

Abstract
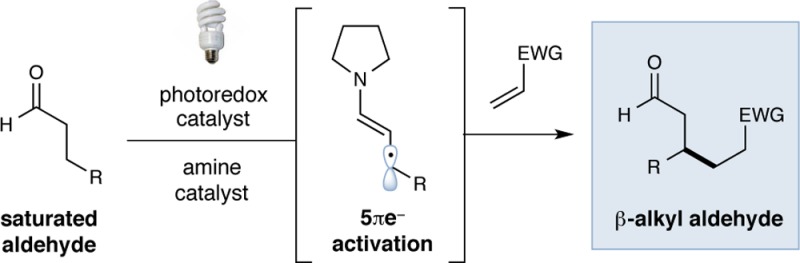
Direct β-alkylation of saturated aldehydes has been accomplished by synergistically combining photoredox catalysis and organocatalysis. Photon-induced enamine oxidation provides an activated β-enaminyl radical intermediate, which readily combines with a wide range of Michael acceptors to produce β-alkyl aldehydes in a highly efficient manner. Furthermore, this redox-neutral, atom-economical C–H functionalization protocol can be achieved both inter- and intramolecularly. Mechanistic studies by various spectroscopic methods suggest that a reductive quenching pathway is operable.
Direct β-functionalization
of saturated carbonyls has recently become an important goal within
the field of new reaction invention.1 While
chemical methods that induce bond formations at the ipso- and α-positions of C=O moieties have long been established
within organic synthesis,2,3 it is remarkable that
the β-functionalization of esters, ketones, aldehydes, and amides
has been effectively limited to the addition of soft nucleophiles
to α,β-unsaturated systems. Recently, our laboratory introduced
a unique 5πe– carbonyl activation mode utilizing
the synergistic merger of organocatalysis and photoredox
catalysis4 to accomplish the direct β-arylation
of saturated ketones and aldehydes (eq 1).1f This strategy employs two catalytically generated
radical species—a β-enaminyl radical formed via oxidation
and β-deprotonation of an enamine, and a radical anion generated
by photocatalytic reduction of a cyanoarene—that
couple to form β-aryl carbonyl products. Furthermore, we recently
demonstrated the generality of this activation platform via direct
β-aldol reaction of ketones with transiently generated aryl
ketyl radicals to form γ-hydroxyketone adducts.1i Here we translate this generic activation mode
to direct β-alkylation of saturated aldehydes with Michael
acceptors. This formal “homo-Michael” transformation
delivers β-alkyl aldehydes by a combination of photoredox
and amine catalysis (eq 2), further emphasizing
the utility of this novel 5πe– carbonyl activation
mode for a broad range of previously unknown transformations.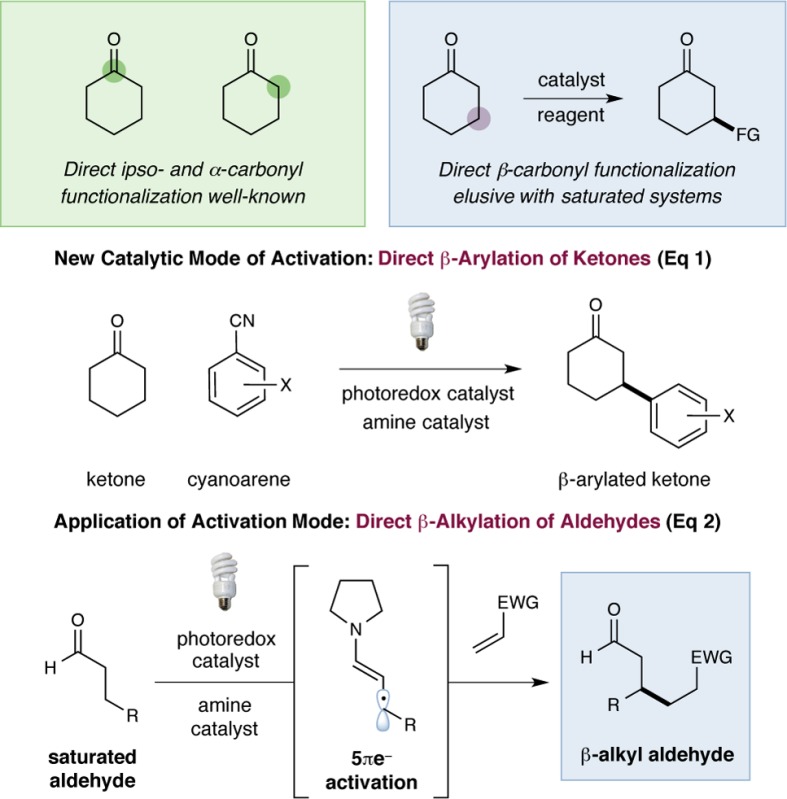
Within the discipline of organic chemistry, the Michael reaction is among the most prevalent and commonly employed strategies to couple electrophilic olefins with enolates or enamines to deliver α-carbonyl alkylated products.7 While 1,4-conjugate addition of α-carbonyl nucleophiles is a well-established transformation,7,8 an analogous “homo-Michael” reaction, in which the β-position of a fully saturated carbonyl species functions as the nucleophile, is essentially