

Abstract
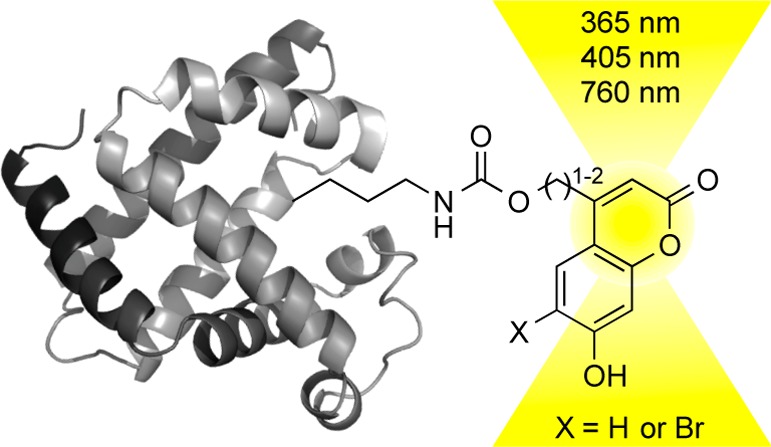
The site-specific incorporation of three new coumarin lysine analogues into proteins was achieved in bacterial and mammalian cells using an engineered pyrrolysyl-tRNA synthetase system. The genetically encoded coumarin lysines were successfully applied as fluorescent cellular probes for protein localization and for the optical activation of protein function. As a proof-of-principle, photoregulation of firefly luciferase was achieved in live cells by caging a key lysine residue, and excellent OFF to ON light-switching ratios were observed. Furthermore, two-photon and single-photon optochemical control of EGFP maturation was demonstrated, enabling the use of different, potentially orthogonal excitation wavelengths (365, 405, and 760 nm) for the sequential activation of protein function in live cells. These results demonstrate that coumarin lysines are a new and valuable class of optical probes that can be used for the investigation and regulation of protein structure, dynamics, function, and localization in live cells. The small size of coumarin, the site-specific incorporation, the application as both a light-activated caging group and as a fluorescent probe, and the broad range of excitation wavelengths are advantageous over other genetically encoded photocontrol systems and provide a precise and multifunctional tool for cellular biology.
Introduction
Good photochemical properties, chemical stability, and ease of synthesis make coumarins an important class of fluorescent probes for biological studies.1−3 In addition to being versatile fluorophores, coumarin chromophores can be used as light-removable protecting groups, so-called “caging groups”, that are photolyzed through one- and two-photon irradiation.4 Caged molecules have been extensively applied in the optical control of cellular processes.5−9 In particular, the 6-bromo-7-hydroxycoumarinmethyl caging group undergoes fast two-photon photolysis at 740 nm and has been used to optically control neurotransmitters, secondary messengers, and oligonucleotides.10−12 Two-photon irradiation enables optical activation of biological processes with enhanced tissue penetration of up to 1 mm. Moreover, two-photon caging groups can be released with greater precision in three-dimensional space than simple one-photon caging groups.4,13
Here we report the site-specific incorporation of three coumarin amino acids into proteins via genetic code expansion with unnatural amino acids (UAAs)14−16 to integrate the optical properties of coumarin probes into cellular systems. Genetic code expansion requires the addition of orthogonal translational machinery to achieve site-specific UAA incorporation into proteins. Recent advances in engineering pyrrolysyl-tRNA synthetase/tRNA pairs for the incorporation of sterically demanding amino acids17−20 prompted us to synthesize coumarin lysines 1–3 (Figure 1A) and to test their incorporation into proteins. The photochemical characteristics of these UAAs complement and enhance the properties of caged and fluorescent amino acids that have been genetically encoded in bacterial and mammalian cells.19−25 Lysines 1–3 were assembled in three steps from their corresponding coumarin alcohols (Supporting Information, Scheme S1). Briefly, the coumarin alcohols were activated with nitrophenyl chloroformate and coupled to commercially available Boc-lysine. A global deprotection under acidic conditions furnished the corresponding coumarin derivatives 1–3 in good yields.
Figure 1.
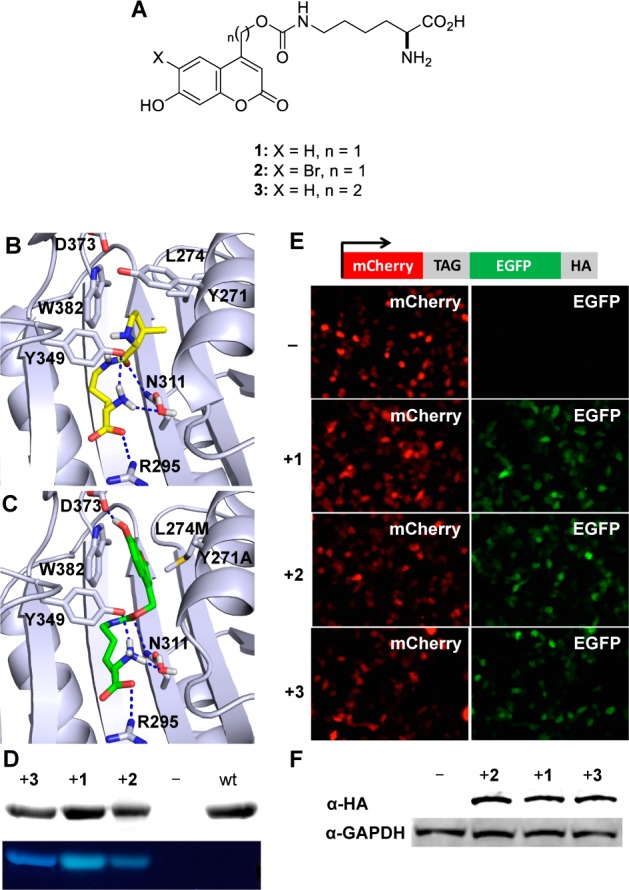
(A) Structures of the genetically encoded coumarin amino acids for fluorescence reporting and light activation of protein function. (B) Crystal structure of PylRS (2Q7H) with the pyrrolysine substrate (yellow) in the active site. (C) Structure of BhcKRS with 1 (green) docked into the active site. Dashed blue lines represent H-bond interactions. (D) SDS-PAGE analysis of sfGFP-Y151TAG containing 1–3 through incorporation in E. coli. The gel was stained with Coomassie blue (top), and coumarin fluorescence was imaged via excitation at 365 nm (bottom). (E) Fluorescence micrographs of HEK 293T cells expressing the BhcKRS/tRNACUA pair and mCherry-TAG-EGFP-HA in the presence or absence of 1–3. (F) Western blot analysis of cell lysates using an anti-HA antibody and a GAPDH antibody as a loading control. Full-length protein expression is only observed in the presence of 1–3, and incorporation efficiency with all three amino acids is similar in mammalian cells.
All three coumarin lysines 1–3 contain identical benzopyrone cores as fluorescent probes. However, subtle substitutions result in a set of coumarin derivatives with unique photochemical properties. Introduction of a bromine at the 6-position enables decaging not only with UV (single photon) light (in case of 1), but also near IR (two-photon) excitation (in case of 2).11 In contrast, extension of the coumarin–carbamate linker by a single carbon atom results in coumarin lysine 3, which does not undergo photolysis and thus represents a stable coumarin amino acid probe. Thus, coumarin lysines 1 and 2 can be used as both fluorescent and light-activated probes for optochemical control of protein function using UV or near-IR light, while coumarin lysine 3 may serve as a stable fluorescent probe that does not decage under UV excitation.
