

Abstract
Attaching and effacing (A/E) pathogens adhere intimately to intestinal enterocytes and efface brush border microvilli. A key virulence strategy of A/E pathogens is the type III secretion system (T3SS)-mediated delivery of effector proteins into host cells. The secreted protein EspZ is postulated to promote enterocyte survival by regulating the T3SS and/or by modulating epithelial signaling pathways. To explore the role of EspZ in A/E pathogen virulence, we generated an isogenic espZ deletion strain (ΔespZ) and corresponding cis-complemented derivatives of rabbit enteropathogenic Escherichia coli and compared their abilities to regulate the T3SS and influence host cell survival in vitro. For virulence studies, rabbits infected with these strains were monitored for bacterial colonization, clinical signs, and intestinal tissue alterations. Consistent with data from previous reports, espZ-transfected epithelial cells were refractory to infection-dependent effector translocation. Also, the ΔespZ strain induced greater host cell death than did the parent and complemented strains. In rabbit infections, fecal ΔespZ strain levels were 10-fold lower than those of the parent strain at 1 day postinfection, while the complemented strain was recovered at intermediate levels. In contrast to the parent and complemented mutants, ΔespZ mutant fecal carriage progressively decreased on subsequent days. ΔespZ mutant-infected animals gained weight steadily over the infection period, failed to show characteristic disease symptoms, and displayed minimal infection-induced histological alterations. Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining of intestinal sections revealed increased epithelial cell apoptosis on day 1 after infection with the ΔespZ strain compared to animals infected with the parent or complemented strains. Thus, EspZ-dependent host cell cytoprotection likely prevents epithelial cell death and sloughing and thereby promotes bacterial colonization.
INTRODUCTION
The attaching and effacing (A/E) group of bacteria includes Gram-negative enteric pathogens that colonize the intestine and cause disease in diverse animals (1, 2). A/E pathogens adhere intimately to intestinal epithelial cells and efface the brush border microvilli (3). Enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli (EHEC) are human-specific A/E pathogens that cause diarrhea and contribute to significant morbidity. EPEC is an important agent of diarrheal disease in children, particularly in developing countries (4). EHEC, a foodborne pathogen that causes watery or bloody diarrhea, is often associated with outbreaks (5). The mouse pathogen Citrobacter rodentium and rabbit EPEC (REPEC) have been used to model EPEC- and EHEC-induced human disease (6–8). C. rodentium is a nonmotile enteric pathogen that causes colonic hyperplasia, but typically not diarrhea, in mice. On the other hand, REPEC causes diarrhea in rabbits and elicits disease that more closely resembles human A/E infections.
All A/E pathogens harbor a type III secretion system (T3SS) that is essential for inducing effacing lesions and causing disease. The T3SS conveys a diverse array of bacterial effector molecules directly into intestinal epithelial cells, where these proteins engage and subvert host cell structural proteins and signaling pathways (9). Studies from several laboratories have uncovered specific host cell-interacting partners of various secreted proteins, and the consequent intestinal cell signaling and structural alterations have been characterized in some detail. In vivo studies have shown the essentiality of the T3SS for virulence, although the requirement for specific secreted effectors is variable (10). While some effector molecules, such as Tir, are critical for virulence (11, 12), deletion mutants lacking other effectors exhibit a colonization defect only in competition studies with the isogenic parent wild-type (WT) strain (13). Colonization by strains mutated for several other effectors, however, is not significantly different from that by the parent strain either alone or in competition studies (14, 15).
EspZ is a 99- to 100-amino-acid secreted effector molecule that was first implicated in virulence in the C. rodentium-mouse model of infection (16, 17). C. rodentium ΔespZ strains colonized the intestines of NIH Swiss mice but failed to cause the characteristic colonic hyperplasia (17). Furthermore, while WT C. rodentium strains caused 100% mortality in the more susceptible C3H/HeJ mice, all animals infected with isogenic ΔespZ strains survived infection (17). Since those observations constituted only a minor aspect of a broader study of C. rodentium virulence factors, complementation data were not provided, and the mechanisms of EspZ action were not explored in that study.
The precise contribution of EspZ to A/E virulence is currently unknown. We and others have demonstrated that EspZ is cytoprotective and that infection with an EPEC ΔespZ mutant, relative to WT EPEC, results in increased epithelial cell death in vitro (18–21). Two mechanisms, perhaps not mutually exclusive, have been proposed to account for the cytoprotective roles of EspZ. The first proposed mechanism is that EspZ, possibly via interactions with host proteins, engages epithelial cell cytoprotective signaling pathways or inhibits processes that trigger the death of infected cells (18–20, 22). The alternate mechanism, proposed by Berger et al., suggests that EspZ may directly modulate the translocation of effector proteins into host cells and that the increased death of ΔespZ mutant-infected cells results from the unregulated delivery of secreted proteins (including cytotoxic effectors) into host cells (21). Furthermore, strain specificity was noted for the EspZ-dependent regulation of type III secretion: EHEC EspZ was shown to block both EPEC- and EHEC-mediated translocation, while EPEC EspZ was able to inhibit translocation from EPEC but not EHEC.
Since A/E pathogen infection in rabbits most closely approximates human disease, the goal of this study was to use this model to determine the role of EspZ in virulence. First, we verified that the in vitro phenotypes attributed to EPEC EspZ held true for the REPEC strain (serotype O103:H2; also known as E22) and that REPEC espZ could complement the EPEC ΔespZ mutant and vice versa. To examine the role of EspZ in virulence, rabbits were mock infected or were infected with the following REPEC strains: the WT, the ΔespZ strain, and single-copy cis-complemented ΔespZ strains (complemented with EPEC or REPEC espZ). Bacterial burden, disease parameters, tissue histological alterations, apoptotic cell death, and host responses of intestinal tissues were monitored for the duration of the study. Our observations reveal that REPEC espZ is essential for virulence in the rabbit model and that the EPEC and REPEC espZ genes exhibit functional similarity under both in vitro and in vivo conditions. We propose that EspZ limits host intestinal epithelial cell death, thereby preserving the substratum and promoting bacterial colonization.
(Portions of this work were presented at the 2012 Digestive Diseases Week in San Diego, CA, and at the 2013 FASEB Summer Conference on Gastrointestinal Mucosa in Steamboat Springs, CO.)
MATERIALS AND METHODS
Bacterial strains and plasmids.
