

Abstract
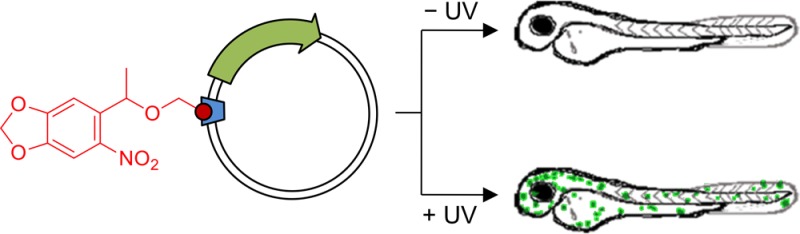
In cell and molecular biology, double-stranded circular DNA constructs, known as plasmids, are extensively used to express a gene of interest. These gene expression systems rely on a specific promoter region to drive the transcription of genes either constitutively (i.e., in a continually “ON” state) or conditionally (i.e., in response to a specific transcription initiator). However, controlling plasmid-based expression with high spatial and temporal resolution in cellular environments and in multicellular organisms remains challenging. To overcome this limitation, we have site-specifically installed nucleobase-caging groups within a plasmid promoter region to enable optochemical control of transcription and, thus, gene expression, via photolysis of the caging groups. Through the light-responsive modification of plasmid-based gene expression systems, we have demonstrated optochemical activation of an exogenous fluorescent reporter gene in both tissue culture and a live animal model, as well as light-induced overexpression of an endogenous signaling protein.
Introduction
Optical control of biological processes conveys unprecedented spatial and temporal control over the activation and deactivation of cellular events, enabling complex investigations that cannot be conducted with other conditional control elements.1−10 Examples of cellular processes that have been engineered to respond to light include ion-channel conductivity,11,12 RNA polymerization,13−17 kinase-catalyzed protein phosphorylation,18−21 and protein translocation.22−24 For transcriptional activation in eukaryotic systems, a gene expression plasmid contains a promoter sequence upstream of the gene of interest, such as the commonly used cytomegalovirus (CMV) promoter,25 which further includes a specific transcription initiator sequence called the “TATA box”.26 Transcription is initiated when a subunit of transcription factor IID (TFIID), referred to as the TATA box binding protein (TBP), binds to the TATA box sequence and recruits additional components of the transcriptional machinery such as RNA polymerase II.27 Since the binding of TBP to the TATA box is the driving force to activate transcription, we hypothesized that this would be an optimal site to optochemically control the transcription of a plasmid by applying nucleobase “caging” technologies.28 Caging involves the synthetic installation of a light-removable protecting group, e.g., an o-nitrobenzyl group, onto a molecule of interest in a way that renders that molecule biologically inactive. Exposure of the molecule to light, e.g., nontoxic UV-A light,29−31 removes the caging groups, and restores the biological activity of the molecule. Since light can be controlled with high spatiotemporal resolution, decaging and biological function can be precisely regulated as well. The optochemical control of oligonucleotide function through installation of nucelobase-caging groups32−38 and photocleavable backbone linkers39−47 has found widespread biological application. In order to develop a generally applicable system for the optical activation of transcription in vivo, we inserted modified short oligomer fragments into plasmids in a site-specific manner48 and introduced caged nucleobases at 1–3 defined sites into the TATA-box promoter region of large (>4 kB) gene expression plasmids. The newly developed caged plasmid system was successfully applied to in vivo optochemical control of gene expression in mammalian cells and in zebrafish.