Results and Discussion
The Methanosarcina barkeri pyrrolysyl tRNA synthetase/tRNACUA (MbPylRS/tRNACUA) is functional and orthogonal in a wide range of organisms, such as Escherichia coli, yeast, mammalian cells, and animals such as Caenorhabditis elegans and Drosophila melanogaster.24,26−31 Furthermore, wild-type PylRS recognizes several unnatural amino acids without accepting any of the 20 common amino acids as a substrate.32 The active site of the PylRS can be further engineered through directed evolution to enable the incorporation of additional unnatural amino acids with new functions, including post-translational modifications, bioconjugation handles, photo-cross-linkers, photocaging groups, and others.14 Thus, we generated and screened a panel of MbPylRS mutants, guided by mutants that were previously reported,14 to direct the incorporation of 2 in response to a TAG amber codon in mammalian cells using a mCherry-TAG-EGFP reporter. Cells containing a MbPylRS mutant with only two amino acid mutations Y271A and L274M showed UAA-dependent expression of full-length mCherry-EGFP-HA. The Y271A mutation has previously been reported to direct the incorporation of Nε-carbamate-linked lysines,33 while the L274M mutation34,35 was discovered to facilitate higher amber suppression activities with 2 in vivo, because it allows greater flexibility of the side chain and imposes less steric bulk at the back of the hydrophobic pocket. This synthetase, termed BhcKRS, enabled the site-specific incorporation of not only 2 but also 1 and 3 in response to the amber codon TAG within sfGFP-Y151TAG-His6 in E. coli (Figure 1). This is not surprising, considering the very similar structures of 1–3 and previous observations of the high promiscuity of PylRS.36,37 To further rationalize the ability of BhcKRS to incorporate 1–3, molecular modeling was employed. The wild-type PylRS structure (PDB: 2Q7H) was used as a starting template for which the Y271A and L274M mutations were introduced using Modeller.38 The mutant structure was energy minimized in Amber molecular dynamics39 before docking 1–3 into the active site pocket using AutoDock4.40 As expected, 1–3 adopt very similar poses, reflecting their similarity in structure (see Supporting Information, Figure S1). The mutated synthetase model reveals that the Y271A and L274M mutations greatly enlarge the binding pocket to accommodate the bulky bicyclic caging group, while also orienting it in a favorable π-stacking interaction with W382. This orientation also benefits from a favorable H-bond interaction between the coumarin hydroxyl group and D373. Similar to published crystal structures, the amino group’s positioning is maintained by interactions with a structural water and Y349.41 It has been previously shown that interactions with N311 and R295 play an important role in amino acid recognition by the PylRS system.31,41,42 The docked structure maintains these key interactions with the carbamate carbonyl forming a H-bond with N311, while the carboxylic acid forms a H-bond with R295 (Figure 1B,C).
SDS-PAGE analysis reveals coumarin fluorescence of the expressed proteins containing the coumarin lysines 1–3. No fluorescence is observed for wild-type sfGFP because its excitation wavelength (488 nm) does not match that of 1–3 (365 nm) and because of the denaturing conditions of the gel. The dependence of protein expression on the presence of 1–3 demonstrates that the engineered BhcKRS synthetase has a high specificity for coumarin lysines and does not significantly incorporate any of the common 20 amino acids. Similar results were obtained for the incorporation of 1–3 into ubiquitin and myoglobin in E. coli. Electrospray ionization mass spectrometry (ESI-MS, Supporting Information, Figures S2–S4) showed that recombinantly expressed sfGFP-1 and -3 have a mass of 28446.22 and 28460.60 Da, in agreement with the expected masses of 28446.03 and 28460.04 Da, respectively. ESI-MS analysis of sfGFP-2 showed a mass of 28445.97 Da, indicating a partial loss of bromine during E. coli expression, possibly due to reductive dehalogenation.43 Overall, these results demonstrate that 1–3 can be incorporated into proteins in E. coli in good yields (8.0 mg/L, 1.6 mg/L, and 2.5 mg/L, respectively, for sfGFP) and with high specificity.
To demonstrate that the coumarin lysines 1–3 can also be genetically incorporated into proteins in mammalian cells, pBhcKRS-mCherry-TAG-EGFP-HA and p4CMVE-U6-PylT were cotransfected into human embryonic kidney (HEK) 293T cells. Cells were incubated for 24 h in the absence of any unnatural amino acid and in the presence of 1–3 (0.25 mM). Fluorescence imaging revealed EGFP expression only in the presence of 1–3, indicating specific incorporation of the coumarin lysines in response to the TAG codon, without measurable incorporation of endogenous amino acids (Figure 1E). This was further confirmed by an anti-HA Western blot on cell lysates from the same experiment (Figure 1F). Furthermore, full-length mCherry-EGFP protein was immunoprecipitated from HEK 293T cells using an immobilized antibody against the HA-tag and mass spectrometry sequencing confirmed that 1–3 are site-specifically incorporated into protein in mammalian cells (Supporting Information, Figure S5). Importantly, the presence of bromine was verified for protein containing 2, confirming the genetic encoding of the Bhc-caged lysine.
Because the coumarin groups on 1 and 2 are caging groups that can be removed via light exposure, loss of their intrinsic fluorescence can be used as an indicator of protein decaging through UV irradiation, as shown in Figure 2A,B. This was demonstrated through a UV exposure time-course of purified sfGFP-1, followed by SDS-PAGE analysis. The coumarin fluorescence intensity of sfGFP-1 gradually decreases with extended UV exposure as more of the coumarin caging group is removed from the protein, while the continued presence of the Coomassie-stained protein band indicates stability of the protein. In a cellular context, this may enable experiments that allow for the determination of protein expression, protein localization, and protein decaging using a single optochemical probe in a single experiment. In contrast, insertion of an extra methylene unit between the lysine and the fluorophore fully abrogates photocleavage and thus establishes 3 as a stable amino acid for the site-specific fluorescent labeling of proteins. No change in coumarin fluorescence is observed after UV exposure of sfGFP-3 for 20 min (Figure 2C,D). Due to the identical fluorophores in 1 and 3 and the stability of 3 to the UV irradiation conditions, the loss of protein fluorescence in sfGFP-1 is due to decaging and not due to photobleaching. This is further supported by mass spectroscopic analysis of the proteins before and after UV exposure (see Supporting Information, Figures S2 and S4).
Figure 2.