unknown.1g Indeed, current methods for installing alkyl groups at the β-position of carbonyls typically require the use of unsaturated carbonyl substrates and stoichiometric organometallic reagents, such as organocuprates.9 Based on the insight gained over the course of our β-arylation program,1f we hypothesized that a transiently generated 5πe– β-enaminyl radical intermediate (formed via an enamine oxidation/deprotonation sequence) could be intercepted by a Michael acceptor, prior to a terminal reduction step.10 Most importantly, this organocatalytic, redox-neutral, and atom-economical approach would provide direct access to a diverse range of β-alkyl aldehydes via a single chemical transformation, requiring no substrate preactivation or use of stoichiometric transition metals.
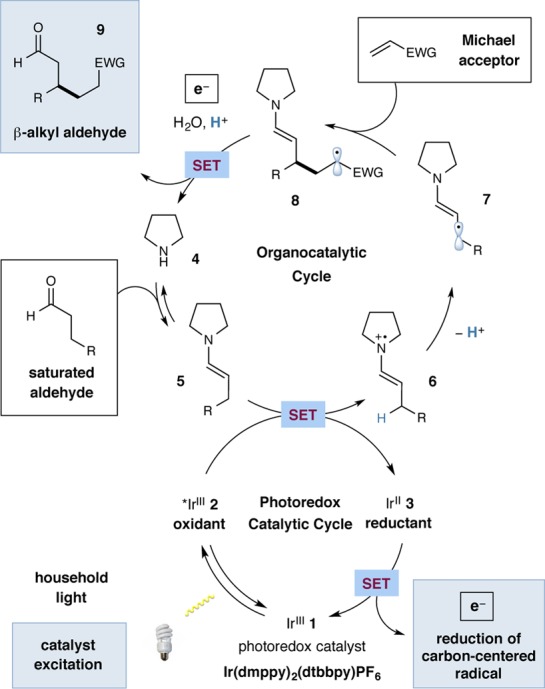
Based on our previous work in organocatalysis and visible light-promoted photoredox catalysis,11 we propose the β-Michael mechanism outlined in Scheme 1. Initial excitation of heteroleptic Ir(III) photocatalyst IrIII(dmppy)2(dtbbpy)PF6 (dmppy = 2-(4-methylphenyl)-4-methylpyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) (1) by visible light produces the photoexcited *IrIII state 2, which can act as both a strong oxidant (E1/2*III/II = +0.55 V vs SCE in MeCN) and reductant (E1/2 = −0.87 V vs SCE) in a single-electron transfer (SET) event with an appropriate substrate quencher.12 Concurrent condensation of a secondary amine catalyst 4 onto an aldehyde forms the enamine intermediate 5. Based on the analysis of standard reduction potentials, we hypothesized that *IrIII2 should readily oxidize the catalytically generated electron-rich enamine 5(13) to form the respective radical cation 6 and the reduced Ir(II) photocatalyst 3. Given the substantial increase in acidity of the β-C–H following enamine oxidation, we presumed that deprotonation of the β-methylene of the radical cation 6 would be facile, forming nucleophilic β-enaminyl radical intermediate 7 (5πe–-activated intermediate).1f This transiently generated 5πe– system could be rapidly intercepted by an electrophilic Michael acceptor, forging the desired C–C bond while producing the α-acyl radical adduct 8. Reducing this 3πe– species 8 (E1/2red = −0.59 to −0.73 V vs SCE)14 with the available IrII species 3 (E1/2 = −1.52 V vs SCE)12 would then return the photocatalyst to its ground state 1, completing the photoredox catalytic cycle. Finally, protonating the enolate along with enamine hydrolysis (thereby completing the organocatalytic cycle by regenerating amine 4) would then deliver the β-alkylated product 9.
Scheme 1. Proposed Mechanism of the β-Alkylation Reaction.