Results and Discussion
The optochemical regulation of an enhanced green fluorescent protein (EGFP) reporter gene was selected as a proof-of-concept model for the application of a caged promoter region. An EGFP plasmid was modified to contain two nicking sites in the CMV promoter flanking the TATA box, in order to remove a short DNA fragment and replace it with a caged DNA insert (Figure 1). We hypothesized that site-specific installation of caged nucleobases within the TATA box will inhibit initiation of EGFP transcription by TBP. Upon UV irradiation, the caging groups would be cleaved and transcription would be activated. To test this hypothesis, 1–3 NPOM-caged thymidine residues49 were site-specifically incorporated into oligonucleotides containing the TATA box sequence via automated solid-phase DNA synthesis (Supporting Table 1). A mutant promoter region that contains three T → C base substitutions (TATATAA → CACACAA) was designed as a negative control, based on our ability to replace the same thymidine residues with caged Ts and based on a previous analysis that showed less than 4% transcriptional efficiency with two or more mutations in the TATA box region.50 This Tmut negative control contains thymidine mutations at the same sites in the TATA box as the designed NPOM-caged promoter constructs. The installation of the caged thymidine nucleotides was predicted to disrupt hydrogen-bonding interactions between base-pairs within the TATA box region; however, nucleotides outside of this region will still be able to hybridize to their complement sequences as needed for ligation of the inserts into the plasmid.48 To ensure that the caged DNA does not completely inhibit hybridization with its reverse complement sequence, melting temperatures (Tm) were determined (Supporting Table 2). With each addition of a caged nucleotide, the Tm slightly decreased, which is reflective of the caging groups interfering with the hydrogen bonding interactions of the nucleobases.51,52 UV irradiation completely restored binding of the caged TATA box sequences to their complement sequences. Importantly, hybridization for all caged primers was still detected at the ligation temperature of 4 °C, a prerequisite for construction of the promoter-caged plasmid.
Figure 1.
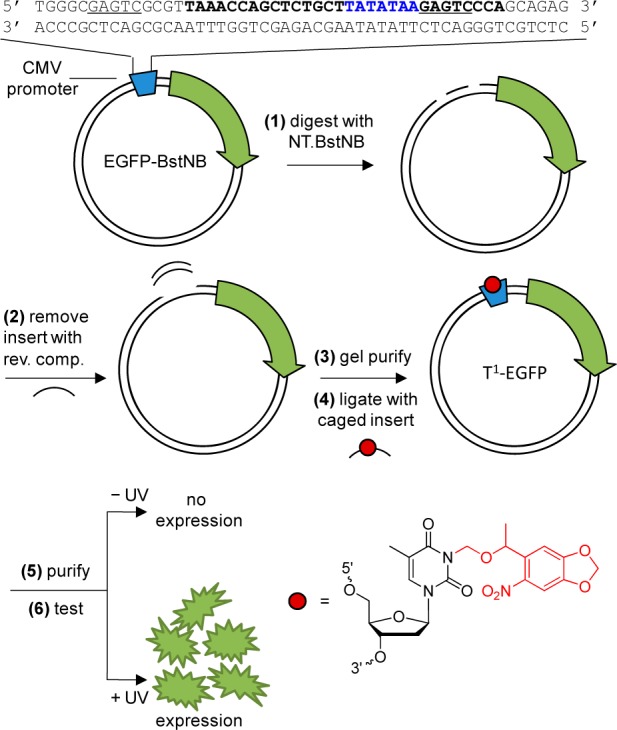
Construction of the site-specifically caged plasmids. (1) pEGFP-Bst is digested with Nt.BstNB and (2) annealed with the reverse complement to remove the TATA box region. (3) The digested plasmid is gel purified and (4) ligated with a phosphorylated caged TATA box insert. (5) The caged plasmid is then column-purified and (6) applied in mammalian cells or live animal models that were either kept in the dark (no expression of EGFP) or irradiated with UV light (EGFP expression). The restriction sites are underlined and the TATA box recognition sequence is shown in blue. The NPOM-caged thymidine is represented by a red circle and the NPOM modification is indicated in red within the nucleotide structure.