The strain designated WT REPEC in this study refers to a Nalr derivative of the sequenced O103:H2 REPEC strain, also known as strain E22 (11). The isogenic nonpolar REPEC ΔespZ strain was generated by using a SacB-based counterselection strategy (23). Fragments flanking the upstream and downstream regions of REPEC espZ were amplified by using primer pairs NoEspZ2/NiEspZR and CoEspZR2/CiEspZ (Table 1), respectively, fused via crossover PCR, and cloned into the SalI site of pTOF24 (23). The resulting plasmid was digested with SmaI and ligated with a similarly digested nonpolar kanamycin resistance cassette amplified from MK41 (16), using primer pair KanF/KanR (Table 1). The final construct was transformed into REPEC E22, and the disruption and loss of espZ in kanamycin-resistant, sucrose-resistant, chloramphenicol-sensitive colonies were verified by PCR.
TABLE 1.
Bacterial strains, plasmids, and primers
| Strain, plasmid, or primer | Genotype and/or description or sequence (5′–3′) | Reference |
|---|---|---|
| Strains | ||
| ECB480 | E22 (WT REPEC; serotype O103:H2) (Nalr) | 11 |
| VK550 | E22 ΔespZ (Nalr Kanr) | This work |
| VK581 | E22 ΔespZΩespZREPEC (Nalr Kanr); REPEC ΔespZ cis-complemented with REPEC espZ | This work |
| VK582 | E22ΔespZΩespZEPEC (Nalr Kanr); REPEC ΔespZ cis-complemented with EPEC espZ | This work |
| Plasmids | ||
| pMB196 | Vector backbone for cloning TEM-1 fusions | 25 |
| pMB201 | Vector control for translocation assays | 25 |
| pSR1 | Expresses the first 20 amino acids of EspZ fused to TEM-1 β-lactamase; used for translocation assays | This work |
| pSR2 | Plasmid for transient expression of REPEC espZ in eukaryotic cells; REPEC espZ allele cloned into pSR4 | This work |
| pSR3 | Plasmid for transient expression of EPEC espZ in eukaryotic cells; EPEC espZ allele cloned into pSR4 | This work |
| pSR4 | Lentiviral expression plasmid based on the pCIP-TTL-HA backbonea | This work |
| pGRG36 | Plasmid used to generate pJSW201 and pJSW101 | 24 |
| pJSW101 | Harbors EPEC espZ; used to generate VK582 | 18 |
| pJSW201 | Harbors REPEC espZ; used to generate VK581 | This work |
| Primers | ||
| Ni-EspZR | AGCGCAGTAACCCGGGGCTGCATCCATTAACTTTTCTCCA | |
| No-EspZ2 | TTTTGTCGACAGCGGGTCAATAACGCCTGC | |
| Ci-EspZ | TGGATGCAGCCCCGGGTTGGCGGCGGATTAGCGATGAAATATGC | |
| Co-EspZR2 | GTTCGTCGACGCCGTTAGCCAGTCACTATC | |
| KanF | CCCGGCATGCTAACTAGGAGGAATAAATG | |
| KanR | ATTACCCGTCCTGTCCTGAG | |
| RespZF | TGCAGGCTCTGAAGTAAGTAT | |
| RespZR | TTAGGCATATTTCATCGCTA | |
| SR1 | TCTAGAATGGAAGCAGCAAATTTAAGCCCTT | |
| SR2 | TCCGGAGGCATATTTCATCGCTAATCCGC | |
| SR3 | TCTAGAATGGATGCAGCAAATTTAAGCCCTT | |
| SR4 | TCCGGAGGCATATTTCATCGCTAATCCGC | |
| SR15 | TCATGAATTCCTGCAGGCTCTGAAGTAAG | |
| SR16 | TTCCGGTACCATTGATTGTGGCTGCCAGTG |
Kind gift from Felicia Goodrum, University of Arizona.
For cis-complementation of the REPEC ΔespZ strain, REPEC espZ, along with its native promoter, was amplified by using primer pair RespZF/RespZR and cloned into pGEM-T Easy (Promega, Madison, WI). The espZ-containing fragment was excised by using NotI and cloned into similarly digested pGRG36 (24) to generate pJSW201. A similar plasmid harboring EPEC espZ, pJSW101, was generated previously (18). To generate REPEC and EPEC espZ complements (VK581 and VK582, respectively), the REPEC ΔespZ strain was transformed with pJSW201 or pJSW101 and screened for the Tn7-mediated transposition of espZ into the neutral site downstream of glmS (24).
To generate a TEM-1 β-lactamase reporter plasmid for the translocation assays, the region encoding the type III secretion signal (first 20 amino acids) of EspZ, along with the native locus of enterocyte effacement 2 (LEE2) promoter, was amplified from EPEC (strain E2348/69) by using primer pair SR15/SR16, digested with KpnI and EcoRI, and cloned into similarly digested pMB196, in frame with β-lactamase (25), to generate plasmid pSR1. Plasmid pMB201 was used as the vector control (25).
For transient expression of espZ in eukaryotic cells, the corresponding REPEC and EPEC isoforms were amplified by using primer pairs SR3/SR4 and SR1/SR2, respectively, and cloned into pGEM-T Easy (Promega, Madison, WI). The espZ fragments were then excised with XbaI and BspEI and cloned into the corresponding sites in pCIP, in frame with the hemagglutinin (HA) tag and upstream of the internal ribosome entry site (IRES) sequence, to generate plasmids pSR2 and pSR3, respectively. pCIP, derived from the lentiviral expression plasmid pCIG (26) by the insertion of a puromycin resistance cassette downstream of the IRES sequence, was a kind gift from Felicia Goodrum, University of Arizona.
Cell lines and in vitro infection studies.
Caco-2 BBE (C2BBE) cells (as well as HeLa and T84 cells [not shown]) were used to monitor infection-induced host cell death, while the translocation assays were performed with HeLa cells. Death of infected host cells was assessed via propidium iodide (PI) uptake assays as described previously (18).
Translocation assays.
