

Abstract
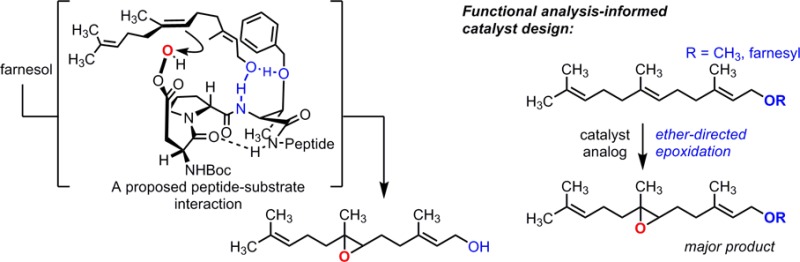
We describe mechanistic investigations of a catalyst (1) that leads to selective epoxidation of farnesol at the 6,7-position, remote from the hydroxyl directing group. The experimental lineage of peptide 1 and a number of resin-bound peptide analogues were examined to reveal the importance of four N-terminal residues. We examined the selectivity of truncated analogues to find that a trimer is sufficient to furnish the remote selectivity. Both 1D and 2D 1H NMR studies were used to determine possible catalyst conformations, culminating in proposed models showing possible interactions of farnesol with a protected Thr side chain and backbone NH. The models were used to rationalize the selectivity of a modified catalyst (17) for the 6,7-position relative to an ether moiety in two related substrates.
Introduction
Directing groups play an important role in complex molecule synthesis.1 Catalytic reactions aided by directing groups are now ubiquitous in the chemical literature. For example, great strides have been made in the area of hydroxyl-directed epoxidation, with enantioselective methods for allylic,2 homoallylic,3 and even bis-homoallylic alcohols now recorded.4 More rare, however, are catalytic reactions directed by a remote chemical moiety—that is, a functional group that is several bonds distal to the reactive site of the substrate.5 Often, these general synthetic strategies rely on conformational restriction or strong molecular interactions to achieve selectivity.6 In contrast, we recently discovered a peptide-based catalyst (1)7 capable of oxidizing an olefin positioned five rotatable bonds (in addition to one C–C double bond) from its directing group. Notably, the selectivity achieved by 1 differs from that observed with farnesol (2) when m-chloroperoxybenzoic acid (mCPBA) or, to our knowledge, any other previously known site-selective oxidation catalyst is used. Despite the remarkable site selectivity exhibited by catalyst 1 (∼1:8:1 for the 2,3-, 6,7-, and 10,11-positions of 2, shown in green, blue, and red, respectively, in Scheme 1A), the observed enantioselectivity for the 6,7-position is low (∼10% ee). Given the peculiar selectivity imparted by this remotely directed catalyst, we sought to understand the molecular underpinnings of the catalyst–substrate interaction.
Scheme 1. Previously Reported 6,7-Oxidation and Proposed Peptide-Catalyzed Epoxidaton Catalytic Cycle.
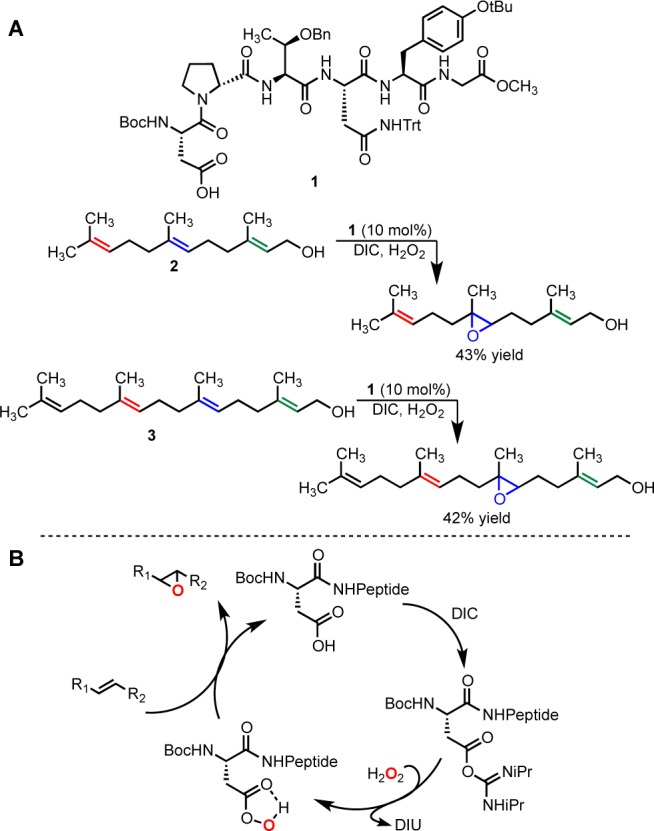
DIU = N,N′-diisopropylurea.
Catalyst 1 was developed using an on-bead screening protocol8 used to evolve combinatorial libraries for site selectivity in the epoxidation of farnesol (Scheme 1A). Each peptide belonging to the initial screening libraries contained a Boc-protected aspartic acid residue at the N-terminus, which possesses a catalytic carboxylic acid moiety in the side chain. In the catalytic cycle (abbreviated in Scheme 1B),9 the carboxylic acid of 1 is activated with N,N′-diisopropylcarbodiimide (DIC) and, in the presence of H2O2, forms a peptidyl peracid that selectively epoxidizes the 6,7-olefin of either 2 or geranylgeraniol (3). Critically, we had established previously that the reaction catalyzed by 1 was predominantly hydroxyl-directed through the observation of selective 6,7-epoxidation of both 2and3. In addition, our preliminary experiments showed that catalysts of this type deliver substantially lower selectivity for the 6,7-olefin of farnesyl methyl ether, comparable to that exhibited by propionic acid [Supporting Information (SI) Figure 9].