The assembled caged plasmids were verified by agarose gel electrophoresis (Supporting Figure 1) and were subsequently assessed for function in mammalian cell culture. A cell viability assay (CellTiter-Glo, Bio-Rad) was performed to analyze the effects of cellular UV-A application with increasing intervals of 365 nm irradiations, demonstrating that UV-A light does not reduce cell viability for exposures of up to 20 min in our irradiation setup (Supporting Figure 2). The caged EGFP plasmids were then cotransfected with a DsRed expression vector as a control, and the transfected HEK293T cells were either irradiated for 5 min (365 nm, 25 W) or kept in the dark, followed by incubation for 48 h. EGFP and DsRed expression were imaged (Supporting Figure 3) and quantified by flow cytometry (Figure 2A and Supporting Figure 4). As expected, Tmut achieved the greatest reduction in EGFP expression from a series of mutants analyzed (data not shown), and the thymidine caging groups were capable of inhibiting EGFP transcription to low basal levels similar to those of the mutated nonfunctional TATA box sequence. With each additional caging group a slight reduction in the EGFP background expression was observed. After UV irradiation, the caged plasmids regained full functionality, showing expression levels virtually identical to the noncaged EGFP expression plasmid (T0), which validates that the incorporation of NPOM-caged thymidine nucleotides within the TATA box sequence can be applied to the optochemical regulation of a plasmid expression vector. The high levels of EGFP expression observed for the T1–T3-caged plasmids after UV irradiation suggest that optical control for each of the constructs results in similar levels of gene activation. The T3-caged plasmid was then applied to all subsequent experiments, since it showed the lowest background EGFP expression before light exposure. One of the main advantages of using nucleobase caging technology to photoregulate gene expression is the ability to perform localized and temporal control over biological activity. To this end, HEK293T cells were cotransfected with the T3-caged EGFP plasmid and the control DsRed expression vector. Following transfection, only a small subset of cells was irradiated with UV light, followed by imaging after a 48 h incubation. As shown in Figure 2B, EGFP expression was localized to the irradiated area, while DsRed expression was observed in all cells. This demonstrates that the developed TATA box-caging methodology can be applied to optochemically regulate gene expression with spatiotemporal control.
Figure 2.
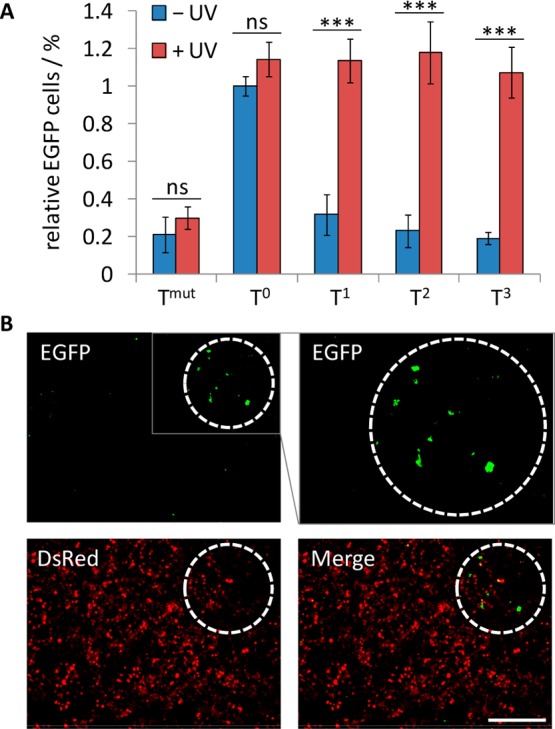
(A) Quantification of light-activated EGFP expression. HEK293T cells were transfected with noncaged and caged EGFP plasmids and a DsRed control plasmid. The cells were irradiated for 5 min (365 nm, 25 W) or kept in the dark. After 48 h incubation, the cells were analyzed by flow cytometry. The number of cells expressing both EGFP and DsRed was normalized to the number of cells expressing only DsRed and set relative to the noncaged plasmid. Standard deviations were calculated form three individual experiments. ns = not significant (P > 0.05), *** = highly significant (P < 0.001). (B) Spatial activation of EGFP expression. HEK293T cells were transfected with T3-caged EGFP and DsRed plasmids. Cells within the white dashed circle were irradiated through a microscope filter cube (DAPI, BP377/28, 40×) for 30 s and were imaged (5× magnification) after 48 h incubation. An enlarged region of the EGFP channel is shown in the gray box. Scale bar indicates 200 μm.