Translocation assays were performed as previously described (21, 27), with some modifications. For the experiments described in the legend of Fig. 2A, HeLa cells were first transiently transfected with pSR2 (harboring REPEC espZ), pSR3 (harboring EPEC espZ), or a lentiviral expression plasmid backbone (pSR4) (control), and translocation of TEM-1 β-lactamase from a subsequent infection with the REPEC strain harboring pSR1 (REPEC/pSR1) was monitored by using a fluorescence-based assay. For the experiments described in the legend of Fig. 2B, HeLa cells were infected with various REPEC strains (“first-wave infection”), and translocation of TEM-1 β-lactamase from a subsequent infection with REPEC/pSR1 was monitored by using the same fluorescence-based assay as the one described above. For transfection studies, HeLa cells seeded into 6-well plates were transfected with pSR2 or pSR3 (REPEC or EPEC espZ) or with pSR4 (vector control) by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 24 h, the transfected cells were seeded into 96-well, black-walled, clear-bottom microplates (PerkinElmer, Waltham, MA) and grown to confluence at 37°C in the presence of 5% CO2. The cells were treated overnight with serum-free Dulbecco's modified Eagle's medium (DMEM) supplemented with mannose and HEPES and then infected with REPEC/pSR1 to monitor translocation. Bacterial cells grown overnight in LB broth were diluted 1:100 in Casamino Acids-DMEM (cDMEM) supplemented with 2.5 mM probenecid until they reached an optical density at 600 nm (OD600) of 0.2 to 0.35. cDMEM is a tissue culture medium with low autofluorescence (28), and probenecid, a nonspecific anion inhibitor, facilitates CCF2-AM [coumarin-cephalosporin-fluorescein (2)-acetoxymethyl] loading by blocking its active efflux from cells (29). HeLa cells preloaded with CCF2-AM (GeneBLAzer In Vivo detection kit; Life Technologies, Carlsbad, CA) were infected with ∼106 bacteria (multiplicity of infection [MOI] of 500 to 700). The cultures were diluted as needed to normalize OD levels. Infection was monitored with a plate reader (Synergy HT; Biotek) set at 37°C for 120 min. Cells were excited at 400/10 nm, and emission was recorded at 460/40 nm and 520/10 nm, respectively, for 2 h at 5-min intervals.
FIG 2.

The presence of EspZ in host cells can limit T3SS-dependent effector translocation. A TEM-1 β-lactamase (encoded on pSR1) reporter system was used to monitor effector translocation from WT REPEC into host cells transiently transfected with an REPEC espZ-containing (pSR2), EPEC espZ-containing (pSR3), or control (pSR4) plasmid (A) or previously infected (first wave) with WT REPEC, the REPEC ΔespZ mutant, or the REPEC ΔespZ mutant complemented with REPEC EspZ (espZ/espZR) or EPEC EspZ (espZ/espZE) (B). TEM-1 β-lactamase translocation-dependent cleavage of the β-lactam-containing compound CCF2 in host cells was monitored via fluorescence measurements over a 120-min time period. Asterisks indicate significant differences (P ≤ 0.05).
For the two-wave infections, HeLa cells were seeded into 96-well, black-walled, clear-bottom microplates (PerkinElmer) and allowed to grow to confluence. The cells were treated overnight with serum-free DMEM supplemented with mannose and HEPES. Bacterial cells grown overnight in LB broth were diluted 1:100 in cDMEM supplemented with probenecid until they reached an OD600 of 0.2 to 0.35. HeLa cells preloaded with CCF2-AM were infected with ∼106 first-wave bacteria and incubated at 37°C for 30 min. The cells were then washed with cDMEM to remove unattached bacteria and infected with ∼106 bacteria (MOI of between 500 and 700) of the second-wave REPEC/pSR1 infection. Translocation of TEM-1 β-lactamase was monitored as described above.
Rabbit infection studies.
All animal experiments and procedures were conducted with IACUC approval from the New Mexico VA Healthcare System (NMVAHCS) IACUC Committee. Groups of 6- to 8-week-old, specific-pathogen-free, New Zealand White (NZW) male rabbits (Charles River, Wilmington, MA), weighing 1.4 to 1.8 kg, were infected with WT REPEC or the ΔespZ or cis-complemented ΔespZ strains (with either REPEC or EPEC espZ) and monitored for a total period of up to 7 days. Briefly, the animals were administered 2 ml 10% sterile sodium bicarbonate via an orogastric tube, followed by 2 ml of a bacterial solution (bacteria suspended in phosphate-buffered saline [PBS]). Bacteria remaining in the orogastric tube were flushed with an additional 1 ml of sterile PBS. Animal weight, clinical signs, and mortality/morbidity were monitored on a daily basis, and rabbits were scored for disease as described in Table S1 in the supplemental material; fecal swabs and/or pellets were monitored for bacterial loads on a daily basis and are reported as CFU/g feces. At the end of the study, infected animals were euthanized by the intramuscular administration of ketamine (35 mg/kg of body weight) and xylazine (7 mg/kg of body weight), followed by an overdose of intracardiac pentobarbital-sodium and phenytoin sodium (Euthasol) (1 ml per rabbit). Animals were necropsied on days 1, 3, and 7, and several samples were collected for subsequent analysis. Intestinal contents and tissues (ileum, cecum, and colon) were homogenized and plated for an estimation of the bacterial burden (intralumenal and attached). For hematoxylin-and-eosin (H&E) staining, intestinal tissues (ileum, cecum, and colon) were fixed in 10% buffered formalin (4% formaldehyde). The sections for histopathology were obtained from identically sampled sites within ceca of all rabbits.
Cytokine measurements.
Cytokine expression in intestinal mucosal scrapings was quantified by quantitative reverse transcription-PCR (qRT-PCR) using rabbit-specific primers for proinflammatory cytokines, as described previously (30). Briefly, intestinal mucosal scrapings were collected on days 3 and 7 postinfection and frozen in RNAlater solution (Life Technologies, Grand Island, NY) at −80°C. Total RNA was extracted by using an RNeasy minikit (Qiagen, Valencia, CA) and reverse transcribed with an RT kit (Qiagen, Valencia, CA), and mRNA was quantitated by using qRT-PCR for interleukin-1β (IL-1β), IL-6, and gamma interferon (IFN-γ). Normalization to the hypoxanthine phosphoribosyltransferase (HPRT) gene was performed.
TUNEL staining of rabbit tissues for evaluation of epithelial apoptosis.
Rabbit ileal tissues were collected and stored in 3% formalin during necropsy. Five-micrometer-thick sections of paraffin-embedded tissues were attached to glass slides. The DeadEnd fluorometric terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) system (Promega), which labels DNA with fluorescein-12-dUTP at the 3′ end, was used to stain apoptotic cells, according to the manufacturer's protocol. Cells were also stained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize DNA and localize both healthy and apoptotic nuclei. Images were collected with a Zeiss 510 confocal microscope and processed by using ImageJ64. The only manipulation of the raw data was changing color of the DAPI pixels to red and the color of fluorescein to green for better image contrast.
Statistical analyses.
A Microsoft-based software package was used for statistical analyses. Significance was determined by using two-tailed Student's t tests (P values of ≤0.05 were considered significant) as well as analysis of variance (ANOVA), where goodness of fit and standardized coefficients of variance were derived in order to facilitate comparisons between multiple groups of independently collected and continuous numerical data. For the translocation studies, and since the number of observations over the various time points was very large (660 total), goodness-of-fit statistical analyses were performed by using ANOVA followed by a stringent Tukey test. P values of <0.05 were considered significant.
RESULTS
REPEC and EPEC EspZ proteins delay death of infected host intestinal epithelial cells.