Other than the apparently fortuitous choice of the amino acids and their position within our screening libraries, we developed 1 without any preconceived notion of how such a catalyst would deliver selectivity. Therefore, an understanding of the mechanism for the selectivity displayed by catalyst 1 demanded further studies, which are presented herein. Simultaneously, a detailed retrospective analysis of the arrival at the best peptide sequence appeared to be inextricably related and has not been described previously. The interplay of these studies is described below.
Results and Discussion
6,7-Selective Catalyst Lineage
A limited understanding of the importance of the residues within peptide 1 was initially derived from studies of the libraries leading to the discovery of 1. The observation of a peptide that exhibited a modicum of selectivity for the 6,7-position of 2 (relative to the 2,3- or 10,11-positions) was unexpected. In fact, the library from which this initial 6,7-selective “hit” derived was generated in order to study 2,3-selectivity. This library had been designed such that two internal residues had been omitted from the original hexameric sequences.7 This shortened library of tetramers was biased to contain a choice of d-amino acids adjacent to the catalytic N-terminal aspartic acid residue and two additional C-terminal variable residues. The first peptide that was found to exhibit the alternate 6,7-selectivity, Boc-Asp-d-Pro-Thr(Bn)-Leu (Boc = tert-butoxycarbonyl, Bn = benzyl), exhibited only modest selectivity (4:5:6 = 1.3:1.5:1.0, point shown in Figure 1A).10 A subsequent library biased toward this initial hit was immediately prepared.7
Figure 1.

(A–C) Ternary plots showing an overlay of peptide selectivities from successive generations of 6,7-selective peptides, where each axis represents the fraction of the total monoepoxide: (A) the first library demonstrating 6,7-selectivity (first generation, red); (B) the first biased library for 6,7-selectivity (second generation, green); (C) second biased library (third generation, blue). Points that are higher (further away from the triangle base) are more 6,7-selective. The highlighted generation from each plot is in the solid color in the foreground atop the other library generations in the background. Solid markers indicate peptides that were sequenced. (D) List of a portion of sequenced peptides from each library generation (shown with solid markers in A–C). Residues shown in blue indicate were picked from the pool of variable residues from their particular library. (E) Selectivity with amino acids at the i + 2 position, which can be compared to the library with i + 2 Thr(Bn) directly to the left (C, third generation, blue). Product ratios were measured by GC. In all of the on-bead screening studies, the formation of diepoxides was intentionally limited through the use of 0.3 equiv of DIC, resulting in analysis of low-conversion reaction mixtures.
Split and pool11,12 libraries biased13 for this study were prepared such that at each varied position in the peptide design, roughly half the library members possess the amino acid of the parent sequence while the remaining library members would have one of six or seven alternative residues. Additionally, each successive library was designed to possess a longer sequence length than the previous generation, with an additional variable residue added to the C-terminal side.7
At the same time that the first directed library was being prepared (second generation), further screening of the initial library (first generation) ultimately led to another more 6,7-selective peptide with an i + 3 trityl (Trt)-protected asparagine [Asn(Trt)] (4:5:6 = 1.2:2.1:1.0). The data from the first-generation library are presented graphically in Figure 1A and discussed in more detail below.
Figure 1 contains ternary plots that illustrate the percentage distribution of the monoepoxides formed by catalysts during on-bead screening experiments. In these plots, the horizontal lines mark the gradations of 6,7-selectivity. Thus, points appearing higher on the graph represent product mixtures with a higher proportion of product 5. Points that appear closer to the other vertices represent peptides that produce a higher proportion of 4 or 6.
Screening of the second generation of catalysts biased toward sequences containing an i + 3 Leu revealed two additional sequences that performed better than the others (4:5:6 = 1.0:1.7:1.1 and 1.5:2.6:1.014). Both of these second-generation hits contained an i + 2 Thr(Bn) and an i +3 Asn(Trt) (Figure 1B).
Finally, a third-generation library was studied wherein the i + 2 Thr(Bn) was fixed and the i + 3 position was biased toward sequences containing Asn(Trt). A number of peptides from this library were found to yield favorable selectivity (Figure 1C), and we subjected many to sequencing. While it is difficult to draw conclusions from any individual peptide sequenced from these libraries without validation, all of the high-performing sequences contained an i + 3 Asn(Trt), suggesting that the Asn(Trt) conferred advantageous selectivity. However, a variety of other residues was found in the i + 4 and i + 5 positions in these high-performing sequences (Figure 1D).
It is interesting to note that the best catalysts from each library generation seem to yield similar positional selectivity when examined on solid supports and under the initial screening conditions (Figure 1A–C). While many of the catalysts found from the later library generations performed similarly, these on-bead screens yielded several leads for peptide validation and optimization in solution, which ultimately led to 1. Nonetheless, the on-bead screening data suggest that a tetramer is sufficient to achieve 6,7-selectivity on-bead (from the first-generation library, observed 4:5:6 = 1.2:2.1:1.0) and that the best sequenced peptides from each on-bead generation contained common elements: an i + 2 Thr(Bn) and an i + 3 Asn(Trt).
With no other structural data but with knowledge of the amino acid sequence of peptide 1, and no expectation that the full diversity of each peptide library had been sampled, we hypothesized that the embedded dipeptide d-Pro-Thr(Bn) might enforce a β-turn. Notably, d-Pro adjacent to a β-branched amino acid is suggestive of a type II′ β-turn,15 an oft-studied motif in a number of peptide-based catalysts from our lab16 and others.17 To examine the existence of this structural element within 1, we resorted to the examination of further altered sequences within on-bead peptide libraries.
If peptide 1 were to exhibit a turn, then perhaps on-bead libraries with different turn-promoting residues at the i + 2 position, such as Val or Aib, might also work at this position. Such substitutions were thought to be a potential avenue for the identification of more selective catalysts.