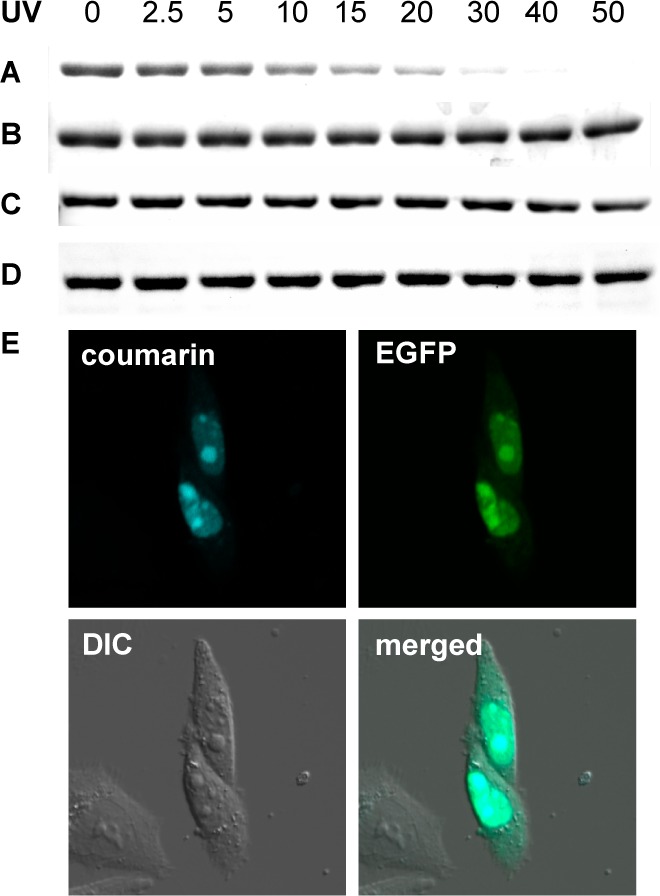
SDS-PAGE fluorescence analysis shows photodecaging of sfGFP-1 while sfGFP-3 is stable to UV exposure. (A) Loss of coumarin fluorescence after extended sfGFP-1 in-gel decaging for 0–50 min (365 nm, transilluminator). (B) Coomassie staining reveals identical sfGFP-1 protein amounts in all lanes. (C) No loss of coumarin fluorescence is observed, since sfGFP-3 does not decage. (D) Coomassie staining reveals identical sfGFP-3 protein amounts in all lanes. (E) Nuclear colocalization of coumarin and EGFP fluorescence in CHO K1 cells cotransfected with pNLS-TAG-EGFP-HA and the BhcKRS/PylT pair (pBhcKRS-4PylT) in the presence of 1 (0.25 mM). A DIC image and a merged image of all three channels are shown as well.
To demonstrate the ability of the genetically encoded coumarin lysines to act as reporters for protein localization in live cells, we investigated their utility as a protein nuclear localization marker. A plasmid was constructed to express EGFP-HA with an N-terminal NLS (nuclear localization signal, pNLS-linker-EGFP-HA),44 which reliably localizes EGFP to the nucleus (Supporting Information, Figure S6). A TAG amber codon was introduced in the linker between the NLS and EGFP, allowing for site-specific unnatural amino acid incorporation without affecting EGFP formation or nuclear translocation. Cells cotransfected with the pNLS-KTAG-EGFP and BhcKRS/PyltRNACUA plasmid pair in the presence of 1 (0.25 mM) were analyzed for coumarin fluorescence (405 nm excitation, 450–480 nm emission) and EGFP fluorescence (488 nm excitation, 490–520 nm emission) by confocal microscopy. The observation of complete colocalization of both fluorophores in the nucleus (merged micrographs) demonstrates the ability to use 1 as a reporter of protein localization (Figure 2E and Supporting Information, movie S1 and Figure S7).
To apply the coumarin lysines 1–3 in the optical control of protein function in live cells, firefly luciferase (Fluc) was selected as an initial target because bioluminescence measurements afford low background, high sensitivity, and easy quantification. On the basis of the Fluc crystal structure, a critical lysine residue, K206, was identified, which is positioned at the edge of the substrate-binding pocket (Figure 3B). It has been proposed that this residue stabilizes and orients ATP in the active site.45,46 The ε-amino group on K206 provides a hydrogen-bond interaction with the γ-phosphate of ATP and promotes the adenylation reaction with luciferin, thus being essential for catalytic activity as shown by the dramatic decrease in enzymatic activity displayed by the K206R mutant.45 Therefore, we hypothesized that a sterically demanding coumarin caging group placed on K206 would prevent the interaction with ATP and limit the overall access of the substrates to the active site (Figure 3A). Photolysis of the coumarin lysine would remove the caging group and produce a native lysine residue, restoring the catalytic activity of the enzyme (Figure 3B). A genetically encoded photocaged lysine at K206 would enable the enhanced regulation of the catalytic activity of firefly luciferase via light activation.
Figure 3.
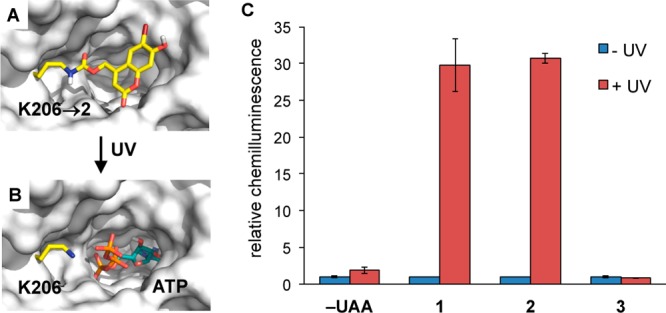
Engineering of an optochemically controlled Photinus pyralis firefly luciferase through unnatural amino acid mutagenesis. (A) Caging groups at position K206 are blocking access to the binding pocket by luciferin and ATP and are disrupting a required hydrogen bonding network. (B) After decaging, wild-type Fluc is generated and the substrates can now enter the active site. PDB: 2D1S. (C) Bright-Glo luciferase assay of cells that were either kept in the dark or irradiated (365 nm, 4 min). Chemiluminescence units were normalized to the −UAA/–UV control. No enzymatic activity was observed for the caged proteins, and significant increases in luminescence were observed after photolysis of luciferase containing 1 or 2, while the K206 → 3 mutant was permanently deactivated, as expected. Error bars represent standard deviations from three independent experiments.
Site-directed mutagenesis of the corresponding K206 residue to the amber codon (TAG) enabled incorporation of 1–3 into firefly luciferase in mammalian cells. HEK 293T cells were cotransfected with the mutated firefly luciferase plasmid (pGL3-K206TAG) and the MbBhcKRS/PyltRNACUA pair (pBhcKRS-4PylT) in the presence of 1–3 (0.25 mM). After 24 h incubation, the cells were either irradiated for 4 min (365 nm, 25 W) or kept in the dark. The incorporation of 1–3 into Fluc caused complete inhibition of luciferase activity before UV irradiation, as determined by a Bright-Glo luciferase assay, comparable to the negative control (no unnatural amino acid). After UV irradiation, 1 and 2 were decaged to produce native lysine, resulting in the activation of firefly luciferase by 34-fold and 31-fold, respectively (Figure 3C). As expected, 3 did not show any activation of luciferase enzymatic activity upon illumination, as it does not undergo decaging. Therefore, the activity of firefly luciferase can be tightly optochemically regulated by incorporation of a coumarin lysine residue into the active site of the luciferase protein. Interestingly, attempts to apply 1 and 2 at position K529, another site that can be used for optical control of luciferase function,47 led to greatly diminished luciferase activity, while introduction of our previously reported o-nitrobenzyl-caged lysine24,48,49 worked at both positions K206 and K529. Western blots confirmed that both Fluc-K206 → 1 and Fluc-K529 → 1 were expressed at similar levels in mammalian cells (Supporting Information, Figures S8 and S9).