In order to demonstrate the general applicability of the developed methodology, the optochemical overexpression of an endogenous gene was investigated. Pololike kinase 3 (Plk3) is a serine/threonine kinase that is essential for cells entering into mitosis, spindle formation, segregation of the chromosomes, and cytokinesis.53 Plk3 is also a tumor suppressor that when overexpressed can induce cell cycle arrest, chromatin condensation, and apoptosis.54 Since the ectopic expression of Plk3 leads to disruption of microtubule integrity, a change in cell morphology occurs, namely cytokinesis defects and formation of binucleated and polynucleated cells.55 Due to its involvement in the cell cycle and the phenotypic change when Plk3 is overexpressed, the optical activation of Plk3 expression was investigated through the engineered caged plasmids. To this end, Plk3 was fused to the C-terminus of the EGFP expression vector based on a previously reported plasmid.55 The EGFP-Plk3 plasmid was then modified with the caged TATA box DNA sequences as previous stated (see Figure 1). HeLa cells were transfected with the modified EGFP-Plk3 plasmids, and the cells were either irradiated for 5 min (365 nm, 25 W) or kept in the dark, followed by incubation for 48 h. This cell line was used to analyze previously reported phenotypic changes55 and to improve single cell imaging capabilities, since HeLa cells have distinct morphology and form cellular monolayers, in contrast to the previously used HEK293T cell line. The cells were fixed and stained to identify actin filaments as well as nuclei, in addition to EGFP expression. As expected, the positive control T0-noncaged EGFP-Plk3 plasmid showed transcription of both genes and a significant change in phenotype, specifically the formation of binucleated cells and a loss in cellular structure, while the negative control Tmut showed normal cellular morphology (Supporting Figure 5). The T3-caged plasmid showed low EGFP expression as well as little morphological change when the cells were kept in the dark. After UV irradiation, the caging groups were removed and transcription of EGFP-Plk3 was activated, as shown by the increase in EGFP expression and the observation of binucleated cells (Figure 3A). Additionally, the overexpression of Plk3 can lead to apoptosis by activating caspase-3.55−57 Thus, the downstream activation of caspase-3 activity was measured in response to the optochemically driven overexpression of Plk3. HeLa cells were transfected with the noncaged or caged EGFP-Plk3 plasmids and either kept in the dark or irradiated (365 nm, 5 min) and caspase-3 activity was measured after 48 h (Ac-DEVD-AFC substrate, Calbiochem). The T0-noncaged TATA box demonstrated a 5-fold increase in caspase-3 activity over nontreated cells. In contrast, the Tmut plasmid only led to basal levels of caspase-3 activity, confirming it as a negative control for Plk3-driven caspase-3 activation (Figure 3B). In the absence of UV light, the T3-caged plasmid was inactive as only basal caspase-3 activity similar to nontreated cell was observed, as expected. However, after light-induced activation an increase in caspase-3 activity was detected, indicative of Plk3-driven downstream pathway regulation. These results demonstrate that the overexpression of an endogenous gene can be optochemically activated through the site-specific incorporation of NPOM-caged thymidine nucleotides within the TATA box transcription regulatory region. Here, this was applied to the induction of a phenotypic change and activation of a downstream signaling pathway; however, broad applicability of the developed methodology to the temporal activation of gene function is conceivable.
Figure 3.