To begin exploring the role of EspZ in the rabbit model of infection, we generated a nonpolar espZ deletion mutant (ΔespZ) of REPEC; this mutant was cis-complemented with REPEC or EPEC espZ by the insertion of the respective genes, along with their native promoter sequences, into the site adjacent to glmS via Tn7 transposition (to generate the ΔespZ/espZR and ΔespZ/espZE strains, respectively). RT-PCR analyses confirmed near-WT levels of espZ expression in the complemented ΔespZ/espZR strain (Fig. 1A). Similarly, espZ expression in the complemented ΔespZ/espZE strain was also verified (not shown). Furthermore, comparable expression levels were observed for the gene downstream of espZ (escI) in the WT, ΔespZ, and complemented strains, confirming that the espZ deletion/disruption did not result in transcriptional polarity (data not shown).
FIG 1.

Both the REPEC and EPEC EspZ proteins protect epithelial cells from REPEC-induced death. (A) Reverse transcription analyses demonstrating comparable espZ expression levels in the REPEC parental (WT) strain and the cis-complemented REPEC ΔespZ strains. Relative gene expression levels are shown and were derived via densitometric analyses of the linear phase of espZ amplicon accumulation. Data are normalized to the expression levels of the E. coli genes escI and groE. (B) PI uptake cell death assay using C2BBE host cells infected with the parent REPEC strain (green bar), the ΔespZ mutant (red bar), and the ΔespZ mutant cis-complemented with espZ from REPEC (ΔespZ/espZR) (dark blue bar) or from EPEC (espZ/espZE) (light blue bar) for 4.5 h. Data are presented as average PI uptake rates ± standard errors of the means for eight technical replicates. Asterisks indicate significant differences (P ≤ 0.05).
We previously demonstrated that EPEC espZ delays the death of infected human intestinal epithelial cells (18). To determine if EspZ played a similar role in the REPEC strain background, C2BBE intestinal epithelial cells were infected with the REPEC WT, ΔespZ, ΔespZ/espZR, or ΔespZ/espZE strain, and host cell survival was quantitated by monitoring propidium iodide uptake. As described above for the corresponding EPEC strains, WT REPEC induced marked death of infected host epithelial cells compared to mock-infected cells. ΔespZ strains induced a greater degree of host cell death than did the isogenic parent strain, and this was reversed by cis-complementation with either REPEC or EPEC espZ (Fig. 1B). Furthermore, both EPEC and REPEC espZ-transfected intestinal epithelial cells were protected from staurosporine-induced apoptosis, suggesting a direct engagement of cytoprotective signaling pathways by these proteins (18) (data not shown).
REPEC EspZ inhibits type III secretion system-mediated effector translocation.
Using a complex experimental setup, Berger et al. observed that the EPEC and EHEC EspZ proteins could interfere with the bacterial T3SS-mediated delivery of effector molecules into host cells (21). Those investigators demonstrated that large amounts of EPEC EspZ in host cells, either expressed from a transfected construct or delivered via a first-wave infection (at a high multiplicity of infection [MOI] of >500), could inhibit T3SS-dependent translocation from a subsequent second wave of EPEC infection. To determine if this function was conserved in REPEC EspZ and if EPEC and REPEC espZ molecules could cross-complement for this phenotype, we performed similar translocation assays. Consistent with the observations for EPEC, REPEC strains were impaired for the translocation of a TEM-1 β-lactamase reporter protein into host cells transfected with REPEC or EPEC espZ compared to vector-transfected controls (Fig. 2A).
A similar conservation of function between the EPEC and REPEC EspZ proteins was observed in the two-wave infection system, which employed a net MOI (number of bacteria/host cell) of >500. The rate of REPEC T3SS-dependent translocation of the reporter protein into epithelial cells (second wave) was significantly lower when host cells were infected previously (first wave) with WT REPEC than when host cells were treated with a first-wave ΔespZ strain infection (Fig. 2B). Surprisingly, however, cis-complementation with either REPEC or EPEC espZ failed to restore the translocation phenotype of the ΔespZ strain (Fig. 2B), even though espZ expression in the ΔespZ/espZR strain was verified to be comparable to that of the parent WT strain (Fig. 1A). The lack of complementation was also specific for the translocation phenotype, since complementation was successful with the same strains in the cell death assays (MOI = 100) (Fig. 1B). Notably, the original report by Berger et al. did not provide complementation data in connection with the EPEC ΔespZ strain's influence on effector translocation (21). The use of extremely high MOIs and the lack of complementation highlight the need for caution in interpreting the results of two-wave infections. Overall, the results suggest that REPEC espZ, like EPEC espZ, may, under certain circumstances (such as transfection-mediated high-level expression), influence the translocation of effector proteins.
EspZ is essential for causing disease in a rabbit model of infection.
To assess the role of EspZ in A/E pathogen virulence, New Zealand White rabbits were infected with WT REPEC or the ΔespZ, ΔespZ/espZR, or ΔespZ/espZE isogenic derivative and monitored for various parameters over 7 days. Clinical signs of disease were pronounced, and the scores progressively increased in WT REPEC-infected animals (Fig. 3A; see also Table S1 in the supplemental material); these signs included moderate-to-severe diarrhea and weight loss (Fig. 3B) over the 7-day period. In contrast, ΔespZ strain-infected animals were clinically indistinguishable from control, uninfected animals and gained weight normally for the duration of the study (Fig. 3). cis-complementation with either REPEC or EPEC espZ partially restored the virulence of the ΔespZ strains, with the animals displaying moderate clinical signs (Fig. 3A) but not the dramatic weight loss noted for WT-infected animals (Fig. 3B). This finding suggests that EspZ is an essential determinant of A/E pathogen virulence and that the REPEC and EPEC EspZ proteins display considerable functional congruence in the rabbit model of infection.
FIG 3.

Rabbits infected with the REPEC ΔespZ strain are clinically indistinguishable from mock-infected animals. Data shown are representative of data from a total of three independent animal studies, each performed with a minimum of eight animals per group on day 1. (A) Clinical sign scores of the animals over 7 days of infection with the parent WT strain, the ΔespZ mutant, and the ΔespZ mutant cis-complemented with REPEC espZ (espZ/espZR) or EPEC espZ (espZ/espZE). Scores are based on four parameters, as detailed in Table S1 in the supplemental material (with “0” referring to no clinical signs), and are for an average of at least three animals per group per time point. The number of animals decreased during the course of the study due to death/euthanasia; therefore, standard statistical analyses were not performed. (B) Weight change(s) of animals over the course of 7 days of infection with the parent WT strain, the ΔespZ mutant, and the ΔespZ mutant cis-complemented with REPEC espZ (espZ/espZR) or EPEC espZ (espZ/espZE).
EspZ is essential for intestinal REPEC colonization.