To observe the optical triggering of protein function via decaging of 1 and 2 in real time, enhanced green fluorescent protein (EGFP) was selected as a second target protein for caging. EGFP consists of an 11-stranded β-barrel and a central α-helix with the Thr65-Tyr66-Gly67 chromophore.50 The chromophore plays a crucial role in EGFP fluorescence and stability.51 Correctly folded EGFP is a prerequisite for mature chromophore formation, with a number of lysine residues being essential to its successful folding.52 Most notable is that only 1 lysine (K85) out of 20 is buried within the protein.52 K85 forms a salt bridge with D82 and H-bonding interactions with the backbone of C70 and S72,52 all of which are in close proximity to the chromophore (Figure 4A). It has been shown that C70, S72, and D82 are key residues for control of chromophore formation and oxidation.53,54 We hypothesized that introduction of coumarin-caged lysines 1 and 2 at K85 would affect D82, C70, and S72, interrupting the α-helix bending and thus indirectly inhibiting chromophore maturation. To this end, we envisioned that UV activation would yield native EGFP that rapidly undergoes maturation. An EGFP mutant with an amber codon at position K85 (pEGFP-K85TAG) was generated as a fusion construct with mCherry, to provide a second reporter for successful plasmid transfection and incorporation of 1 and 2. HEK 293T cells were cotransfected with pEGFP-K85TAG-mCherry and the BhcKRS/PyltRNA pair in the presence of 1 and 2 (0.25 mM). After 24 h, the cells were washed and incubated in fresh media for 1 h. Cells expressing mCherry were observed by fluorescence imaging to confirm that EGFP-1/2-mCherry is generated in the presence of 1 or 2. Cells were irradiated for 30 s at 365 nm, and fluorescence was imaged by time-lapse microscopy. After photolysis of EGFP-1, green fluorescence started to appear around 10 min, and over time the fluorescence intensity gradually increased, reaching a plateau at 120 min (Figure 4D and Supporting Information, movie S2). A half-life of 49 min was observed, matching reports of EGFP chromophore maturation as the rate-limiting step.55 Previous measurements of EGFP folding and maturation have been exclusively performed in test tubes.56 No cellular studies have been conducted, as a precise starting point for kinetic analysis could not be provided.
Figure 4.
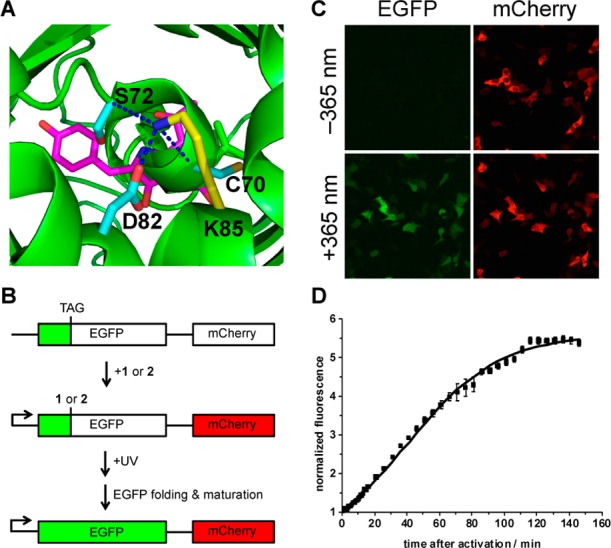
(A) Location of K85 (yellow) and interactions with D82, C70, and S72 in EGFP. The chromophore is shown in magenta. (B) Schematic of the pEGFP-K85TAG-mCherry construct and its application in light activation studies. (C) Fluorescence imaging of HEK 293T cells expressing EGFP-K85TAG-mCherry, 90 min after irradiation at 365 nm (30 s, DAPI filter, 358–365 nm) in the presence of 1 (Nikon A1R confocal microscope, 20× objective, 2-fold zoom). (D) Normalized EGFP fluorescence as a function of time after 365 nm light activation (error bars represent standard deviations from the measurement of three independent cells, t1/2 = 49 min).
Given that the coumarin lysines have relatively broad absorption bands in the 300–420 nm range that enable decaging at longer wavelengths, we speculated that irradiation at 405 nm1,57 may efficiently activate 1 and 2. Thus, activation through blue light irradiation using a standard laser-scanning confocal microscope was tested. As expected, exposure at 405 nm induced fluorophore formation of EGFP-1 and -2 (Figure 5A,B). Because attempts to decage a previously incorporated nitrobenzyloxycarbonyl lysine24,48,49 and nitrobenzyl tyrosine19,58,59 at 405 nm were not successful on comparable time scales and at comparable illumination power (data not shown), the caged lysines 1 and 2 may enable multiwavelength activation of proteins caged with the two different optical probes.
Figure 5.
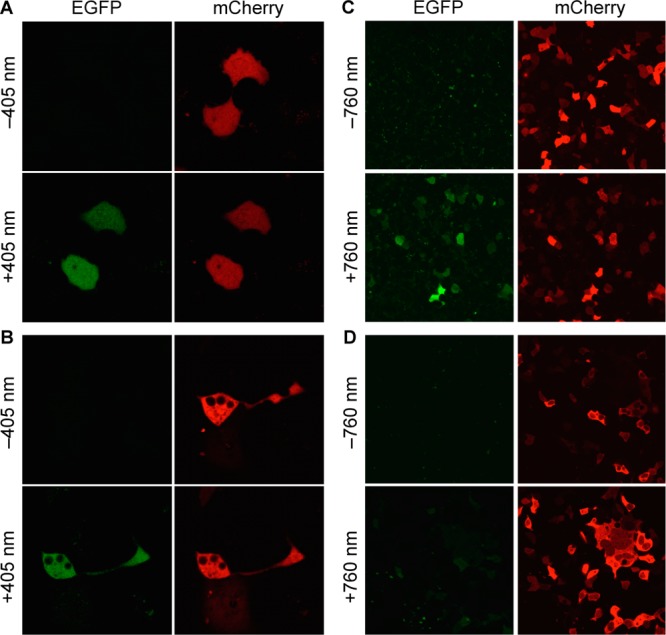
Fluorescence confocal imaging of COS-7 cells expressing EGFP-KTAG-mCherry, before and after irradiation at 405 nm (30 mW diode laser, 20% laser power, 12.6 μs dwell time, 8 cycles) in the presence of 2 (A) or 1 (B) (Zeiss confocal LSM710 microscope, 40× water objective). Similar light-activation experiments before and after irradiation of HEK 293T cells at 760 nm (130 mW, 2 μm/s dwell time, 30 cycles, Olympus Fluoview FV1000 MPE, MaiTai DSBB-OL IR pulsed laser), in the presence of 2 (C) or 1 (D), imaged with a Olympus Fluoview1000, 40× oil objective.
Taking advantage of the two-photon decaging feature of 2,11 photocontrol of EGFP folding by two-photon activation of EGFP-2 was performed. HEK 293T cells were cotransfected with pEGPF-K85TAG-mCherry and pBhcKRS-4PylT in the absence or presence of 1 and 2 (0.25 mM). After a 24 h incubation, the cells were washed and incubated in fresh media for 1 h and irradiated with a multiphoton laser (760 nm, 130 mW, 2 μm/s dwell time, 30 cycles, Olympus Fluoview FV1000 MPE Multiphoton laser scanning microscope FV10-ASW, MaiTai DSBB-OL IR pulsed laser). Images were acquired before and after two-photon irradiation using both EGFP (488 nm) and mCherry (561 nm) excitation. Gratifyingly, an EGFP fluorescent signal was observed after photolysis of 2 at 760 nm (Figure 5C). The cells expressing EGFP containing 1, as a control, were also exposed to two-photon excitation (760 nm) and imaged in the same fashion (Figure 5D); no EGFP activation was observed. In addition to the increased three-dimensional resolution that is provided through two-photon excitation, effectively shifting the activation wavelength to the near-IR will enable multiwavelength activation in conjunction with other optically triggered biological processes, while also preventing any overlap with established fluorescent reporter proteins.