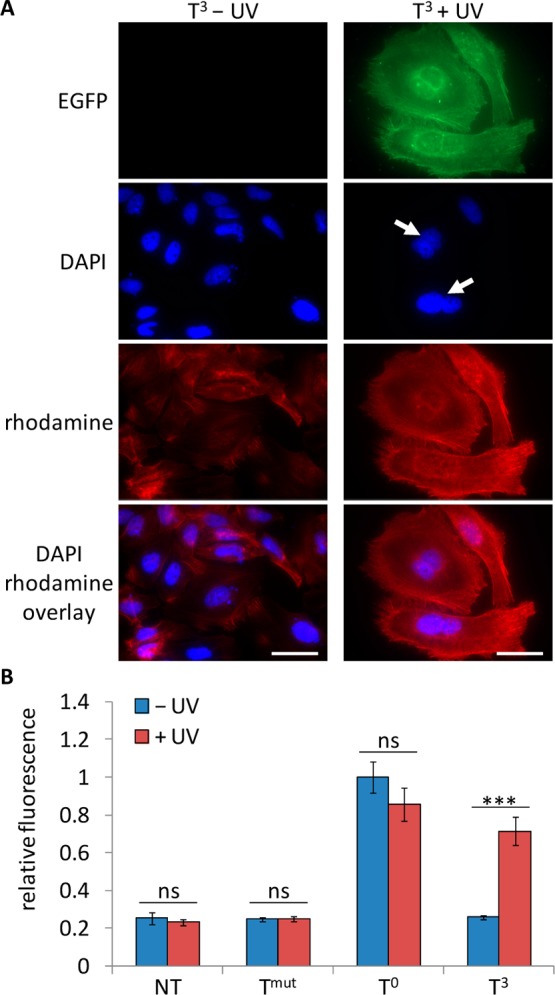
(A) Light-induced expression of Plk3. HeLa cells were transfected with the T3-caged EGFP-Plk3 plasmid followed by irradiation of the caged construct (365 nm, 5 min, 25 W) and incubated for 48 h. The cells were then fixed and stained with DAPI (nuclei) and rhodamine phalloidin (actin filaments) prior to imaging (63× magnification). White arrows indicate binucleated cells, and scale bars indicate 50 μm. (B) Light-induced activation of caspase-3. HeLa cells were transfected with the Tmut negative control, T0-noncaged, and T3-caged EGFP-Plk3 plasmids. The cells were either irradiated (365 nm, 5 min, 25 W) or kept in the dark and lysed after 48 h. The lysate was assayed with a fluorogenic caspase-3 substrate (Calbiochem). Fluorescence units were normalized to the noncaged control, and standard deviations were calculated from three individual experiments. ns = not significant (P > 0.05), *** = highly significant (P < 0.001).
In order to demonstrate the applicability of the caged TATA box construct to the optochemical control of gene function in an animal, the expression of a fluorescent reporter was tested in zebrafish embryos. The zebrafish was selected because it is a common model organism for developmental studies, its transparency facilitates irradiation, and plasmid-driven gene expression has been well documented via direct microinjection into fertilized eggs.58−60 Microinjections with modified EGFP plasmids were performed at the 1-cell stage, and embryos were either irradiated (365 nm, 2 min) or kept in the dark. After 24 h incubation the embryos were dechorionated and imaged for EGFP expression directly and 24 h later. The T0-noncaged plasmid was used as a positive control, exhibiting mosaic EGFP expression patterns commonly observed with DNA injection60 due to differential plasmid content in each cell (Supporting Figure 6). When the TATA box region of the vector was caged, transcription was deactivated and only a minimal level of EGFP expression was observed. However, after UV exposure, the embryos injected with the T3-caged plasmid showed activation of EGFP expression, conferring the optical control observed in a single cell environment to gene expression in a multicellular animal (Figure 4A; see Supporting Figure 7 for additional images). More than 45% of live embryos injected with the caged plasmid construct expressed EGFP after UV irradiation, with levels of transcriptional activation similar to the noncaged control (Figure 4B). Although a small population of embryos exhibited low levels of EGFP expression in the absence of UV exposure, a 5-fold increase in EGFP expressing embryos was observed. Additionally, a UV irradiation time course was performed, indicating that longer exposures do not significantly enhance the frequency of EGFP expressing embryos (Supporting Figure 8A). Late-stage irradiations at 8 hpf (75% epiboly stage) were also performed to examine caged plasmid activation during gastrulation, and EGFP expression was observed at 24 hpf (Supporting Figure 8B). Although the total number of EGFP positive embryos was slightly lower compared to irradiations at earlier stages, the presence of similar mosaic expression patterns as observed in the case of 1 hpf irradiation shows that caged plasmids can provide a means to activate gene expression through UV irradiation later in development (Supporting Figure 8C). These results demonstrate that the caged promoter sequence allows for the construction of plasmid-based optochemical gene expression that can be readily applied to live aquatic embryos for the regulation of gene function.