To determine if disease progression correlated with intestinal colonization of the different strains, stool and cecal tissue samples from infected animals were processed and plated onto selective media. We focused on cecal colonization since a pilot experiment revealed maximal bacterial burden at this site compared to the ileum and the rectum (not shown). WT colonization and expansion were evident in ceca of infected animals (Fig. 4C), and bacteria were shed at high levels in the stool throughout the duration of the study (Fig. 4A and B). For the ΔespZ strain-colonized animals, stool ΔespZ cell counts were >10-fold lower than those of the WT on day 1 postinfection and steadily decreased to a level ∼100-fold lower than those of the WT by day 7 postinfection. Cecal ΔespZ cell counts were comparable to those of the parent WT strain only on day 1 postinfection and decreased to levels ∼15-fold lower than those of WT REPEC on day 7 postinfection (Fig. 4C). cis-complementation of the mutant with REPEC or EPEC espZ increased bacterial loads in both the cecum and the stool relative to the ΔespZ strain, although colonization was less robust than that by the parent WT strain. Taken together, these data suggest that EspZ is essential for REPEC intestinal colonization and expansion and that both the REPEC and EPEC espZ genes can partially restore function to a REPEC ΔespZ strain in vivo.
FIG 4.

The REPEC ΔespZ strain does not expand in, and is rapidly cleared from, infected rabbits. The data shown are representative of a total of three independent animal studies, each performed with a minimum of eight animals per group on day 1. (A) The bacterial burden throughout rabbit infection with the parent WT strain, the ΔespZ mutant, or the ΔespZ mutant cis-complemented with REPEC espZ (espZ/espZR) or EPEC espZ (espZ/espZE) or in mock-infected animals was enumerated. Fecal pellets were collected from a minimum of three animals per group per day and plated onto medium selective for the infecting REPEC strain, as described in Materials and Methods. Data shown are average CFU per gram of fecal matter for each group of animals over time. (B) Bacterial burden data from panel A highlighting pathogen accumulation at the first (day 1) and last (day 7) time points following infection of rabbits with the parent WT strain, the ΔespZ mutant, or the ΔespZ mutant cis-complemented with espZ from REPEC (espZ/espZR) or EPEC (espZ/espZE). Data shown are average CFU per gram of fecal matter for each group of animals. (C) Enumeration of bacteria adherent to the cecum over time for rabbits infected with the parent WT strain, the ΔespZ mutant, or the ΔespZ mutant cis-complemented with REPEC espZ (espZ/espZR) or EPEC espZ (espZ/espZE). Ceca from euthanized animals were dissected, and REPEC bacteria were recovered as described in Materials and Methods. Data shown are average CFU per gram of cecal tissue for each group of animals. The limit of detection was 100 CFU/mg tissue. Since the number of animals in each group decreased variably over time due to the need to euthanize ill or moribund animals, statistical analyses were not performed on these data.
EspZ is essential for REPEC to cause heightened and sustained tissue injury.
Histopathological examination of hematoxylin-and-eosin (H&E)-stained sections of the ceca of rabbits infected with REPEC, the complemented mutants, or the ΔespZ strain revealed mild hyperemia and a mild heterophilic infiltrate in the lamina propria at day 1 postinfection (Fig. 5). Slight to moderate multifocal enterocyte death with mild mucosal attenuation was also observed for the ΔespZ strain-infected rabbits. At day 3 postinfection, the severity of inflammation had progressed in the ceca of rabbits infected with REPEC or the complemented mutants but not in the ΔespZ strain-infected rabbits. By day 7 postinfection, there was a moderate to multifocally severe heterophilic and lymphoplasmacytic infiltrate in the lamina propria and submucosa, accompanied by multifocal proprial and submucosal microhemorrhage in the ceca of rabbits infected with REPEC or the complemented mutants. Multifocal mucosal attenuation progressing to ulceration with fibrinous exudation and accumulation of cellular debris in cecal crypts were also evident in these animals (Fig. 5). In contrast, by day 7 postinfection, cecal damage was considerably mitigated in the ΔespZ strain-infected animals, with only mild hyperemia, scattered proprial microhemorrhage, and a mild heterophilic and lymphoplasmacytic infiltrate limited to the lamina propria being observed. Mild multifocal enterocyte death was also observed for the ΔespZ strain-infected animals, but unlike for the WT- and complemented mutant-infected animals, no ulceration was identified.
FIG 5.
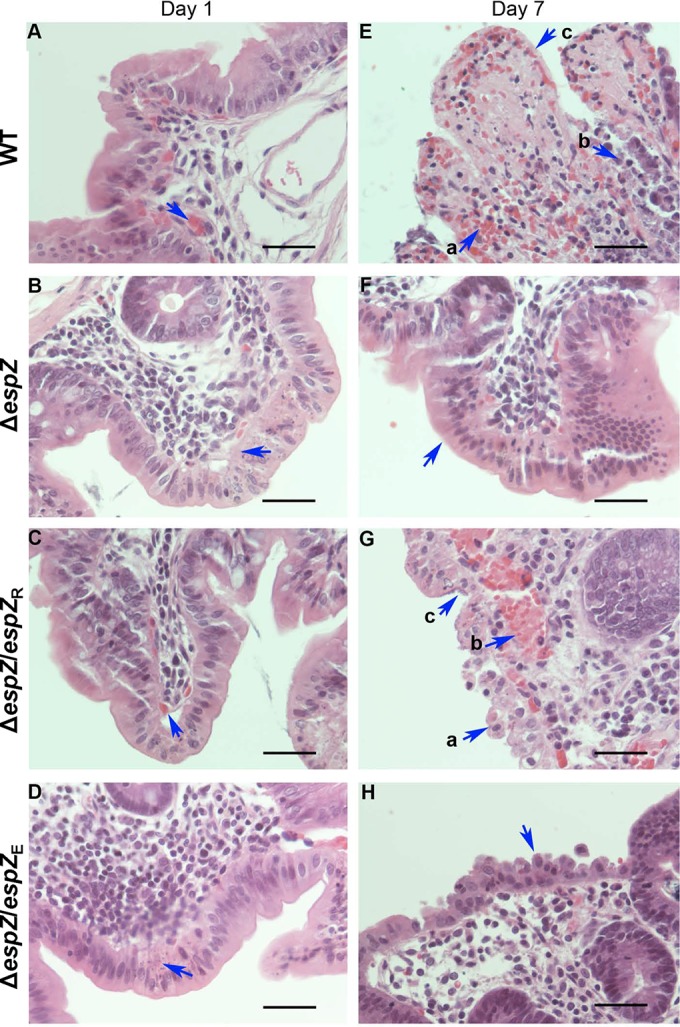
Infection with the ΔespZ REPEC strain causes minimal and unsustained gut tissue damage. (A to D) H&E-stained sections of rabbit cecum on day 1 postinfection. Shown are representative images from a minimum of three animals per group infected with WT REPEC (A), the ΔespZ mutant (B), or the ΔespZ mutant cis-complemented with REPEC espZ (espZ/espZR) (C) or EPEC espZ (espZ/espZE) (D). Arrows are positioned over sites of proprial or submucosal hemorrhage (a to c) or over apoptosing enterocytes (d). Bar, 50 μm. (E to H) H&E-stained sections of rabbit cecum on day 7 postinfection. Shown are representative images from a minimum of three animals per group infected with either WT REPEC (E), the ΔespZ mutant (F), or the ΔespZ mutant cis-complemented with REPEC espZ (espZ/espZR) (G) or EPEC espZ (espZ/espZE) (H). Arrows are positioned over sites of hemorrhage (Ea, Ec, and Gb), breached or damaged epithelial cells (Eb, Ga, and H), epithelial cells coated with bacteria (Gc), or epithelial cells with normal morphology (F). Bar, 50 μm. Tissue alterations were scored by an American College of Veterinary Pathologists board-certified pathologist (M. W. Riggs) blind to treatment conditions.