Summary
The site-specific genetic incorporation of three new coumarin lysine analogues 1–3 into proteins was achieved in bacterial and mammalian cells using an engineered BhcKRS synthetase system. The genetically encoded coumarin lysines were successfully applied as fluorescent cellular probes for protein localization, and the small size of these coumarin lysines is expected to minimally perturb protein structure and function, unless they are placed at critical sites. In addition to their small size, the spectral properties of 1–3 do not interfere with common fluorescent proteins (e.g., EGFP). While the amino acid 3 showed stability under irradiation conditions, the coumarins 1 and 2 were readily decaged, generating wild-type lysine residues. As a proof-of-principle, photoregulation of firefly luciferase was achieved in live cells by caging a key lysine residue, and excellent OFF to ON light-switching ratios were observed for 1 and 2. As expected, the stable fluorescent amino acid 3 did not undergo photolysis. Furthermore, two-photon and single-photon optochemical control of EGFP maturation was demonstrated, enabling the use of different, potentially orthogonal, excitation wavelengths (365, 405, and 760 nm) for the sequential activation of protein function in live cells. While the caged lysine 2 could be activated using two-photon irradiation at 760 nm, the lysine 1 was stable under these conditions. However, decaging of 1 was readily achieved with blue light of 405 nm, while a previously encoded o-nitrobenzyl-caged lysine requires UV activation.24,48,49 These results demonstrate that coumarin lysines are a new and valuable class of optical probes that can potentially be used for the investigation and regulation of protein structure, dynamics, function, and localization in live cells. The small size of coumarin, the application as both a light-activated caging group and a fluorescent probe, and the broad range of excitation wavelengths are advantageous over other genetically encoded photocontrol systems and provide a unique and multifunctional tool for cellular biology. The ability to incorporate all three coumarin lysines with the same PylRS/tRNACUA pair further facilitates their application.
Experimental Section
Cloning
(1) Construction of pNLS-TAG-EGFP-HA: The pTAG-EGFP-HA fragment was amplified from pmCherry-TAG-EGFP-HA using the PCR primers G1/G2, digested with HindIII and BglII, and ligated into pEGFP-N1 (Clontech), generating the pTAG-EGFP-HA plamid. The pNLS PCR fragment was obtained by using primers N1/N2 and then ligated into the HindIII and XbaI sites of pTAG-EGFP-HA to generate the pNLS-TAG-EGFP-HA plasmid. (2) Construction of pNLS-WT-EGFP-HA: Plasmids were obtained by converting the TAG codon of pNLS-TAG-EGFP-HA into an AAG (Lys) codon using primers QC1/QC2 and a QuikChange site-directed mutagenesis kit (Agilent). (3) Construction of pBhcKRS-4PylT: The plasmid was obtained by ligating the p4CMVE-U6-PylT fragment from pMbPylT between the restriction sites NheI and MfeI sites of pMbBhcKRS.
Expression and Purification of Proteins in E. coli
The plasmid, pBAD-sfGFP-Y151TAG-pylT was cotransformed with pBK-BhcKRS24 into E. coli Top10 cells. A single colony was grown in LB media overnight, and 500 μL of the overnight culture was added to 25 mL of LB media, supplemented with 1 mM of the designated unnatural amino acid and 25 μg/mL of tetracycline and 50 μg/mL of kanamycin. Cells were grown at 37 °C, 250 rpm, and protein expression was induced with 0.1% arabinose when the OD600 reached ∼0.6. After overnight expression at 37 °C, cells were harvested and washed by PBS. The cell pellets were resuspended in 6 mL of phosphate lysis buffer (50 mM, pH 8.0) and Triton X-100 (60 μL, 10%), gently mixed, and incubated for 1 h at 4 °C. The cell mixtures were sonicated, and the cell lysates were centrifuged at 4 °C, 13 000 g, for 10 min. The supernatant was transferred to a 15 mL conical tube, and 100 μL of Ni-NTA resin (Qiagen) was added. The mixture was incubated at 4 °C for 2 h under mild shaking. The resin was then collected by centrifugation (1000g, 10 min), washed twice with 400 μL of lysis buffer, and followed by two washes with 400 μL of wash buffer containing 20 mM imidazole. The protein was eluted with 400 μL of elution buffer containing 250 mM imidazole. The purified proteins were analyzed by 10% SDS-PAGE and stained with Coomassie Blue.
Protein Analysis by ESI-MS
Two different instruments were used: (A) Protein samples were analyzed using capillary LC ESI-TOF MS. The protein samples were loaded onto a PRLP-S column (Thermo Fisher 5 μm, 1000 A, 300 μm i.d. × 100 mm) on an LC system (Ultimate 3000, Dionex, Sunnyvale, CA). The LC system was directly coupled to an electrospray ionization time-of-flight mass spectrometer (microTOF, BrukerDaltonics, Billerica, MA). Chromatographic separation was performed at a constant flow rate of 3.5 μL/min using a binary solvent system (solvent A: 2.5% acetonitrile and 0.1% formic acid; solvent B: 80% acetonitrile and 0.1% formic acid) and a linear gradient program (0–5 min, 5% B; 5–10 min, 5–30% B; 10–30 min, 30–75% B; 30–35 min, 75–100% B; 35–45 min, 100–5% B; 45–60 min, 5% B). Mass spectra were acquired in positive ion mode over the mass range m/z 50 to 3000. ESI spectra were deconvoluted with the MaxEnt algorithm (Data Analysis 3.3, Bruker Daltonics, Billerica, MA), obtaining molecular ion masses with a mass accuracy of 1–2 Da. (B) High-resolution exact mass measurement were conducted on an Agilent Technologies (Santa Clara, CA) 6210 LC-TOF mass spectrometer. Samples were analyzed via a 1 μL flow injection at 300 μL/min in a water:methanol mixture (25:75 v/v) with 0.1% formic acid. The mass spectrometer was operated in positive ion mode with a capillary voltage of 4 kV, nebulizer pressure of 35 psi, and a drying gas flow rate of 12 L/min at 350 °C. The fragmentor and skimmer voltages were 200 and 60 V, respectively. Reference ions of purine at m/z 121.0509 and HP-0921 at m/z 922.0098 were simultaneously introduced via a second orthogonal sprayer and used for internal calibration.