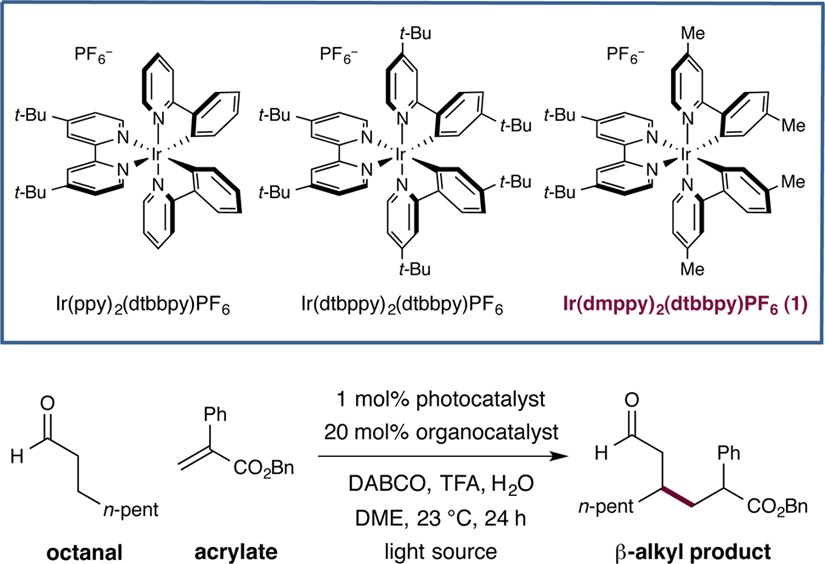
We initiated our examination of the proposed β-alkylation protocol using benzyl 2-phenyl acrylate as the electrophilic coupling partner and octanal as the saturated carbonyl component. To our delight, we observed the desired β-alkylation product (albeit in a modest 7% yield) using Ir(ppy)3 as photocatalyst and diisobutylamine as the amine organocatalyst (Table 1, entry 1). From an early stage we identified that the use of 1,4-diazabicyclo[2.2.2]octane (DABCO) as an organic base and DME as solvent was essential for the desired bond formation to be realized. Early comparisons of photocatalysts revealed noticeable improvements in efficiency when switching to more oxidizing photocatalysts, such as Ru(bpy)3Cl2 and Ir(ppy)2(dtbbpy)PF6 (cf. entries 1–3). Tuning the light source to the maximum absorption wavelength of the photocatalyst (λmax = 450 nm)12 via the use of blue LEDs resulted in further improvements in efficiency (entry 4). At this point, we next examined the influence of the organocatalyst in this β-alkylation protocol. As might be expected, employing a more nucleophilic amine catalyst dramatically diminished reaction yields due to a competing 1,4-heteroconjugate addition with the acrylate electrophile (entry 5), a problem that is commonly confronted in prototypical Michael reactions with organocatalysts.15 In contrast, increasing the steric bulk on the secondary amine catalyst by installing α-branched alkyl groups adjacent to the nitrogen position provided superior efficiency (entries 6 and 7). Indeed, the use of the modified photocatalyst Ir(dmppy)2(dtbbpy)PF6 with dicyclohexylamine was found to be optimal, providing the β-alkylated product in 84% yield (entry 9). Last, control experiments revealed the requirement for base, light, photocatalyst, and organocatalyst in this new β-alkylation protocol (entries 10–13).
Table 1. Initial Studies toward the β-Alkylation Reaction.
| entry | photocatalyst | organocatalyst | light source | yielda (%) |
|---|---|---|---|---|
| 1 | Ir(ppy)3 | i-Bu2NH | 26 W CFL | 7 |
| 2 | Ru(bpy)3Cl2 | i-Bu2NH | 26 W CFL | 50 |
| 3 | Ir(ppy)2(dtbbpy)PF6 | i-Bu2NH | 26 W CFL | 52 |
| 4 | Ir(ppy)2(dtbbpy)PF6 | i-Bu2NH | blue LEDs | 64 |
| 5 | Ir(ppy)2(dtbbpy)PF6 | pyrrolidine | blue LEDs | 6 |
| 6 | Ir(ppy)2(dtbbpy)PF6 | i-Pr2NH | blue LEDs | 77 |
| 7 | Ir(ppy)2(dtbbpy)PF6 | Cy2NH | blue LEDs | 80 |
| 8 | Ir(dtbppy)2(dtbbpy)PF6 | Cy2NH | blue LEDs | 56 |
| 9b | Ir(dmppy)2(dtbbpy)PF6 | Cy2NH | blue LEDs | 84 |
| 10c | Ir(dmppy)2(dtbbpy)PF6 | Cy2NH | blue LEDs | 0 |
| 11 | Ir(dmppy)2(dtbbpy)PF6 | Cy2NH | none | 0 |
| 12 | none | Cy2NH | blue LEDs | 0 |
| 13 | Ir(dmppy)2(dtbbpy)PF6 | none | blue LEDs | 0 |
Yield determined by 1H NMR analysis using methyl benzoate as internal standard. Reactions performed with 2.0 equiv of octanal and 1.0 equiv of DABCO.