Four distinct biased split-and-pool libraries of on-bead peptides derived from libraries that initially led to 1 were therefore compared for site selectivity in the epoxidation of 2 (compare panels C and E of Figure 1). Each library possessed either Val, Aib, Thr(tBu), or Thr(Bn) in the i + 2 position. Although the data in Figure 1E represent only a limited sampling of three libraries, the testing of different i + 2 residues while simultaneously evaluating other residues at adjacent positions can potentially reveal epistatic interactions. That is, in theory, this experiment allows for the possibility of finding better peptides that might only be found through multiple amino acid substitutions, where the better selectivity results from some combination of changes. Nonetheless, peptides containing protected Thr derivatives [Thr(Bn) or Thr(tBu)] were more 6,7-selective than those containing either Val or Aib. These findings suggest that even if the i + 2 Thr(Bn) is turn-promoting, it likely plays an additional role in delivering selectivity for the 6,7-olefin of 2.
Peptides with either an i + 2 Thr(Bn) or Thr(tBu) demonstrated favorable 6,7-selectivity on-bead and in solution. During the optimization studies of the 6,7-selective catalysts in solution, we generally observed that the site and enantioselectivity afforded by the catalysts for the 6,7-position were uncorrelated. For example, i + 2 Thr(Bn)-containing peptide 7 gave 4:5:6 = 1.0:4.1:1.3 with 5 in 1.6:1.0 er, yet the Thr(tBu)-containing analogue of 7 gave 4:5:6 = 1.0:3.7:1.1 with 5 in 1.9:1.0 er. These findings underline the potentially complicated relationship between the different rates of epoxidation leading to either site and/or enantioselectivity.
On-Bead Analogue Studies
Substitutions of other residues within 1 were studied by examining specific analogues on-resin. These peptide analogues were examined on-resin for convenience of synthesis and ease of screening, and they yielded reduced selectivity in comparison with their solution-phase counterparts. Additionally, the on-bead reactions were performed under unoptimized conditions with substoichiometric oxidant, restricting the conversion.
An on-bead analogue of 1 furnished a 4:5:6 product mixture of 1.2:2.3:1.0 (Table 1, entry 1). Modifications to the i + 2 Thr side chains, such as replacement of the benzyl protecting group with a trityl group (entry 2) or removal of the γ-methyl (entry 3) resulted in diminished selectivity. Replacement of the ethereal O-benzyl with an ethyl group (Ile; entry 4) still furnished some selectivity for 5, although it was attenuated.
Table 1. Product Distributions Resulting from Oxidation of 2 with a Number of On-Bead Peptide Analogues of 1a,b.
| entry | i | i + 1 | i + 2 | i + 3 | i + 4 | 10,11- (4) | 6,7- (5) | 2,3- (6) |
|---|---|---|---|---|---|---|---|---|
| 1 | Boc-Asp | d-Pro | Thr(Bn) | Asn(Trt) | Tyr(tBu) | 1.2 | 2.3 | 1.0 |
| 2 | Boc-Asp | d-Pro | Thr(Trt) | Asn(Trt) | Tyr(tBu) | 1.1 | 1.5 | 1.0 |
| 3 | Boc-Asp | d-Pro | Ser(Bn) | Asn(Trt) | Tyr(tBu) | 1.1 | 1.7 | 1.0 |
| 4 | Boc-Asp | d-Pro | Ile | Asn(Trt) | Tyr(tBu) | 1.0 | 1.5 | 1.1 |
| 5 | Boc-Asp | d-Pro | Thr(Bn) | Asp(tBu) | Tyr(tBu) | 1.0 | 1.2 | 1.1 |
| 6 | Boc-Asp | d-Pro | Thr(Bn) | Hse(Trt) | Tyr(tBu) | 1.1 | 1.1 | 1.0 |
| 7 | Boc-Asp | d-Pro | Thr(Bn) | Ser(Trt) | Tyr(tBu) | 1.1 | 1.1 | 1.0 |
| 8 | Boc-Asp | d-Pro | Thr(Bn) | Leu | Tyr(tBu) | 1.2 | 1.5 | 1.0 |
| 9 | Boc-Asp | d-Pro | Thr(Bn) | Asn(Trt) | Phe | 1.2 | 2.2 | 1.0 |
| 10 | Boc-Asp | d-Pro | Thr(Bn) | Asn(Trt) | hPhe | 1.2 | 2.1 | 1.0 |
| 11 | Boc-Asp | d-Pro | Thr(Bn) | Asn(Trt) | 2-Nal | 1.2 | 2.3 | 1.0 |
| 12 | Boc-Asp | d-Pro | Thr(Bn) | Asn(Trt) | Cha | 1.2 | 2.2 | 1.0 |
Reactions were conducted with 2 (1 μmol, 1.0 equiv), DIC (0.3 equiv), 1-hydroxybenzotriazole (HOBt) (0.1 equiv), 4-dimethylaminopyridine (DMAP) (0.1 equiv), H2O2 (1.0 equiv), dichloromethane (DCM) (0.2 M), and a single bead bound to peptide.
2-Nal = 2-naphthylalanine, Cha = cyclohexylalanine, hPhe = homophenylalanine, Hse = homoserine.
Modifications to the i + 3 Asn(Trt) also resulted in reduced selectivity. When other trityl-containing amino acids [Hse(Trt) and Ser(Trt); Table 1, entries 6 and 7] or Asp(tBu) (entry 5) were studied in this position, the selectivity was nearly entirely lost. In comparison with other substitutions, Leu yielded a decent level of selectivity (entry 8), though less than the parent peptide, which is potentially meaningful because Leu is sometimes thought to be an Asn isostere,18 though without a trityl protecting group. These observations are consistent with those made during the screening of on-bead libraries: both the i + 2 Thr(Bn) and i + 3 Asn(Trt) are important and sensitive to substitution.