Coumarin Lysine Incorporation in Human Cells
Human embryonic kidney (HEK) 293T cells were grown in DMEM (Dulbecco’s Modified Eagle Medium, Gibco) supplemented with 10% FBS (Gibco), 1% Pen-Strep (Corning Cellgro), and 2 mM l-glutamine (Alfa Aesar) in 96-well plates (Costar) in a humidified atmosphere with 5% CO2 at 37 °C. HEK 293T cells were transiently transfected with the pMbBhcKRS-mCherry-TAG-EGFP-HA and p4CMVE-U6-PylT24 at ∼75% confluency in the presence or absence of 1, 2, and 3 (0.25 mM) in 96-well plates. Double transfections were performed with equal amounts of both plasmids. After an overnight incubation at 37 °C, the cells were washed by PBS and imaged with a Zeiss Axio Observer.Z1Microscope (10× objective). To confirm the expression of the fusion protein and also differentiate between expression levels, a Western blot was performed. HEK 293T cells were cotransfected with pMbBhcKRS-mCherry-TAG-EGFP-HA and p4CMVE-U6-PylT in the presence or absence of 1, 2, and 3 (0.25 mM) in six-well plates. After 24 h of incubation, the cells were washed by chilled PBS, lysed in mammalian protein extraction buffer (GE Healthcare) with complete protease inhibitor cocktail (Sigma) on ice, and the cell lysates were cleared at 13 200 rpm centrifugation (4 °C, 20 min). The protein lysate was boiled with loading buffer and then analyzed by 10% SDS-PAGE. After gel electrophoresis and transfer to a PVDF membrane (GE Healthcare), the membrane was blocked in TBS with 0.1% Tween 20 (Fisher Scientific) and 5% milk for 1 h. The blots were probed and incubated with the primary antibody, α-HA-probe (Y-11) rabbit polyclonal lgG (sc-805, Santa Cruz Biotech), overnight at 4 °C, followed by a fluorescent secondary antibody, goat-α-rabbit lgG Cy3 (GE Healthcare), for 1 h at room temperature. The binding and washing steps were performed in TBS with 0.1% Tween 20.
Protein Sequencing by LC-MS/MS
HEK 293T cells were transfected with pBhcKRS-mCherry-TAG-EGFP-HA and p4CMVE-U6-PylT in a 10 cm Petri dish and incubated with DMEM containing 1, 2, or 3 (0.25 mM) for 24 h. Cells were lysed with extraction buffer (GE Healthcare) and the mCherry-1/2/3-EGFP-HA protein was immunoprecipitated using the Pierce HA Tag IP/Co-IP kit (Pierce) according to manufacturer’s protocol. The proteins were separated on SDS-PAGE gels and stained with silver stain. Regions corresponding to the expected molecular weight of mCherry-EGFP-HA were excised, washed with HPLC water, and destained with 50% acetonitrile/25 mM ammonium bicarbonate until no visible staining. Gel pieces were dehydrated with 100% acetonitrile and reduced with 10 mM dithiothreitol at 56 °C for 1 h, followed by alkylation with 55 mM iodoacetamide at room temperature for 45 min in the dark. Gel pieces were then again dehydrated with 100% acetonitrile to remove excess alkylating and reducing agents and rehydrated with 20 ng/μL trypsin/25 mM ammonium bicarbonate and digested overnight at 37 °C. The resultant tryptic peptides were extracted with 70% acetonitrile/5% formic acid, speed-vac dried, and reconstituted in 18 μL of 0.1% formic acid. Tryptic digests were analyzed by reverse-phased LC-MS/MS using a nanoflow LC (Waters nanoACQUITY UPLC system, Waters Corp., Milford, MA) coupled online to an LTQ/Orbitrap Velos hybrid mass spectrometer (Thermo-Fisher, San Jose, CA). Separations were performed using a C18 column (PicoChip column packed with 10.5 cm Reprosil C18 3 μm 120 Å chromatography media with a 75 μm ID column and a 15 μm tip, New Objective, Inc., Woburn, MA). Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. Samples were injected onto a trap column (nanoACQUITY UPLC trap column, Waters Corp., Milford, MA) and washed with 1% mobile phase B at a flow rate of 5 μL/min for 3 min. Peptides were eluted from the column using a 90 min gradient running at 300 nL/min (5% B for 3 min, 5–36% B in 62 min, 36–95% B in 2 min, 95% B for 8 min, 95%–5% B in 1 min, 5% B for 16 min). The LTQ/Orbitrap instrument was operated in a data-dependent MS/MS mode in which each high resolution broad-band full MS spectra (R = 60 000 at mass to charge (m/z) 400, precursor ion selection range of m/z 300 to 2000) was followed by 13 MS/MS scans in the linear ion trap where the 13 most abundant peptide molecular ions dynamically determined from the MS scan were selected for tandem MS using a relative collision-induced dissociation (CID) energy of 35%. Dynamic exclusion was enabled to minimize redundant selection of peptides previously selected for CID. MS/MS spectra were searched with the MASCOT search engine (version 2.4.0, Matrix Science Ltd.) against a UniProt jellyfish proteome database (June 2014 release) from the European Bioinformatics Institute (http://www.ebi.ac.uk/integr8) combined with endogenous mCherry-EGFP fasta sequences. The following modifications were used: static modification of cysteine (carboxyamidomethylation, +57.0214 Da) and variable modification of methionine (oxidation, +15.9949 Da) for all searches, variable modifications of lysine for mCherry-EGFP-HA (1, +218.17 Da; 2, +295.93 Da; 3, +231.03 Da). The mass tolerance was set at 20 ppm for the precursor ions and 0.8 Da for the fragment ions. Peptide identifications were filtered using PeptideProphet and ProteinProphet algorithms with a protein threshold cutoff of 99% and peptide threshold cutoff of 95% implemented in Scaffold (Proteome Software, Portland, OR).
Expression of Caged Firefly Luciferase and Light Activation
HEK 293T cells were cultured in DMEM (Dulbecco’s Modified Eagle Medium, Gibco) supplemented with 10% FBS (Gibco), 1% Pen-Strep (Gibco), and 2 mM l-glutamine (Alfa Aesar) in 96-well plates (BD Falcon) in a humidified atmosphere with 5% CO2 at 37 °C. At 80–90% confluency, cells seeded on plates were transfected and the medium was changed to fresh DMEM supplemented without or with 1, 2, or 3 (0.25 mM). The plasmid pMbBhcKRS-4PylT was constructed containing both CMV-MbBhcKRS and 4CMVE-U6-PylT. A TAG amber stop codon was introduced at the K206 site using primers GL1/GL2 and a QuikChange mutagenesis kit (Agilent Technologies). A pGL3-control plasmid containing the gene encoding P. pyralis firefly luciferase with the TAG amber mutation at residue K206 (pGL3-K206TAG) was cotransfected into cells with the plasmid pBhcKRS-4PylT using linear PEI according to the manufacturer’s protocol (Millipore). After double transfection and 24 h incubation, the medium was changed to DMEM without phenol red, and the cells were irradiated with UV light (365 nm) for 4 min using a 365 nm UV lamp (high performance UV transilluminator, UVP, 25 W) or kept in the dark. Cells were lysed by addition of 100 μL of substrate solution (Promega) in a 96-well plate (BD Falcon), and luminescence was measured on a Synergy 4 multimode microplate reader with an integration time of 2 s and a sensitivity of 150 or on a Tecan M1000 microplate reader with an integration time of 1 s.
Visualization of Nuclear Localization through Coumarin Lysine Incorporation
CHO K1 cells were plated into a polylysine-coated four-well chamber slide (Lab-Tek) and, after incubation to 75% confluency, were transfected with 1 μg of pNLS-KTAG-EGFP and pBhcKRS-4PylT each. After 16 h incubation at 37 °C/5% CO2 in DMEM with 10% FBS in the presence of 1 (0.25 mM), cells were washed with DMEM without phenol red and then incubated for 2 h. The cells were washed with PBS, fixed with 4% formaldehyde, and stained with rhodamine–phalloidin (Life Technologies). The chamber slide was dried in the dark overnight and cells were imaged on a Zeiss 710 confocal microscope (40× water objective).