Reaction complete after 12 h.
Reaction performed in the absence of DABCO. CFL = compact fluorescent light.
With the optimal β-alkylation conditions in hand, we sought to determine the generality of this direct β-Michael addition. As shown in Table 2, we identified a broad range of electrophilic olefin acceptors as effective alkylation partners for this protocol. Notably, aryl substitution at the α-position of acrylate olefins proved highly effective for both benzyl and methyl ester systems (entries 1 and 2, 83% and 79% yield), presumably due to formation of a benzylic radical in the key C–C bond-forming step (radical 8, Scheme 1). Sterically demanding arenes are readily accommodated on the acrylate coupling partner (entry 3, 77% yield). Electron-rich and electron-deficient arenes on the olefin are also tolerated (entries 4 and 5, 69% and 79% yield), including a series of halogen-substituted phenyl rings (entries 5–7, 69–79% yield). Importantly, unsubstituted acrylates, vinyl sulfones, acryloyl oxazolidinones, and acrylonitriles are also competent electrophiles in this direct β-alkylation reaction (entries 8–12, 50–80% yield). Interestingly, highly electrophilic Michael acceptors such as alkylidene malonates do not participate in this β-coupling reaction. Remarkably, these reaction partners form aldehyde α-alkylation products exclusively, a regiochemical outcome that is not observed for other Michael acceptors shown in Table 2 (e.g., acrylates, vinyl sulfones, and acrylonitriles).16
Table 2. Scope of the Michael Acceptor Coupling Partnera.

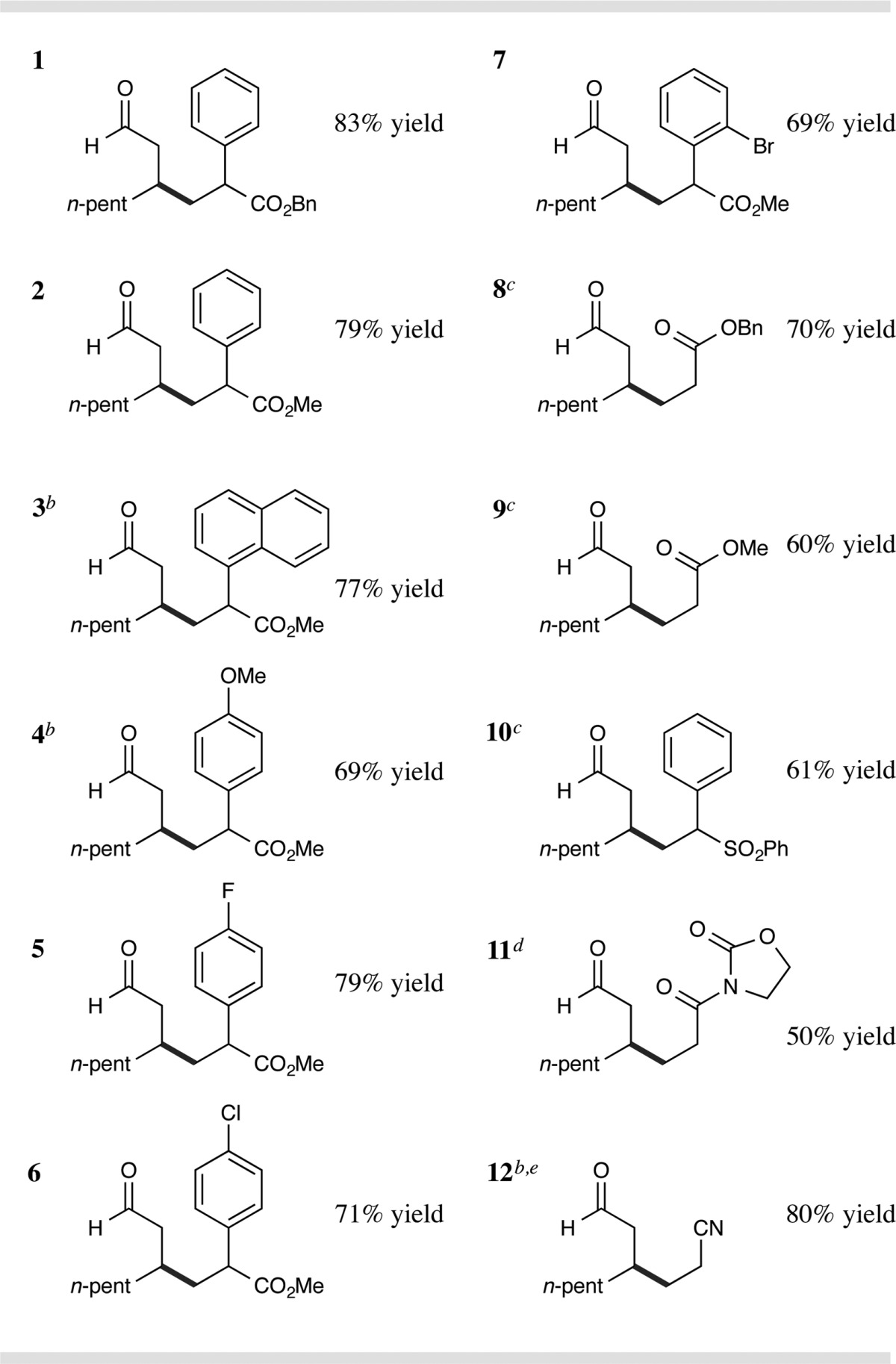
Isolated yields, see SI for experimental details. Diastereomeric ratios (dr) 1–1.3:1, determined by 1H NMR analysis.
Reaction time = 24 h.
5.0 equiv of DABCO and 3.0 equiv of octanal for 30 h.
5.0 equiv of DABCO, 5.0 equiv of octanal, 40 mol% Cy2NH, and HOAc instead of TFA.
3.0 equiv of octanal.