EspZ is essential for inducing proinflammatory cytokine production from infected tissues in vivo.
We next sought to determine if the impaired colonization and decreased tissue injury observed for ΔespZ strain-infected animals correlated with altered proinflammatory cytokine production. Infection with the WT, but not the ΔespZ strain, resulted in significantly elevated levels of IL-6, but not IL-1β or IFN-γ, as early as day 1 postinfection (Fig. 6). By day 7 postinfection, however, WT REPEC induced significant increases in the levels of all three cytokines. In contrast, the production of each of these cytokines by ΔespZ strain-infected animals was indistinguishable from that in the PBS-treated control animals. Taken together, these data suggest that EspZ promotes intestinal bacterial colonization, which in turn results in increased epithelial tissue injury and increased cytokine production.
FIG 6.

Proinflammatory cytokine levels remain unaltered over time in rabbits infected with the ΔespZ REPEC strain. Intestinal mucosal scrapings were used for quantitation of multiple cytokines on day 3 (A) or day 7 (B) postinfection in rabbits gavaged with PBS or the WT or isogenic ΔespZ strain. RNA was isolated from the samples as described in Materials and Methods and subjected to reverse transcription and quantitative PCR to determine IL-1β, IL-6, and IFN-γ levels. The relative gene expression level was obtained by normalizing the specific cytokine gene expression level to that of the control hypoxanthine-guanine phosphoribosyltransferase gene.
EspZ limits intestinal epithelial cell apoptosis during the early stages of infection.
Based on the observed in vitro cytoprotective phenotype for EspZ, we hypothesized that the ΔespZ strain induced heightened enterocyte death in the rabbit intestine early (days 1 to 2) in infection. We further hypothesized that the sloughing of apoptotic epithelial cells, along with the attached bacteria, would limit colonization. To test this hypothesis, cecal tissues from WT-infected rabbits at day 1 of infection, as well as from PBS-treated control animals, were subjected to semiquantitative terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining. Compared to PBS-treated animals, intestinal tissue from WT-infected animals displayed a modest increase in the number of TUNEL-positive cells (Fig. 7). In ΔespZ mutant-infected animals, despite a bacterial burden comparable to (or lower than) that in the WT-infected animals (Fig. 4), there was a marked increase in the number of apoptotic epithelial cells; consistent with the disease phenotype, epithelial cells from animals infected with the complemented strains displayed an intermediate level of TUNEL-positive cells. Thus, consistent with the in vitro observations, EspZ has a cytoprotective role in infected intestinal epithelial tissues of rabbits.
FIG 7.

Infection with the REPEC ΔespZ strain results in significant intestinal cell apoptosis in vivo. Shown are images from TUNEL staining of cecal tissues from rabbits mock gavaged with PBS or infected with the parent WT strain, the ΔespZ mutant, or the ΔespZ mutant cis-complemented with REPEC espZ (espZ/espZR) or EPEC espZ (espZ/espZE). Images are representative of those from multiple fields and multiple animals and highlight the intestinal cell apoptotic status on day 1 postinfection (yellow arrows), when histopathological observations revealed alterations for all groups (Fig. 5). Host cell nuclei were stained red by using the fluorescent marker DAPI, and TUNEL-positive apoptotic cells were stained green by using resorcinol phthalein (fluorescein). Light blue arrows pointing to diffuse staining are subepithelial autofluorescing erythrocytes. Bar, 100 μm.
DISCUSSION
The primary findings of this study are that EspZ promotes host epithelial cell survival in vitro and in vivo and that it is essential for virulence in a rabbit model of infection. REPEC EspZ recapitulates the in vitro functional attributes of EPEC EspZ, and both isoforms partially complement the in vivo virulence defects of a REPEC ΔespZ strain. The differential abilities of the two isoforms to complement the REPEC ΔespZ strain, particularly in vivo, could be due to differences in the primary sequence, although a potential contribution of expression variability cannot be ruled out. The two isoforms display 82% identity and 92% similarity at the amino acid level.
EspZ is unique to A/E pathogens and does not display similarity to proteins outside this group. The EPEC and REPEC EspZ proteins promote the survival of infected intestinal epithelial cells in vitro and in vivo (in rabbit intestines). Accordingly, Heczko et al. reported decreased apoptosis in the ileum and ileal Peyer's patches of rabbits with severe REPEC infection relative to uninfected control animals (31).
Two nonexclusive mechanisms have been proposed to explain the cytoprotective effect of EspZ. Roxas et al. and Shames et al. presented data to suggest that EspZ directly engages epithelial cell proteins and influences host signaling pathways to prevent the premature death of infected cells (18–20). Importantly, we demonstrated that transfected REPEC espZ protected both C2BBE and HeLa cells from staurosporine-induced intrinsic apoptosis (18). Shames et al. identified two EspZ-interacting epithelial cell proteins, the transmembrane glycoprotein CD98 and the inner mitochondrial protein TIM17b, and implied a role for these proteins in the survival of infected host cells. The EspZ interaction with CD98 was hypothesized to induce focal adhesion kinase (FAK)- and phosphatidylinositol 3-kinase (PI3K)/Akt-dependent prosurvival signaling (19); however, we reported that the inhibition of FAK phosphorylation decreased the survival of EPEC-infected HeLa cells but not that of intestinal epithelial cell lines (18). For TIM17b, consistent with a role in EspZ-dependent cytoprotection, small interfering RNA (siRNA)-mediated depletion of this protein resulted in an increased death of WT EPEC-infected, but not ΔespZ strain-infected, HeLa cells (20). A role for TIM17b in infected intestinal epithelial cells remains to be established.