Substitution of the i + 4 Tyr(tBu) side chain with other aryl- (Table 1, entries 9–11) and alkyl-containing (entry 12) side chains did not seem to lead to drastic changes in selectivity. These observations largely agree with those made from studies of substitutions of the i + 4 and i + 5 residues with peptides under more homogeneous conditions (i.e., not resin-bound).19
Studies with Truncated Peptides
To explore the necessity of each amino acid, the selectivities of a number of truncated analogues of 1 (7–12) were evaluated in solution. The results, presented in Figure 2, underline the complicated function that each amino acid of 1 plays in delivering optimal selectivity for the 6,7-olefin. Notably, these reactions were performed at ca. 5 °C lower than the previously reported epoxidation conditions.7 At −20 °C, 1 furnishes slightly higher selectivity (4:5:6 = 1.1:8.6:1.0; Figure 2A).
Figure 2.

(A) Selectivities observed with truncated peptides. (B) Abbreviated 1D 1H NMR spectra of 1 and its truncated analogues acquired at −20 °C at 20 mM.
The truncated peptides reveal the interesting roles played by the i + 3 Asn(Trt) and i + 4 Tyr(tBu). Replacement of the C-terminal Gly-OCH3 of 1 with a methyl ester, as in 7, results in roughly the same 6,7:10,11 selectivity but decreases the 6,7:2,3 selectivity by about half. Deletion of each sequential amino acid from the C-terminus seems to decrease the overall selectivity for the 6,7-olefin relative to the 10,11-olefin. Moreover, through each truncation, the selectivity for 6 increases relative to 5, yet peptides 7, 8, and 9 display roughly the same 5:6 selectivity of 3:1 to 4:1.
Consistent with the hypothesis of an interaction of the Thr(Bn) region of the peptide with farnesol, tripeptide 9 with a C-terminal Thr(Bn)-NHCH3 (entry 4) is sufficient to furnish some 6,7-selectivity, though reduced from the parent peptide (entry 1). However, tripeptide 10 with a C-terminal methyl ester lacks selectivity entirely. Additionally, the selectivity actually decreases substantially and begins to favor the 2,3-olefin within farnesol in the case of 11 and 12, where the Thr(Bn) is lacking. We were surprised to find that the simple aspartic acid derivative 12 slightly favors 6 given that simpler peracids (e.g., propionic acid) favor oxidation of the most electron-rich olefin to make 4. Perhaps these observations underscore the surprising frequency with which catalysts selective for the olefin most proximal to a directing group are found. Indeed, our experience exploring catalysts for the oxidation of 2 has been rife with allylic epoxidation catalysts.
1H NMR-Assisted Analysis of Catalyst Structure
The intriguing transition from the observed 6,7-selectivity to an absence of selectivity with the change of an amide to an ester in the trimer may result from a variety of scenarios. The NH of the methyl amide in 9 may interact with farnesol to deliver selectivity. Alternatively, the change from amide to ester (9 to 10) may result in a number of conformational changes, which are manifest in the 1H NMR spectrum. Compared with the other peptides, some proton signals in the spectra of 10 are doubled, whereas the longer truncated variants seem to display less conformational heterogeneity. One possible conformation available to 9 but not 10 is a β-turn with a 10-membered ring formed through hydrogen bonding of the methyl amide.
The selectivity data from the truncated analogues combined with the 1D 1H NMR spectra (Figure 2B) suggest that a β-turn may form in trimer 9 and the longer peptides. In particular, the 1H NMR spectrum of tetramer 8 reveals significant downfield shifts of the Boc-NH and NH-Asn signals, corresponding to protons that would participate in the turn, while the NH-Thr signal shifts upfield.
While the NH-Asn signals shift back upfield in the spectra of pentamer 7 and hexamer 1 (relative to 8), the observed Boc-NH shifts appear further downfield for both. Indeed, the spectra of the longer peptides are difficult to interpret given the broadening of some peaks at this low temperature. Nonetheless, 2D 1H–1H rotating-frame nuclear Overhauser effect spectroscopy (ROESY) data corroborate the interpretation of a β-turn (Figure 3A). The spectra suggest a through-space correlation between the β-protons of the i Asp and i + 3 Asn(Trt) in spectra of 1 and 7 acquired at 25 and −20 °C, respectively. Additionally, these data suggest that the Hα-d-Pro is close to the amide protons of both the i + 2 Thr(Bn) and i + 3 Asn(Trt). Notably, many of the correlations found in 1 at 25 °C are also found in truncated peptide 7 at −20 °C, indicating that the room-temperature data for 1 are likely relevant.
Figure 3.
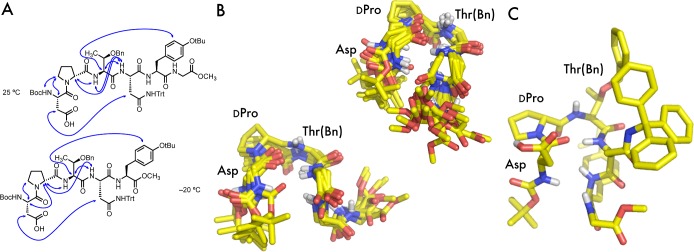
(A) Selected 1H–1H ROESY correlations found in 1 at 25 °C and truncated peptide 7 at −20 °C. (B) Two structural ensembles from the 20 CNS-generated structures of 1 computed using 25 °C ROESY data. (C) One structure chosen from the 20 structures shown with side chains.