One-Photon Light Activation of EGFP
HEK 293T cells were plated into a poly-d-lysine-coated eight-well chamber slide (Lab-Tek). After incubation to 70% confluency, cells were transfected with pEGFP-K85TAG-mCherry and pBhcKRS-4PylT (200 ng each). After a 20 h incubation at 37 °C/5% CO2 in DMEM with 10% FBS in the presence of 1 (0.25 mM), cells were washed with DMEM without phenol red and then incubated for 1 h. Before light activation, mCherry-expressing cells were identified using the TXRED channel, and imaged with a Nikon A1Rsi confocal microscope (20× objective, 2-fold zoom, EGFP (ex. 488 nm) and mCherry (ex. 560 nm) channels). Subsequently, cells were illuminated for 15 s at 365 nm light (DAPI filter, 358–365 nm), and EGFP and mCherry fluorescence was acquired by time-lapse imaging (every 1 min for the first 15 min, every 5 min for the following 150 min, scan resolution 512 × 512, scan zoom 2× , dwell time 1.9 ms). The mean EGFP fluorescence intensities were quantified using Nikon Elements software.
Two-Photon Light Activation of EGFP
HEK 293T cells were plated into a polylysine-coated μ-dish (ibidi), and after incubation to 50% confluency, the cells were transfected with 1 μg each of pEGFP-KTAG-mCherry and pBhcKRS-4PylT. After a 20 h incubation at 37 °C/5% CO2 in DMEM with 10% FBS in the presence of 1 or 2 (0.25 mM, 0.5% DMSO), cells were washed with DMEM without phenol red and then incubated for 1 h. Cells were imaged with an Olympus Fluoview confocal microscope before two-photon irradiation (40× oil objective, EGFP (ex. 488 nm) and mCherry (ex. 560 nm) channels), imaging positions for mCherry-expressing cells were recorded, and the cell μ-dish was transferred to an Olympus multiphoton microscope for irradiation (Olympus Fluoview FV1000 MPE). Cells were localized at the previously recorded positions, focused using the mCherry channel, and then irradiated using a 760 nm laser (130 mW, 5% of laser power, 30 cycles of scanning, 2 μm/s dwell time, MaiTai DSBB-OL IR pulsed laser). After irradiation, the cell μ-dish was transferred back to the original microscope for imaging.
Mutant PylRS Structure Modeling and Energy Minimization
The initial template structure of PDB 2Q7H was chosen as a starting point for all modeling. The missing loops were remodeled using MODELER and the two point mutations (Y271A and L274M) were constructed using the mutate_model.py script provided by MODELER. Superposition of PDB 2Q7G on top of 2Q7H provided the coordinates for the incorporation of ATP and magnesium ions into the newly mutated structure. The ATP and magnesium ions were parametrized in antechamber using previously developed parameters.60,61 The mutated structure was imported into AMBER12 software using the AMBER FF99SBILDN force field.62 The protein was placed into a cubic box with a 12.0-Å border, solvated with 17 316 water molecules, and charge neutralized with the addition of six sodium ions. This system was energy minimized first with 5000 steps steepest descent method, followed by 15 000 steps conjugate gradient method with 5 kcal/mol restraints on all atoms. This was followed by another 5000 steps steepest descent method, followed by 15 000 steps conjugate gradient method with 2 kcal/mol restraints on all atoms except Y271A and L274M. The resulting energy-minimized structure was used as the starting structure for all our docking experiments. All AMBER12 computational experiments were completed on the Center for Simulation and Modeling (SAM) Frank supercomputer at the University of Pittsburgh.
Molecular Docking Experiments
The energy-minimized mutant structure was prepared for docking with AutoDock4 by removing all sodium ions, and all water molecules except for a single water molecule which exists in the active site pocket of the protein. This structural water molecule is present in all available crystal structures and plays an important role in amino acid recognition. The receptor input file was prepared using AutoDock Tools software.40 The side chains for residue L274M were treated as flexible, while all other side chains were kept rigid. The unnatural amino acid ligands were constructed using ChemBioDraw3D, and the molecular geometry was optimized using the MMFF94 force field.63 The ligand input files were prepared for docking using AutoDock Tools as well. Lamarkian genetic algorithm was used for docking with the following parameters: number of runs: 75, ga_pop_size 150, ga_num_evals 250 000 000, ga_num_generations 27 000 were set, all other parameters were kept default. Docking results were clustered based on RMSD of each pose. Each coumarin lysine yielded a low energy cluster with binding scores of −9.63 kJ/mol, −6.18 kJ/mol, and −6.14 kJ/mol for 1, 2, and 3, respectively.
Acknowledgments
We thank Dr. Dustin Lockney for preparation of the pEGFP-K85TAG plasmid. This work was supported in part by the National Science Foundation (MCB-1330746, CHE-0848398) and the University of Pittsburgh. This research used the Center for Biologic Imaging, the Biomedical Mass Spectrometry Center and UPCI Cancer Biomarker Facility that are supported in part by the National Institutes of Health (P30CA047904). J.W.C. is supported by the Medical Research Council (U105181009, UD99999908). D.P.N. was supported by a fellowship from Trinity College.
Supporting Information Available
Protein mass spectrometry, additional micrographs, NMR spectra, oligonucleotide sequences, and synthesis protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bort G.; Gallavardin T.; Ogden D.; Dalko P. I. Angew. Chem., Int. Ed. 2013, 52, 4526–4537. [DOI] [PubMed] [Google Scholar]
- Krueger A. T.; Imperiali B. ChemBioChem 2013, 14, 788–799. [DOI] [PubMed] [Google Scholar]
- Goncalves M. S. Chem. Rev. 2009, 109, 190–212. [DOI] [PubMed] [Google Scholar]
- Klan P.; Solomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Chem. Rev. 2013, 113, 119–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Angew. Chem., Int. Ed. 2012, 51, 8446–8476. [DOI] [PubMed] [Google Scholar]
- Riggsbee C. W.; Deiters A. Trends Biotechnol. 2010, 28, 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiters A. Curr. Opin. Chem. Biol. 2009, 13, 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. M.; Larson D. R.; Lawrence D. S. ACS Chem. Biol. 2009, 4, 409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A. S.; Deiters A. ACS Chem. Biol. 2014, 9, 1398–1407. [DOI] [PubMed] [Google Scholar]
- Furuta T.; Takeuchi H.; Isozaki M.; Takahashi Y.; Kanehara M.; Sugimoto M.; Watanabe T.; Noguchi K.; Dore T. M.; Kurahashi T.; Iwamura M.; Tsien R. Y. ChemBioChem. 2004, 5, 1119–1128. [DOI] [PubMed] [Google Scholar]
- Furuta T.; Wang S. S.; Dantzker J. L.; Dore T. M.; Bybee W. J.; Callaway E. M.; Denk W.; Tsien R. Y. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H.; Furuta T.; Tsien R. Y.; Okamoto H. Nat. Genet. 2001, 28, 317–325. [DOI] [PubMed] [Google Scholar]