Figure 4.
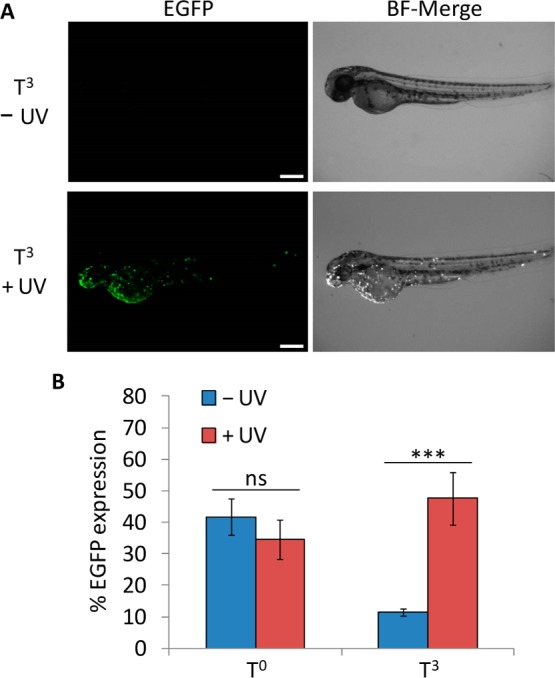
(A) Light-induced EGFP expression in zebrafish. Embryos were microinjected at the 1-cell stage with the T3-caged EGFP expression plasmid or the noncaged T0 plasmid. Embryos were then irradiated (2 min, 365 nm) or kept in the dark and incubated at 28 °C for 24 hpf, followed by dechorionation. Imaging was performed at 48 hpf. Scale bars indicate 250 μm. (B) Frequency of the EGFP phenotype for each condition. Error bars represent standard deviations from three (T0) or four (T3) independent experiments. N = 9–24. ns = not significant (P > 0.05), *** = highly significant (P < 0.001).
Summary
In summary, we have engineered a system in which plasmid function can be optochemically regulated with high spatial and temporal resolution. A site-specifically caged promoter region was inserted into expression plasmids via ligation with synthetic nucleobase-caged DNA strands. By installing NPOM-caged thymidine nucleotides within the TATA box promoter region, transcription was inhibited and activity was not observed until the caging groups were removed through a brief exposure to UV light. The optical OFF → ON switching of plasmid function was assessed using a fluorescent reporter gene in live cells, and spatial control of activation was demonstrated for TATA box-driven gene expression in human tissue culture. Additionally, we were able to show that the engineered system could be used to regulate cellular signaling cascades by optochemically triggering overexpression of an endogenous gene, polo-like kinase 3 (Plk3). The effect of Plk3 overexpression was observed by a phenotype change leading to binucleation only in irradiated cells and through upregulation of caspase-3 activity after light-induced Plk3 activation. Lastly, we were able to apply the caged vector methodology to the optochemical triggering of gene expression in live animals. Specifically, light-activated gene expression was achieved in the zebrafish embryo, a multicellular model organism that is extensively used for genetic studies. In contrast to caging of the oligonucleotide phosphate backbone61−63 our approach is completely site-specific, generally applicable, and does not require auxiliary proteins, as only 1–3 NPOM-caging groups are synthetically incorporated onto nucleobases in the TATA-box region of the expression plasmids. Thus, only a few photolysis reactions are required to optically activate gene expression from an otherwise inactive expression vector. This method adds a new and precise synthetic biology tool to the light-regulation of gene function in cells and organism and has broad applicability in the regulation of plasmid-encoded protein expression, as demonstrated in mammalian cell culture and zebrafish embryos.