To determine whether the NMR data fit our qualitative inferences about secondary structure, we estimated and binned distance restraints from the ROESY data for 1 acquired at 25 °C for structure calculations using the program CNS.20 The interpretation of these structures is accompanied by the usual caveats. For example, as with any structural study of a catalyst, observations of the resting or intermediate state may differ completely from those of the active catalyst.21 Additionally, calibration of distance restraints is usually performed by comparison to an experimentally determined distance (e.g., two ortho aryl protons), but we were unable to find pairs of protons that could confidently be used for this purpose, so we looked at two different calibration sets between Hα-Asp and Hδ-d-Pro (see the Supporting Information). From each calibrated distance, the other correlations were assigned to different bins as appropriate, and 10 structures that best fit the data were analyzed from each data set. An overlay of the combined 20 structures suggested the existence of two structural ensembles, each of which contained members derived from both of the distance restraint sets. The two ensembles are shown in Figure 3B.
The calculated structures agree with other experimental observations presented herein and provide insight into some possible structural features. Many of the structures that best fit the data are suggestive of a β-turn characterized by an apparent hydrogen bond between the i Asp-CO and the i + 3 NH-Asn(Trt) (the distance between the backbone i CO and i + 3 Nα ranged from 2.8 to 3.7 Å across the 20 structures). Indeed, these observations are not surprising given the correlation between the β-protons of those residues in the NMR spectra. A smaller set of the calculated structures may also contain a hydrogen bond between the i + 2 NH-Thr(Bn) and the i CO, indicative of a γ-turn.22 Additionally, the data suggest that the i + 5 Tyr(tBu) side chain is oriented underneath the turn, as is evident from a representative structure picked from the ensembles (Figure 3C).
On the basis of the aforementioned experimental evidence, we hypothesized that the interactions of 1 and 2 defined in Figure 4 are consistent with the selective oxidation of the 6,7-olefin. We consider on the basis of analogue and structural studies that the NH of the i + 2 Thr(Bn) could interact with the hydroxyl (as shown). The data suggest that a type II′ β-turn can form, which orients this NH of the i + 2 Thr(Bn) in the same direction as the aspartic acid side chain (upward as in Figure 4A,B). A γ-turn may also form in addition to or in combination with the β-turn (Figure 4C). Furthermore, the i + 2 Thr(Bn) side chain Oγ and the backbone NH may serve as hydrogen-bond acceptor and donor, respectively, with the hydroxyl of farnesol, leading to the proper proximal position of the 6,7-olefin relative to the modeled peracid (Figure 4A–C). While the NMR experiments suggest a β-turn, which is often accompanied by the discussed i → i + 3 hydrogen bond, it is possible that under the reaction conditions, the NH of the i + 3 Asn(Trt) is oriented such that a hydrogen bond forms with the substrate hydroxyl as well (Figure 4B). It is not clear whether the intramolecular i → i + 3 hydrogen bond is beneficial to the selectivity or whether the i + 3 NH is important for binding with the substrate, or both, a detail that is difficult to discern experimentally. Regardless, it appears that this NH is important for one or both hydrogen-bonding interactions. Of note, our data support models wherein at least two loci contribute to the hydrogen-bonding network between the catalyst and substrate. That is, ablation of the ethereal hydrogen-bond acceptor in the i + 2 position is not enough to induce a complete loss of 6,7-selectivity (Table 1, entry 4; see also Table 2, entry 4). Analysis of the other libraries is also internally consistent (Figure 1E; Val and Aib libraries).
Figure 4.
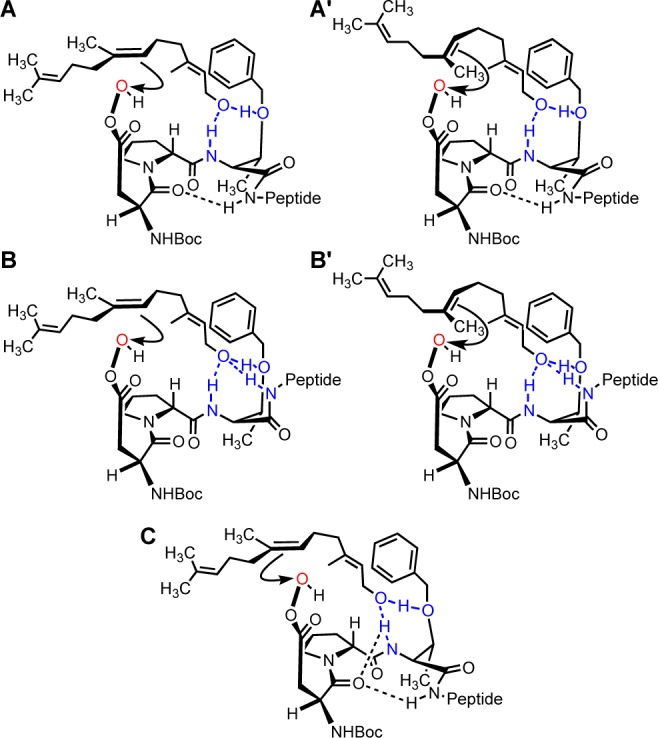
Hypothesized models for selectivity toward 5.
Table 2. Correlation of the i + 3 Position with Hydroxyl and Methoxyl Directing Groups.
The hypothetical transition-state interactions between the catalyst and substrate in Figure 4 are consistent with the observed selectivity with trimer 9 and are supported by the NMR studies of the longer peptides. Interestingly, the minimal enantioselectivity in the formation of 5 can be explained by the accessibility of either face of the olefin through a number of possible σ-bond rotations in the substrate (different rotational configurations are shown in Figure 4A′,B′).