We next focused our attention on the scope of the aldehydic coupling partner, as exemplified in Table 3. Aliphatic aldehydes function broadly, regardless of the inherent steric bulk positioned around the reactive β-C site (entries 1 and 2, 79% and 72% yield). Importantly, a variety of functional groups are tolerated on the alkanal system, including ethers, esters, alkynes, and alkenes (entries 3–6, 66–83% yield). Perhaps most notably, quaternary C centers can be formed in a facile manner utilizing this new transformation, with rapid alkylation of β-sites that are found within cyclic (tetrahydropyran and piperidine) and acyclic (gem-dimethyl) systems (entries 7–10, 72–78% yield). β-Amino aldehydes are competent substrates for formation of stereogenic amines with good levels of reaction efficiency (entry 11, 66% yield). Intriguingly, propionaldehyde undergoes β-alkylation at the terminal methyl site using these photoredox conditions (entry 12, 59% yield), indicating that primary β-enaminyl radicals can be generated in this protocol.
Table 3. Scope of the Aldehyde in the β-Alkylation Reactiona.

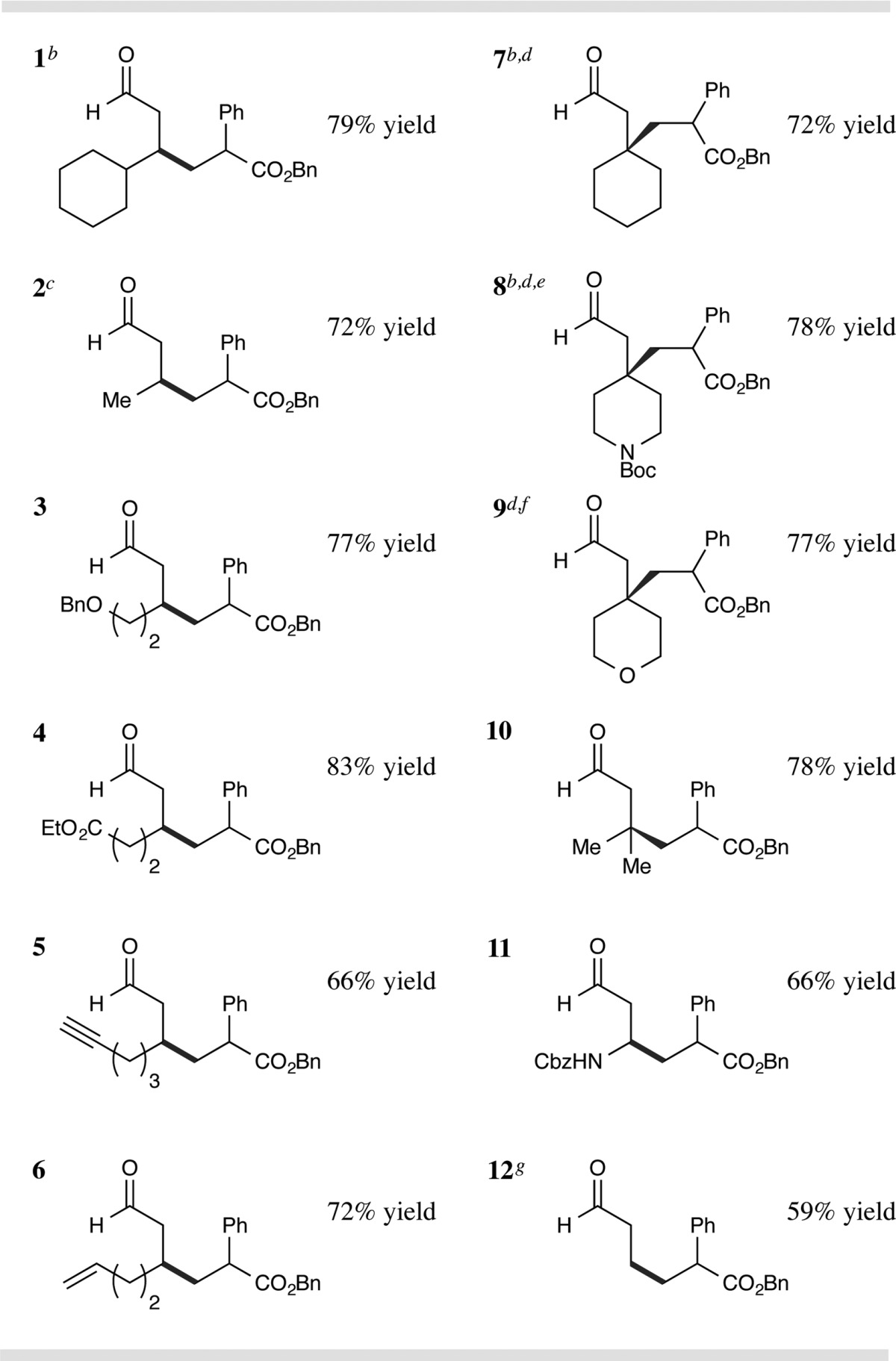
Isolated yields, see SI for experimental details. Diastereomeric ratios 1–2:1, determined by 1H NMR analysis.
Reaction time = 24 h.
5.0 equiv of butanal.
3.0 equiv of aldehyde.
HOAc used instead of TFA.
Reaction time = 36 h.
10 equiv of propionaldehyde.
A series of Stern–Volmer fluorescence quenching experiments were performed in an effort to provide evidence for the mechanistic proposal outlined in Scheme 1. Indeed, we have determined that the emission intensity of *IrIII(dmppy)2(dtbbpy)PF6 is dramatically diminished in the presence of the operating enamine (formed in situ from dicyclohexylamine and octanal), thereby indicating that enamine oxidation is likely the first step in the photoredox cycle.17 Comparatively, there is no fluorescence quenching when the amine