On the other hand, Berger et al. demonstrated that EspZ limits the translocation of bacterial effector proteins into host epithelial cells, possibly via interactions with components of the T3SS (21). According to this model, the protective effects of EspZ can be explained by its ability to limit the translocation of cytotoxic effector proteins during an infection. Our studies similarly confirmed that REPEC and EPEC espZ-transfected host cells were refractory to translocation from infecting REPEC strains. While we were also able to confirm that a first wave of WT infection was more effective (than the isogenic ΔespZ strain) in inhibiting translocation from a subsequent second wave of REPEC infection (net MOI of >500), the failure of complementation of the ΔespZ strain infection suggests caution in interpreting this phenotype.
Notably, since complementation was demonstrated for the cell death phenotype with the same strains (even at a “low” MOI of ∼100), the increased cytotoxicity of the ΔespZ strain is likely not due to an enhanced translocation of other effectors. Additionally, our studies call into question the implicit assumption that the first-wave infection with the WT and ΔespZ strains differed in only one aspect: the presence or absence of EspZ. We noted clear differences in cell death between the two conditions, consistent with the cytoprotective role of EspZ (data not shown). The fluorescence readout for the translocation reporter does not factor into such alterations.
Irrespective of the underlying mechanisms, the cytoprotective effects of EspZ likely contribute to enhanced colonization in vivo. Thus, while the REPEC ΔespZ strain induces heightened enterocyte death in rabbits on day 1, bacterial colonization is inefficient relative to that of the WT strain, and the animals do not display the characteristic pathologies associated with infection. Previous studies by Mills et al. suggest that EspZ is secreted into host cells early in infection (27). Protection of infected epithelial cells during the initial stages of infection could therefore facilitate bacterial colonization by stabilizing the underlying epithelial substratum. Indeed, such protection may be achieved via cooperative, or additive, actions of many effectors. NleB1, another effector protein common to A/E pathogens, inhibits death receptor signaling by engaging and modifying host cell death domain-containing proteins (32, 33). C. rodentium mutants lacking NleB1 were impaired for colonization of mouse intestines.
More broadly, other pathogens also appear to employ the strategy of host epithelial cell preservation as a means to increase the efficiency of colonization. The Shigella flexneri effector protein OspE binds to host cell integrin-linked kinase (ILK), promotes FAK phosphorylation, and inhibits the detachment of infected cells from the basement membrane (34). OspE-deficient bacteria fail to colonize or cause disease in a guinea pig model of infection.
The significantly diminished proinflammatory cytokine responses in ΔespZ strain-infected animals throughout the course of infection suggest that the decreased virulence of this strain results from impaired colonization, which is supported by the decreased bacterial burdens observed, rather than heighted bacterial clearance via host immune responses. We cannot, however, rule out a broader role for EspZ in limiting host immune responses, particularly during a prolonged infection. Interleukin-17-producing T helper 17 (Th17) cells play an essential role in the clearance of the A/E pathogen C. rodentium (35). Phagocytosis of infected apoptotic cells by dendritic cells uniquely triggers the release of both interleukin-6 and transforming growth factor β, which is essential for Th17 development. Blocking apoptosis during C. rodentium infection impairs Th17 development. It is possible, therefore, that the EspZ-dependent inhibition of apoptosis delays Th17 development and the consequent clearance of the bacteria.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a National Institutes of Health grant to V.K.V. (NIAID1R01AI081742). Work in the Viswanathan laboratory is also supported by the U.S. Department of Agriculture (CSREES Hatch Program grant ARZT-570410-A-02-140 to V.K.V.).
We thank Nancy Craig for the Tn7-targeting plasmid pGRG36, John Leong for the translocation reporter and control plasmids pMB196 and pMB201, and Felicia Goodrum for the eukaryotic expression vector pCIP-TTL-HA. We gratefully acknowledge expert technical assistance from Jennifer Roxas, Gresa Sylejmani, and Ross Monasky.
We confirm that we do not have any conflict of interest related or relevant to the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02876-14.
REFERENCES
- 1.Wales AD, Woodward MJ, Pearson GR. 2005. Attaching-effacing bacteria in animals. J Comp Pathol 132:1–26. doi: 10.1016/j.jcpa.2004.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nataro JP, Kaper JB. 1998. Diarrheagenic Escherichia coli. Clin Microbiol Rev 11:142–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai Y, Rosenshine I, Leong JM, Frankel G. 2013. Intimate host attachment: enteropathogenic and enterohaemorrhagic Escherichia coli. Cell Microbiol 15:1796–1808. doi: 10.1111/cmi.12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ochoa TJ, Contreras CA. 2011. Enteropathogenic Escherichia coli infection in children. Curr Opin Infect Dis 24:478–483. doi: 10.1097/QCO.0b013e32834a8b8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pennington H. 2010. Escherichia coli O157. Lancet 376:1428–1435. doi: 10.1016/S0140-6736(10)60963-4. [DOI] [PubMed] [Google Scholar]
- 6.Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, Frankel G. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12:612–623. doi: 10.1038/nrmicro3315. [DOI] [PubMed] [Google Scholar]
- 7.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. 2005. Citrobacter rodentium of mice and man. Cell Microbiol 7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 8.Law RJ, Gur-Arie L, Rosenshine I, Finlay BB. 2013. In vitro and in vivo model systems for studying enteropathogenic Escherichia coli infections. Cold Spring Harb Perspect Med 3:a009977. doi: 10.1101/cshperspect.a009977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL. 2011. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol 80:1420–1438. doi: 10.1111/j.1365-2958.2011.07661.x. [DOI] [PubMed] [Google Scholar]
- 10.Deng W, de Hoog CL, Yu HB, Li Y, Croxen MA, Thomas NA, Puente JL, Foster LJ, Finlay BB. 2010. A comprehensive proteomic analysis of the type III secretome of Citrobacter rodentium. J Biol Chem 285:6790–6800. doi: 10.1074/jbc.M109.086603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marches O, Nougayrede JP, Boullier S, Mainil J, Charlier G, Raymond I, Pohl P, Boury M, De Rycke J, Milon A, Oswald E. 2000. Role of tir and intimin in the virulence of rabbit enteropathogenic Escherichia coli serotype O103:H2. Infect Immun 68:2171–2182. doi: 10.1128/IAI.68.4.2171-2182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng W, Vallance BA, Li Y, Puente JL, Finlay BB. 2003. Citrobacter rodentium translocated intimin receptor (Tir) is an essential virulence factor needed for actin condensation, intestinal colonization and colonic hyperplasia in mice. Mol Microbiol 48:95–115. doi: 10.1046/j.1365-2958.2003.03429.x. [DOI] [PubMed] [Google Scholar]