It is not clear from these data what the additional amino acids that are potentially disposed farther away from the Thr(Bn) and catalytic Asp residue are doing to improve the 6,7-selectivity of 1. We wonder whether there may be a number of peptide–farnesol interactions that lead to allylic alcohol epoxidation, as the 2,3-position is most proximal to the putative directing group. Our studies have suggested that the mode by which these catalysts achieve selectivity is quite delicate, which is perhaps underlined by the limited solvent compatibility. Whereas other peptidyl peracids that our group has studied have generally yielded some selectivity in a range of solvents, we have observed substantial selectivity of 1 toward 5 only in halogenated solvents. Moreover, our data are consistent with several possible transition-state models (e.g., Figure 4) that cannot be resolved at this time.
Experimental Interrogation of the Proposed Models
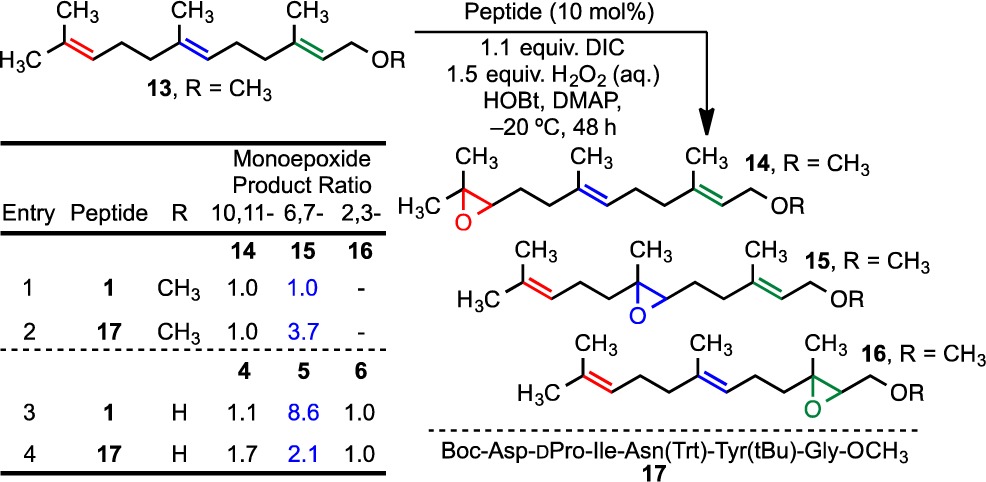
To test our hypothetical models, we re-examined a substrate that we had studied earlier, farnesyl methyl ether (13). As noted in the Introduction, we found that a 6,7-selective pentamer affords no appreciable selectivity under a previous set of reaction conditions, commensurate with loss of a hydroxyl-group-directed mechanism. Under the updated reaction conditions of Table 2, we found once again that peptide 1 affords no selectivity between the 10,11-epoxide 14 and the 6,7-epoxide 15 (entry 1). Moreover, when the terminus is functionalized (e.g., 13), we observe little formation of the 2,3-epoxide (e.g., 16); the reactivity of the allylic olefin in the electrophilic epoxidation is attenuated by the electron-withdrawing oxygen-containing substituent, and there is no compensatory hydroxyl direction.23 However, the lack of selectivity between the 10,11-epoxide 14 and the 6,7-epoxide 15 observed with catalyst 1 allows a test of our models with an opportunity to enhance selectivity for one isomer.
The models in Figure 4 are consistent with the methyl group of farnesyl methyl ether (13) obstructing the highly orchestrated hydrogen-bonding array that accommodates the hydroxyl of farnesol (one scenario is depicted in Figure 5A). Notably, each of the arrays in Figure 4 is buttressed by an attractive interaction between the Thr(Bn) γ-oxygen atom and the OH group. Substitution of the i + 2 Thr(Bn) of 1 with an Ile, as in peptide 17, eradicates this attractive interaction. Its absence could then make space available for the methyl group while retaining a less elaborate but still functional group-directing array for ether-directed epoxidation (Figure 5B). Consistent with this notion, catalyst 17 delivers an increase in the product ratio to nearly 1:4 for the 6,7-position (14:15; Table 2, entry 2), instead of the 1:1 ratio observed with catalyst 1 (entry 1). As emphasized in Figure 5A, it is possible that with 1, unfavorable steric or electronic interactions exclude ether-directed chemistry. However, with the alkyl side chain of Ile in 17, the peptide is still able to accommodate hydrogen-bonding interactions with the ethereal oxygen of the substrate (Figure 5B), albeit with a perhaps reduced efficiency as one of several hydrogen-bond loci is removed. In this vein, catalyst 17 is indeed quite a bit less selective than catalyst 1 for farnesol itself (4:5:6 = 1.1:8.6:1.0 for 1 and 1.7:2.1:1.1 for 17; Table 2, entries 3 and 4). Taken together, the picture that emerges as farnesol (2) and its derived methyl ether (13) are evaluated with catalysts 1 and 17 is one that links the models in Figures 4 and 5 in a self-consistent manner.
Figure 5.
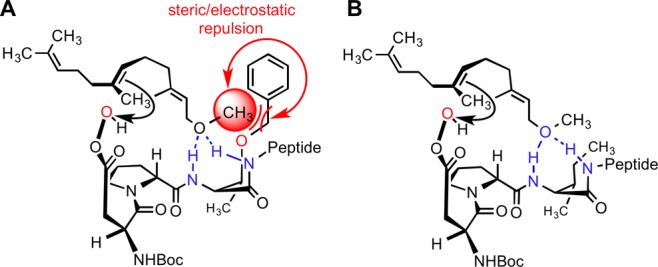
Hypothesized interactions of 13 with i + 3 side-chain variants of 1 and 17. One of the several scenarios based on Figure 4 is depicted.