catalyst, aldehyde donor, or benzyl 2-phenyl acrylate acceptor is exposed separately to the photoexcited *IrIII species. In addition, electron paramagnetic resonance (EPR) spectroscopy has revealed the existence of an organic radical (giso = 1.9858) following excitation of the photocatalyst in the presence of enamine; this signal is absent if either aldehyde or amine is removed.17 It is important to consider that an alternative catalysis mechanism might involve single-electron reduction of the Michael acceptor prior to coupling with the β-enaminyl radical (a radical–radical combination that would be consistent with our previous β-arylation and β-aldol studies). However, this pathway would depend on a facile reduction of benzyl 2-phenyl acrylate (E1/2red = −1.97 V vs SCE),12 which is thermodynamically unfavorable for either the *IrIII or IrII oxidation state of photocatalyst 1. Indeed, EPR studies indicate that no organic radical is generated with benzyl 2-phenyl acrylate in the presence of either photocatalyst 1 or Ir(ppy)3—a strongly reducing *IrIII complex (E1/2 = −1.73 V vs SCE)18—further indicating that acrylate reduction is not likely involved in the catalytic cycle.17 As such, we conclude that addition of a 5πe– β-enaminyl radical (such as 7) to the ground state of the Michael acceptor coupling partner (as shown in Scheme 1) is the operating C–C bond-forming step.19
Last, to further explore the utility of this new β-alkylation reaction, we have investigated intramolecular variants as a mechanism to rapidly access ring systems of various formats. As shown in Scheme 2, both 6-exo and 5-exo cyclizations are accomplished with useful efficiencies and diastereocontrol (47–54% yield, 4–9:1 dr). This provides further evidence that the critical key step does not involve radical–radical coupling, given the low probability of generating two radicals simultaneously on the same molecule.