- Helmchen F.; Denk W. Nat. Methods 2005, 2, 932–940. [DOI] [PubMed] [Google Scholar]
- Wan W.; Tharp J. M.; Liu W. R. Biochim. Biophys. Acta 2014, 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. Annu. Rev. Biochem. 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- Chin J. W. Annu. Rev. Biochem. 2014, 83, 379–408. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Peters F. B.; Yang P. Y.; Reed S.; Chittuluru J. R.; Schultz P. G. ACS Chem. Biol. 2014, 9, 1092–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K.; Davis L.; Torres-Kolbus J.; Chou C.; Deiters A.; Chin J. W. Nat. Chem. 2012, 4, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Deiters A.; Groff D.; Ryu Y.; Xie J.; Schultz P. G. Angew. Chem., Int. Ed. 2006, 45, 2728–2731. [DOI] [PubMed] [Google Scholar]; b Arbely E.; Torres-Kolbus J.; Deiters A.; Chin J. W. J. Am. Chem. Soc. 2012, 134, 11912–11915. [DOI] [PubMed] [Google Scholar]
- Chatterjee A.; Guo J.; Lee H. S.; Schultz P. G. J. Am. Chem. Soc. 2013, 135, 12540–12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. R.; Wang Y. S.; Wan W. Mol. BioSyst. 2011, 7, 38–47. [DOI] [PubMed] [Google Scholar]
- Lemke E. A.; Summerer D.; Geierstanger B. H.; Brittain S. M.; Schultz P. G. Nat. Chem. Biol. 2007, 3, 769–772. [DOI] [PubMed] [Google Scholar]
- Summerer D.; Chen S.; Wu N.; Deiters A.; Chin J. W.; Schultz P. G. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9785–9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A.; Nguyen D. P.; Lusic H.; An W.; Deiters A.; Chin J. W. J. Am. Chem. Soc. 2010, 132, 4086–4088. [DOI] [PubMed] [Google Scholar]
- a Uprety R.; Luo J.; Liu J.; Naro Y.; Samanta S.; Deiters A. ChemBioChem 2014, 15, 1793–1799. [DOI] [PubMed] [Google Scholar]; b Nguyen D. P.; Mahesh M.; Elsaesser S. J.; Hancock S. M.; Uttamapinant C.; Chin J. W. J. Am. Chem. Soc. 2014, 136, 2240–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wu N.; Deiters A.; Cropp T. A.; King D.; Schultz P. G. J. Am. Chem. Soc. 2004, 126, 14306–14307. [DOI] [PubMed] [Google Scholar]
- Greiss S.; Chin J. W. J. Am. Chem. Soc. 2011, 133, 14196–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco A.; Townsley F. M.; Greiss S.; Lang K.; Chin J. W. Nat. Chem. Biol. 2012, 8, 748–750. [DOI] [PubMed] [Google Scholar]
- Parrish A. R.; She X. Y.; Xiang Z.; Coin I.; Shen Z. X.; Briggs S. P.; Dillin A.; Wang L. ACS Chem. Biol. 2012, 7, 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. Y.; Kawaguchi D.; Coin I.; Xiang Z.; O’Leary D. D. M.; Slesinger P. A.; Wang L. Neuron 2013, 80, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock S. M.; Uprety R.; Deiters A.; Chin J. W. J. Am. Chem. Soc. 2010, 132, 14819–14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T.; Ishii R.; Fukunaga R.; Kobayashi T.; Sakamoto K.; Yokoyama S. Chem. Biol. 2008, 15, 1187–1197. [DOI] [PubMed] [Google Scholar]
- Polycarpo C. R.; Herring S.; Berube A.; Wood J. L.; Soll D.; Ambrogelly A. FEBS Lett. 2006, 580, 6695–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T.; Ishii R.; Fukunaga R.; Kobayashi T.; Sakamoto K.; Yokoyama S. J. Mol. Biol. 2008, 378, 634–652. [DOI] [PubMed] [Google Scholar]
- Lang K.; Davis L.; Wallace S.; Mahesh M.; Cox D. J.; Blackman M. L.; Fox J. M.; Chin J. W. J. Am. Chem. Soc. 2012, 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. J.; Borbas J.; Drescher M.; Summerer D. J. Am. Chem. Soc. 2014, 136, 1238–1241. [DOI] [PubMed] [Google Scholar]
- Wang Y. S.; Fang X. Q.; Chen H. Y.; Wu B.; Wang Z. Y. U.; Hilty C.; Liu W. S. R. ACS Chem. Biol. 2013, 8, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharp J. M.; Wang Y. S.; Lee Y. J.; Yang Y.; Liu W. R. ACS Chem. Biol. 2014, 9, 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M. Y.; Pieper U.; Sali A.. Current Protocols in Bioinformatics; Wiley: New York,2006; Chapter 5, Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. J. Comput. Chem. 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavran J. M.; Gundllapalli S.; O’Donoghue P.; Englert M.; Soll D.; Steitz T. A. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S.; Gattner M. J.; Vrabel M.; Flugel V.; Lopez-Carrillo V.; Prill S.; Carell T. ChemBioChem. 2013, 14, 2114–2118. [DOI] [PubMed] [Google Scholar]
- Mohn W. W.; Tiedje J. M. Microbiol. Rev. 1992, 56, 482–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y.; Mi J.; Cui J.; Lu D.; Zhang X.; Guo C.; Gao G.; Liu Q.; Chen B.; Shao C.; Gong Y. J. Biol. Chem. 2009, 284, 33320–33332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti E.; Franks N. P.; Brick P. Structure 1996, 4, 287–298. [DOI] [PubMed] [Google Scholar]
- Fraga H.; Fernandes D.; Novotny J.; Fontes R.; Esteves da Silva J. C. ChemBioChem. 2006, 7, 929–935. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Lin S.; Huang Y.; Zhao J.; Chen P. R. J. Am. Chem. Soc. 2013, 135, 7410–7413. [DOI] [PubMed] [Google Scholar]
- Gautier A.; Deiters A.; Chin J. W. J. Am. Chem. Soc. 2011, 133, 2124–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill J.; Chou C.; Chin J. W.; Deiters A. J. Am. Chem. Soc. 2013, 135, 13433–13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormo M.; Cubitt A. B.; Kallio K.; Gross L. A.; Tsien R. Y.; Remington S. J. Science 1996, 273, 1392–1395. [DOI] [PubMed] [Google Scholar]
- Stepanenko O. V.; Stepanenko O. V.; Kuznetsova I. M.; Verkhusha V. V.; Turoverov K. K. Int. Rev. Cell Mol. Biol. 2013, 302, 221–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokalingam S.; Raghunathan G.; Soundrarajan N.; Lee S. G. PLoS One 2012, 7, e40410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye S.; Tsuji F. I. FEBS Lett. 1994, 351, 211–214. [DOI] [PubMed] [Google Scholar]
- Pletnev S.; Subach F. V.; Dauter Z.; Wlodawer A.; Verkhusha V. V. J. Am. Chem. Soc. 2010, 132, 2243–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka R.; Yamagishi-Shirasaki M.; Funatsu T. Anal. Biochem. 2011, 414, 173–178. [DOI] [PubMed] [Google Scholar]
- Kutrowska B. W.; Narczyk M.; Buszko A.; Bzowska A.; Clark P. L. J. Phys.: Condens. Matter 2007, 28, 285223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroberardino P. G.; Orr A. L.; Hu X. P.; Na H. M.; Greenamyre J. T. Free Radic. Biol. Med. 2008, 45, 971–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.; Deiters A. Angew. Chem., Int. Ed. 2011, 50, 6839–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards W. F.; Young D. D.; Deiters A. ACS Chem. Biol. 2009, 4, 441–445. [DOI] [PubMed] [Google Scholar]
- Allnér O.; Nilsson L.; Villa A. J. Chem. Theory Comput. 2012, 8, 1493–1502. [DOI] [PubMed] [Google Scholar]
- Meagher K. L.; Redman L. T.; Carlson H. A. J. Comput. Chem. 2003, 24, 1016–1025. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Proteins 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.