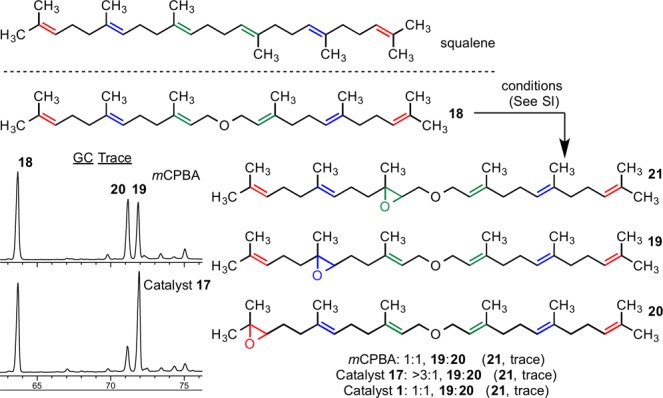
A thought-provoking and also consistent experiment was performed with difarnesyl ether (18). Of note, this compound is an oxide of the venerable natural product squalene, with a sole oxygen atom inserted within its central C–C bond. When catalyst 17 was examined with substrate 18, once again we observed that the catalytic formation of monoxides was appreciably enhanced in the anticipated internal olefin 19 (19:20 > 3:1; Scheme 2 inset), with minimal allylic oxidation detected. This result contrasts with the virtual lack of selectivity observed with mCPBA (19:20 = 1:1) or catalyst 1 (19:20 = 1:1). The selective formation of 19 also differs markedly from the apparent statistical distribution of epoxides observed for squalene itself under the various conditions we have examined. Taken together, these results represent a self-consistent data set for the models presented in Figures 4 and 5, ultimately derived from NMR structural data, extensive SAR, and challenging of the model with a new substrate type.
Scheme 2. Comparison of Reactions with Difarnesyl Ether.
Concluding Perspectives
We close by noting a potential irony emerging from this study. Although we employed a diversity-based approach to catalyst discovery, we found that the catalyst that provides selectivity for the 6,7-position of farnesol exhibits a propensity for β-turn formation, a motif that our lab has studied on numerous occasions. However, the structural analysis of 1 and its large extended family of variants led to new insights. Notably, an interaction between the backbone NH and the side-chain oxygen of Thr is thought to form part of the oxyanion hole of lipases.24 Indeed, the comparative behavior of catalysts 1 and 17 provides some analogy for this feature.25 Indeed, since our experiments with catalysts 1 and 17 provide evidence for remote directing effects with different substrate types, the observations highlight the tunability of catalyst hydrogen-bond arrays to accommodate different substrate-directing groups.
Substrates like farnesol, its derived ethers, and many other polyenes remain great challenges for total control of site selectivity through catalysis. Nevertheless, the mechanistic hypotheses derived from the experimental data presented herein have suggested several opportunities for further catalyst development. Future efforts in our lab will be focused on developing analogues of catalyst 1 (and 17) for remote oxidation of other substrates and other positions.
Acknowledgments
The authors thank Dr. Michael Giuliano and Dr. Eric Paulson for discussions about NMR structure elucidation and Prof. Matthew Sigman and Elizabeth Bess (University of Utah) for helpful discussions. This work was supported by the National Institutes of Health (NIH) (R01-GM096403 to S.J.M). Additionally, P.A.L. was partially supported by the NIH (CBI-TG-GM-067543).
Supporting Information Available
Additional figures, experimental details, characterization data, and calculated structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Hoveyda A. H.; Evans D. A.; Fu G. C. Chem. Rev. 1993, 93, 1307–1370. [Google Scholar]; b Rousseau G.; Breit B. Angew. Chem., Int. Ed. 2011, 50, 2450–2494. [DOI] [PubMed] [Google Scholar]
- For example, see:; a Katsuki T.; Martin V. S. Org. React. 1996, 48, 1–299. [Google Scholar]; b Zhang W.; Basak A.; Kosugi Y.; Hoshino Y.; Yamamoto H. Angew. Chem., Int. Ed. 2005, 44, 4389–4391. [DOI] [PubMed] [Google Scholar]; c Malkov A. V.; Czemerys L.; Malyshev D. A. J. Org. Chem. 2009, 74, 3350–3355. [DOI] [PubMed] [Google Scholar]; d Egami H.; Oguma T.; Katsuki T. J. Am. Chem. Soc. 2010, 132, 5886–5895. [DOI] [PubMed] [Google Scholar]
- a Zhang W.; Yamamoto H. J. Am. Chem. Soc. 2007, 129, 286–287. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhi L.; Wei Z.; Yamamoto H. Angew. Chem., Int. Ed. 2008, 47, 7520–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Olivares-Romero J. L.; Zhi L.; Yamamoto H. J. Am. Chem. Soc. 2013, 135, 3411–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Yamamoto H. J. Am. Chem. Soc. 2010, 132, 7878–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Leow D.; Li G.; Mei T.-S.; Yu J.-Q. Nature 2012, 486, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tang R.-Y.; Li G.; Yu J.-Q. Nature 2014, 507, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For some representative examples, see:; a Breslow R.; Maresca L. M. Tetrahedron Lett. 1977, 18, 623–626. [Google Scholar]; b Breslow R.; Maresca L. M. Tetrahedron Lett. 1978, 19, 887–890. [Google Scholar]; c Das S.; Incarvito C. D.; Crabtree R. H.; Brudvig G. W. Science 2006, 312, 1941–1943. [DOI] [PubMed] [Google Scholar]; d Li J.-J.; Giri R.; Yu J.-Q. Tetrahedron 2008, 64, 6979–6987. [Google Scholar]
- Lichtor P. A.; Miller S. J. Nat. Chem. 2012, 4, 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtor P. A.; Miller S. J. ACS Comb. Sci. 2011, 13, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris G.; Jakobsche C. E.; Miller S. J. J. Am. Chem. Soc. 2007, 129, 8710–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This peptide was validated on-bead and found to exhibit a 4:5:6 product ratio of 1.5:1.9:1.0 (see ref (7)).