Scheme 2. Intramolecular β-Alkylation Cyclization Reaction.
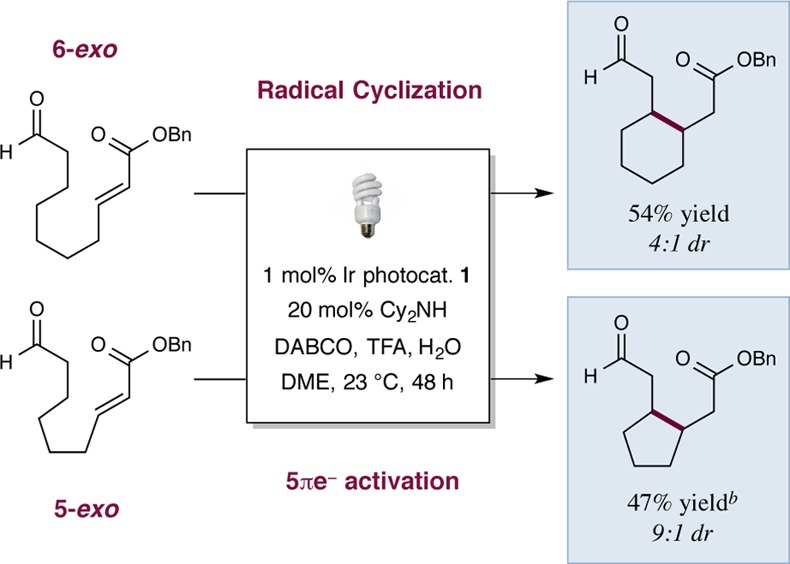
Isolated yields, see SI for experimental details. Diastereomeric ratios determined by 1H NMR analysis.
40 mol% isopropylbenzylamine as organocatalyst for 72 h.
In summary, through the synergistic combination of organocatalysis and photoredox catalysis, we have accomplished the first direct β-alkylation of fully saturated aldehydes with Michael acceptors. We have further demonstrated the utility of a 5πe– β-enaminyl activation platform as a general approach to direct β-functionalization of carbonyls. Importantly, this C–H bond activation method is entirely redox-neutral and atom-economical, and it requires no preactivation of either coupling partner. Mechanistic studies have provided spectroscopic evidence supporting a reductive quenching pathway, in which C–C bond formation occurs by β-enaminyl radical addition into a ground-state Michael acceptor. Efforts toward expanding the scope of the carbonyl coupling partner as well as developing an asymmetric variant are currently underway and will be reported in due course.
Acknowledgments
Financial support was provided by NIHGMS (R01 01 GM093213-01) and gifts from Merck and Amgen.
Supporting Information Available
Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Wasa M.; Engle K. M.; Yu J.-Q. J. Am. Chem. Soc. 2009, 131, 9886. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Renaudat A.; Jean-Gérard L.; Jazzar R.; Kefalidis C. E.; Clot E.; Baudoin O. Angew. Chem., Int. Ed. 2010, 49, 7261. [DOI] [PubMed] [Google Scholar]; c Zhang S.-L.; Xie H.-X.; Zhu J.; Li H.; Zhang X.-S.; Li J.; Wang W. Nat. Commun. 2011, 2, 211. [DOI] [PubMed] [Google Scholar]; d Hayashi Y.; Itoh T.; Ishikawa H. Angew. Chem., Int. Ed. 2011, 50, 3920. [DOI] [PubMed] [Google Scholar]; e Stowers K. J.; Kubota A.; Sanford M. S. Chem. Sci. 2012, 3, 3192. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Pirnot M. T.; Rankic D. A.; Martin D. B. C.; MacMillan D. W. C. Science 2013, 339, 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Fu Z.; Xu J.; Zhu T.; Leong W. W. Y.; Chi Y. R. Nat. Chem. 2013, 5, 835. [DOI] [PubMed] [Google Scholar]; h Mo J.; Shen L.; Chi Y. R. Angew. Chem., Int. Ed. 2013, 52, 8588. [DOI] [PubMed] [Google Scholar]; i Petronijević F. R.; Nappi M.; MacMillan D. W. C. J. Am. Chem. Soc. 2013, 135, 18323. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Huang Z.; Dong G. J. Am. Chem. Soc. 2013, 135, 17747. [DOI] [PubMed] [Google Scholar]
- Carey F. A.; Sundberg R. J.. Advanced Organic Chemistry: Part B: Reactions and Synthesis; Springer: New York, 2001. [Google Scholar]