- 13.Lee SF, Kelly M, McAlister A, Luck SN, Garcia EL, Hall RA, Robins-Browne RM, Frankel G, Hartland EL. 2008. A C-terminal class I PDZ binding motif of EspI/NleA modulates the virulence of attaching and effacing Escherichia coli and Citrobacter rodentium. Cell Microbiol 10:499–513. doi: 10.1111/j.1462-5822.2007.01065.x. [DOI] [PubMed] [Google Scholar]
- 14.Mundy R, Petrovska L, Smollett K, Simpson N, Wilson RK, Yu J, Tu X, Rosenshine I, Clare S, Dougan G, Frankel G. 2004. Identification of a novel Citrobacter rodentium type III secreted protein, EspI, and roles of this and other secreted proteins in infection. Infect Immun 72:2288–2302. doi: 10.1128/IAI.72.4.2288-2302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly M, Hart E, Mundy R, Marches O, Wiles S, Badea L, Luck S, Tauschek M, Frankel G, Robins-Browne RM, Hartland EL. 2006. Essential role of the type III secretion system effector NleB in colonization of mice by Citrobacter rodentium. Infect Immun 74:2328–2337. doi: 10.1128/IAI.74.4.2328-2337.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanack KJ, Crawford JA, Tatsuno I, Karmali MA, Kaper JB. 2005. SepZ/EspZ is secreted and translocated into HeLa cells by the enteropathogenic Escherichia coli type III secretion system. Infect Immun 73:4327–4337. doi: 10.1128/IAI.73.7.4327-4337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vazquez A, Barba J, Ibarra JA, O'Donnell P, Metalnikov P, Ashman K, Lee S, Goode D, Pawson T, Finlay BB. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci U S A 101:3597–3602. doi: 10.1073/pnas.0400326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roxas JL, Wilbur JS, Zhang X, Martinez G, Vedantam G, Viswanathan VK. 2012. The enteropathogenic Escherichia coli-secreted protein EspZ inhibits host cell apoptosis. Infect Immun 80:3850–3857. doi: 10.1128/IAI.00335-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shames SR, Deng W, Guttman JA, de Hoog CL, Li Y, Hardwidge PR, Sham HP, Vallance BA, Foster LJ, Finlay BB. 2010. The pathogenic E. coli type III effector EspZ interacts with host CD98 and facilitates host cell prosurvival signalling. Cell Microbiol 12:1322–1339. doi: 10.1111/j.1462-5822.2010.01470.x. [DOI] [PubMed] [Google Scholar]
- 20.Shames SR, Croxen MA, Deng W, Finlay BB. 2011. The type III system-secreted effector EspZ localizes to host mitochondria and interacts with the translocase of inner mitochondrial membrane 17b. Infect Immun 79:4784–4790. doi: 10.1128/IAI.05761-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger CN, Crepin VF, Baruch K, Mousnier A, Rosenshine I, Frankel G. 2012. EspZ of enteropathogenic and enterohemorrhagic Escherichia coli regulates type III secretion system protein translocation. mBio 3(5):e00317-12. doi: 10.1128/mBio.00317-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roxas JL, Ryan K, Vedantam G, Viswanathan VK. 2014. Enteropathogenic Escherichia coli dynamically regulates EGFR signaling in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 307:G374–G380. doi: 10.1152/ajpgi.00312.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merlin C, McAteer S, Masters M. 2002. Tools for characterization of Escherichia coli genes of unknown function. J Bacteriol 184:4573–4581. doi: 10.1128/JB.184.16.4573-4581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenzie GJ, Craig NL. 2006. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol 6:39. doi: 10.1186/1471-2180-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vingadassalom D, Campellone KG, Brady MJ, Skehan B, Battle SE, Robbins D, Kapoor A, Hecht G, Snapper SB, Leong JM. 2010. Enterohemorrhagic E. coli requires N-WASP for efficient type III translocation but not for EspFU-mediated actin pedestal formation. PLoS Pathog 6:e1001056. doi: 10.1371/journal.ppat.1001056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jabbour M, Campbell EM, Fares H, Lybarger L. 2009. Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. J Immunol 183:6500–6512. doi: 10.4049/jimmunol.0901521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills E, Baruch K, Aviv G, Nitzan M, Rosenshine I. 2013. Dynamics of the type III secretion system activity of enteropathogenic Escherichia coli. mBio 4(4):e00303-13. doi: 10.1128/mBio.00303-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shifrin Y, Peleg A, Ilan O, Nadler C, Kobi S, Baruch K, Yerushalmi G, Berdichevsky T, Altuvia S, Elgrably-Weiss M, Abe C, Knutton S, Sasakawa C, Ritchie JM, Waldor MK, Rosenshine I. 2008. Transient shielding of intimin and the type III secretion system of enterohemorrhagic and enteropathogenic Escherichia coli by a group 4 capsule. J Bacteriol 190:5063–5074. doi: 10.1128/JB.00440-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinberg TH, Newman AS, Swanson JA, Silverstein SC. 1987. Macrophages possess probenecid-inhibitable organic anion transporters that remove fluorescent dyes from the cytoplasmic matrix. J Cell Biol 105:2695–2702. doi: 10.1083/jcb.105.6.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crane JK, Byrd IW, Boedeker EC. 2011. Virulence inhibition by zinc in Shiga-toxigenic Escherichia coli. Infect Immun 79:1696–1705. doi: 10.1128/IAI.01099-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heczko U, Carthy CM, O'Brien BA, Finlay BB. 2001. Decreased apoptosis in the ileum and ileal Peyer's patches: a feature after infection with rabbit enteropathogenic Escherichia coli O103. Infect Immun 69:4580–4589. doi: 10.1128/IAI.69.7.4580-4589.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearson JS, Giogha C, Ong SY, Kennedy CL, Kelly M, Robinson KS, Lung TW, Mansell A, Riedmaier P, Oates CV, Zaid A, Muhlen S, Crepin VF, Marches O, Ang CS, Williamson NA, O'Reilly LA, Bankovacki A, Nachbur U, Infusini G, Webb AI, Silke J, Strasser A, Frankel G, Hartland EL. 2013. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501:247–251. doi: 10.1038/nature12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao X, Wang X, Pham TH, Feuerbacher LA, Lubos ML, Huang M, Olsen R, Mushegian A, Slawson C, Hardwidge PR. 2013. NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-kappaB activation. Cell Host Microbe 13:87–99. doi: 10.1016/j.chom.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, Koyama T, Nagai S, Lange A, Fassler R, Sasakawa C. 2009. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 459:578–582. doi: 10.1038/nature07952. [DOI] [PubMed] [Google Scholar]
- 35.Torchinsky MB, Garaude J, Martin AP, Blander JM. 2009. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature 458:78–82. doi: 10.1038/nature07781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.