulence regulator reveals a role for the C-terminal region in oligomerization and transcriptional activation. Mol Microbiol 83:953–967. doi: 10.1111/j.1365-2958.2012.07980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribardo DA, McIver KS. 2006. Defining the Mga regulon: comparative transcriptome analysis reveals both direct and indirect regulation by Mga in the group A streptococcus. Mol Microbiol 62:491–508. doi: 10.1111/j.1365-2958.2006.05381.x. [DOI] [PubMed] [Google Scholar]
- 49.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fogg GC, Gibson CM, Caparon MG. 1994. The identification of rofA, a positive-acting regulatory component of prtF expression: use of an m gamma delta-based shuttle mutagenesis strategy in Streptococcus pyogenes. Mol Microbiol 11:671–684. doi: 10.1111/j.1365-2958.1994.tb00345.x. [DOI] [PubMed] [Google Scholar]
- 51.Granok AB, Parsonage D, Ross RP, Caparon MG. 2000. The RofA binding site in Streptococcus pyogenes is utilized in multiple transcriptional pathways. J Bacteriol 182:1529–1540. doi: 10.1128/JB.182.6.1529-1540.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neely MN, Olson ER. 1996. Kinetics of expression of the Escherichia coli cad operon as a function of pH and lysine. J Bacteriol 178:5522–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dell CL, Neely MN, Olson ER. 1994. Altered pH and lysine signalling mutants of cadC, a gene encoding a membrane-bound transcriptional activator of the Escherichia coli cadBA operon. Mol Microbiol 14:7–16. doi: 10.1111/j.1365-2958.1994.tb01262.x. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y, Zeng L, Burne RA. 2009. AguR is required for induction of the Streptococcus mutans agmatine deiminase system by low pH and agmatine. Appl Environ Microbiol 75:2629–2637. doi: 10.1128/AEM.02145-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y, Burne RA. 2009. Multiple two-component systems of Streptococcus mutans regulate agmatine deiminase gene expression and stress tolerance. J Bacteriol 191:7363–7366. doi: 10.1128/JB.01054-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yesilkaya H, Spissu F, Carvalho SM, Terra VS, Homer KA, Benisty R, Porat N, Neves AR, Andrew PW. 2009. Pyruvate formate lyase is required for pneumococcal fermentative metabolism and virulence. Infect Immun 77:5418–5427. doi: 10.1128/IAI.00178-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cusumano ZT, Watson ME Jr, Caparon MG. 2013. Streptococcus pyogenes arginine and citrulline catabolism promotes infection and modulates innate immunity. Infect Immun 82:233–242. doi: 10.1128/IAI.00916-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.