- a Mukherjee S.; Yang J. W.; Hoffmann S.; List B. Chem. Rev. 2007, 107, 5471. [DOI] [PubMed] [Google Scholar]; b Allen A. E.; MacMillan D. W. C. Chem. Sci. 2012, 3, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Evans D. A.; Helmchen G.; Rüping M.. Asymmetric Synthesis—The Essentials; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- For reviews on photoredox catalysis, see:; a Zeitler K. Angew. Chem., Int. Ed. 2009, 48, 9785. [DOI] [PubMed] [Google Scholar]; b Yoon T. P.; Ischay M. A.; Du J. Nat. Chem. 2010, 2, 527. [DOI] [PubMed] [Google Scholar]; c Inagaki A.; Akita M. Coord. Chem. Rev. 2010, 254, 1220. [Google Scholar]; d Teply F. Collect. Czech. Chem. Commun. 2011, 76, 859. [Google Scholar]; e Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102. [DOI] [PubMed] [Google Scholar]; f Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Schultz D. M.; Yoon T. P. Science 2014, 343, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Krause N.; Hoffmann-Röder A. Synthesis 2001, 171. [Google Scholar]; b Tsogoeva S. B. Eur. J. Org. Chem. 2007, 11, 1701. [Google Scholar]
- Bressy C.; Dalko P. I.. Enantioselective Organocatalysis: Reactions and Experimental Procedures; Wiley-VCH: Weinheim, 2007. [Google Scholar]
- a Rossiter B. E.; Swingle N. E. Chem. Rev. 1992, 92, 771. [Google Scholar]; b Sibi M. P.; Manyem S. Tetrahedron 2000, 56, 8033. [Google Scholar]
- For examples of photoredox-catalyzed reductive quenching mechanisms involving radical additions into Michael acceptors, see:; a Miyake Y.; Ashida Y.; Nakajima K.; Nishibayashi Y. Chem. Commun. 2012, 48, 6966. [DOI] [PubMed] [Google Scholar]; b Miyake Y.; Nakajima K.; Nishibayashi Y. J. Am. Chem. Soc. 2012, 134, 3338. [DOI] [PubMed] [Google Scholar]; c Kohls P.; Jadhav D.; Pandey G.; Reiser O. Org. Lett. 2012, 14, 672. [DOI] [PubMed] [Google Scholar]
- a Nicewicz D. A.; MacMillan D. W. C. Science 2008, 322, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nagib D. A.; Scott M. E.; MacMillan D. W. C. J. Am. Chem. Soc. 2009, 131, 10875. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shih H.-W.; Vander Wal M. N.; Grange R. L.; MacMillan D. W. C. J. Am. Chem. Soc. 2010, 132, 13600. [DOI] [PMC free article] [PubMed] [Google Scholar]; d McNally A.; Prier C. K.; MacMillan D. W. C. Science 2011, 334, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Nagib D. A.; MacMillan D. W. C. Nature 2011, 480, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See SI for cyclic voltammetry and UV/vis data.
- a Schoeller W. W.; Niemann J. J. Chem. Soc., Perkin Trans. 2 1988, 369. [Google Scholar]; b Renaud V. P.; Schubert S. Angew. Chem. 1990, 102, 416. [Google Scholar]
- Bortolamei N.; Isse A. A.; Gennaro A. Electrochim. Acta 2010, 55, 8312. [Google Scholar]
- Kano T.; Shirozu F.; Akakura M.; Maruoka K. J. Am. Chem. Soc. 2012, 134, 16068. [DOI] [PubMed] [Google Scholar]
- At this stage we have found Michael acceptors bearing β-substitution to be less competent in the described intermolecular β-alkylation protocol.
- See SI for fluorescence quenching and EPR data.
- Flamigni L.; Barbieri A.; Sabatini C.; Ventura B.; Barigelletti F. Top. Curr. Chem. 2007, 281, 143. [Google Scholar]
- When coupling partners that undergo facile single-electron reduction, such as vinyl ketones, are employed, no desired β-alkylated product is formed, and only dimerization of the coupling partner is observed. This suggests against a radical–radical coupling event in the key C–C bond-forming step of this protocol.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.