- a Lam K. S.; Lebl M.; Krchnák V. Chem. Rev. 1997, 97, 411–448. [DOI] [PubMed] [Google Scholar]; b Furka A.; Sebestyen F.; Asgedom M.; Dibo G. Int. J. Pept. Protein Res. 1991, 37, 487–493. [DOI] [PubMed] [Google Scholar]; c Lam K. S.; Salmon S. E.; Hersh E. M.; Hruby V. J.; Kazmierski W. M.; Knapp R. J. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- For other selected applications of the split-and-pool method to catalyst discovery, see:; a Taylor S. J.; Morken J. P. Science 1998, 280, 267–270. [DOI] [PubMed] [Google Scholar]; b Krattiger P.; McCarthy C.; Pfaltz A.; Wennemers H. Angew. Chem., Int. Ed. 2003, 42, 1722–1724. [DOI] [PubMed] [Google Scholar]; c Lingard I.; Bhalay G.; Bradley M. Chem. Commun. 2003, 2310–2311. [DOI] [PubMed] [Google Scholar]; d Clouet A.; Darbe T.; Reymond J.-L. Angew. Chem., Int. Ed. 2004, 43, 4612–4615. [DOI] [PubMed] [Google Scholar]; e Krattiger P.; Kovasy R.; Revell J. D.; Ivan S.; Wennemers H. Org. Lett. 2005, 7, 1101–1103. [DOI] [PubMed] [Google Scholar]
- Copeland G. T.; Miller S. J. J. Am. Chem. Soc. 2001, 123, 6496–6502. [DOI] [PubMed] [Google Scholar]
- This peptide contained an i + 4 Tyr(tBu) and, when validated on-bead, was found to exhibit a 4:5:6 product ratio of 1.5:2.9:1.0 (see ref (7)).
- a Wilmot C. M.; Thornton J. M. J. Mol. Biol. 1988, 203, 221–232. [DOI] [PubMed] [Google Scholar]; b Haque T. S.; Littler J. C.; Gellman S. H. J. Am. Chem. Soc. 1996, 118, 6975–6985. [Google Scholar]
- a Miller S. J. Acc. Chem. Res. 2004, 37, 601–610and references therein. [DOI] [PubMed] [Google Scholar]; b Angione M. C.; Miller S. J. Tetrahedron 2006, 62, 5254–5261. [Google Scholar]; c Blank J. T.; Miller S. J. Biopolymers 2006, 84, 38–47. [DOI] [PubMed] [Google Scholar]; d Lewis C. A.; Miller S. J. Angew. Chem., Int. Ed. 2006, 45, 5616–5619. [DOI] [PubMed] [Google Scholar]; e Cowen B. J.; Saunders L. B.; Miller S. J. J. Am. Chem. Soc. 2009, 131, 6105–6107. [DOI] [PubMed] [Google Scholar]; f Lewis C. A.; Longcore K. E.; Miller S. J.; Wender P. A. J. Nat. Prod. 2009, 72, 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Fowler B. S.; Mikochik P. J.; Miller S. J. J. Am. Chem. Soc. 2010, 132, 2870–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Fowler B. S.; Laemmerhold K. M.; Miller S. J. J. Am. Chem. Soc. 2012, 134, 9755–9761. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Barrett K. T.; Miller S. J. J. Am. Chem. Soc. 2013, 135, 2963–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Linton B. R.; Reutershan M. H.; Aderman C. M.; Richardson E. A.; Brownell K. R.; Ashley C. W.; Evans C. A.; Miller S. J. Tetrahedron Lett. 2007, 48, 1993–1997. [Google Scholar]; b Akagawa K.; Akabane H.; Sakamoto S.; Kudo K. J. Org. Chem. 2008, 10, 2035–2037. [DOI] [PubMed] [Google Scholar]; c Wu F.-C.; Da C.-S.; Du Z.-X.; Guo Q.-P.; Li W.-P.; Yi L.; Jia Y.-N.; Ma X. J. Org. Chem. 2009, 74, 4812–4818. [DOI] [PubMed] [Google Scholar]; d Akagawa K.; Suzuki R.; Kudo K. Adv. Synth. Catal. 2012, 354, 1280–1286. [Google Scholar]; e Mahajan M.; Bhattacharjya S. Angew. Chem., Int. Ed. 2013, 52, 6430–6434. [DOI] [PubMed] [Google Scholar]; f Du Z.-X.; Zhang L.-Y.; Fan X.-Y.; Wu F.-C.; Da C.-S. Tetrahedron Lett. 2013, 54, 2828–2832. [Google Scholar]
- Bryan P.; Pantoliano M. W.; Quill S. G.; Hsiu H.-Y.; Poulos T. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 3743–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supplementary Table S1 of ref (7).
- a Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J.-S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Acta Crystallogr. 1998, D54, 905–921. [DOI] [PubMed] [Google Scholar]; b Brünger A. T. Nat. Protoc. 2007, 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- Halpern J. Science 1982, 217, 401–407. [DOI] [PubMed] [Google Scholar]
- Milner-White E. J.; Ross B. M.; Ismail R.; Belhadj-Mostefa K.; Poet R. J. Mol. Biol. 1988, 204, 777–782. [DOI] [PubMed] [Google Scholar]
- Henbest H. B.; Wilson R. A. L. Chem. Ind. 1956, 26, 659. [Google Scholar]
- a Uppenberg J.; Öhrner N.; Norin M.; Hult K.; Kleywegt G. J.; Patkar S.; Waagen V.; Anthonsen T.; Jones T. A. Biochemistry 1995, 34, 16838–16851. [DOI] [PubMed] [Google Scholar]; b Magnusson A.; Hult K.; Holmquist M. J. Am. Chem. Soc. 2001, 123, 4354–4355. [DOI] [PubMed] [Google Scholar]
- Knowles R. R.; Jacobsen E. N. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.