Methods
Construction of the Caged Plasmids
The Nt.BstNB restriction sites were cloned into the pEGFP-N1 19 bases upstream and immediately downstream of the TATA box through PCR amplification: Forward Primer: 5′ TATATAAGACCGAGTCCCGTCGTCAGATCCGC. Reverse Primer: 5′ AGCAGAGCTGGTTTAACGCGACTCGCCCAACCGC. The created plasmid, pEGFP-BstNB (40 μg), was digested with Nt.BstNB (New England Biolabs, Buffer 3.1) at 55 °C for 2 h (500 μL). The enzyme was heat inactivated at 80 °C for 20 min. The reverse complement to the 34 bp DNA fragment was added to the digestion reaction (25 μL of a 100 μM solution) and then annealed with 80 °C for 5 min and slowly cooled to rt. The digested plasmid was gel purified (0.8% agarose gel) with the E.Z.N.A. gel extraction kit (Omega) and eluted in 50 μL of water. The noncaged and caged TATA box sequences (see Supporting Table 1) were 5′ phosphorylated with T4 polynucleotide kinase (New England Biolabs) at 50 μM (50 μL) and were ligated (10 μL insert, 60 μL reaction) into the purified plasmid using Quick ligase (New England Biolabs). The ligated product was column purified with E.Z.N.A. plasmid purification kit (Omega) and quantified with a Nanodrop spectrometer.
DNA Synthesis Protocol
DNA synthesis was performed using an Applied Biosystems (Foster City, CA) Model 394 automated DNA/RNA Synthesizer using standard β-cyanoethyl phosphoramidite chemistry. The caged DNA oligonucleotides were synthesized using 40 nmol scale solid-phase supports obtained from Glen Research. Reagents for automated DNA synthesis were also obtained from Glen Research. Specialized NPOM-caged thymidine phosphoramidite was synthesized as previously described and dissolved in anhydrous acetonitrile to a final concentration of 0.05 M. Standard synthesis cycles provided by Applied Biosystems were used for all normal bases using 2 min coupling times. The coupling time was increased to 10 min for the positions at which the caged thymidine phosphoramidites were incorporated. Each synthesis cycle was monitored by following the release of dimethoxytrityl (DMT) cations after each deprotection step. No significant loss of DMT was noted following the addition of the caged-T to the DNA, thus 10 min was sufficient to allow maximal coupling of the caged thymidine. Oligonucleotides were eluted from the solid-phase supports with 1 mL ammonium hydroxide methylamine (AMA, 1:1) and deprotected at 65 °C for 2 h. The full-length caged oligonucleotides were purified with Nap-10 columns (GE Healthcare).
Melting Temperatures
The melting temperature (Tm) of each TATA box duplex was measured using a CFX96 Touch Real Time PCR Detection System (Bio-Rad). TATA box DNA duplexes (20 μL, 1 μM) were incubated in TAE/Mg2+ buffer (0.04 M tris-acetate, 1 mM EDTA, and 12.5 mM magnesium acetate) and annealed over a temperature gradient from 95 to 4 °C over 10 min. The samples were then heated in the presence of SYBR green (1 μL of 20× SsoFast EvaGreen Supermix, Bio-Rad) from 0 to 100 °C, at a rate of 0.5 °C/min, with a dwell time of 10 s and the fluorescence measured every 0.5 °C. The Tm was determined by the maximum of the first derivative of the fluorescence vs temperature plot. Standard deviations were calculated from three individual experiments.
Analysis of Cell Viability with UV-A Exposure
Human embryonic kidney (HEK) 293T cells were grown at 37 °C, 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone), supplemented with 10% fetal bovine serum (Hyclone) and 10% streptomycin/penicillin (MP Biomedicals). Cells were passaged into a 96-well plate (200 μL per well, ∼1 × 104 cells per well) and grown to ∼70% confluence within 24 h. Cells were then irradiated for 0–20 min (365 nm, 25 W), followed by incubation for 24 h at 37 °C, 5% CO2. After the overnight incubation, 150 μL of the cellular media was removed and 50 μL CellTiter-Glo (Bio-Rad) reagent was added. Chemiluminescence was measured on a BioTek Synergy 4 plate reader at 10 min. Standard deviations were calculated from three individual experiments.
Light Activation of EGFP Expression
HEK293T cells were passaged into 96-well plates (200 μL per well, ∼1 × 104 cells per well) and grown to ∼70% confluence within 24 h. The media was replaced with DMEM without antibiotics, and the cells were transfected with pEGFP-BstNB (150 ng/well) and pDsRed-N1 (300 ng/well) plasmids using branched polyethylene imine (bPEI, 0.5 μL/well) in an overnight experiment. The following morning, the cells were either irradiated for 5 min (365 nm, 25 W) or kept in the dark, followed by incubation for 48 h at 37 °C, 5% CO2. The cells were imaged on a Zeiss Observer Z1 microscope (5× magnification objective, filter sets 43 HE DsRed and 38 HE EGFP).
Flow Cytometry Analysis
HEK293T cells were passaged into 24-well plates (1 mL per well, ∼4 × 104 cells per well) and grown to ∼70% confluence within 24 h. The media was replaced with DMEM without antibiotics, and the cells were transfected with pEGFP-BstNB (750 ng/well) and pDsRed-N1 (1500 ng/well) plasmids using branched polyethylene imine (bPEI, 0.5 μL/well) in an overnight experiment. The following morning, the cells were either irradiated for 5 min (365 nm, 25 W) or kept in the dark, followed by incubation for 48 h at 37 °C, 5% CO2. The cells were trypsinized and resuspended in DMEM media. A total of 20000 cells/events were gated by flow cytometry. Analysis was performed on a FACSCalibur (Becton-Dickinson) instrument, using a 488 nm excitation laser with a 530 nm band-pass filter (EGFP) and a 633 nm excitation argon laser with a 671 nm band-pass filter (DsRed). Fluorescence was analyzed using the Cellquest Pro Software. For each of the triplicates, the data were averaged, normalized to the T0-noncaged control, and standard deviations were calculated. P values were calculated from unpaired t tests.
Spatial Activation of Gene Expression
HEK293T cells were passaged into 96-well plates (200 μL per well, ∼1 × 104 cells per well) and grown to ∼70% confluence within 24 h. Cells were transfected with the T3-caged EGFP (50 ng) and pDsRed-N1 (300 ng) plasmids using 1 μL of lipofectamine transfection reagent (Invitrogen) in 200 μL Opti-Mem media (Invitrogen) at 37 °C for 4 h. The media was removed and replaced with DMEM growth media. Localized irradiation was performed with a Zeiss Observer Z1 microscope (40X objective, NA 0.75 plan-apochromat; Zeiss) and a DAPI filter (68 HE, ex:BP377/28) to irradiate a specific subset of cells for 30 s. The cells were then incubated at 37 °C, 5% CO2 for 48 h and imaged on a Zeiss Observer Z1 microscope (5× magnification objective, filter sets 43 HE DsRed and 38 HE EGFP).
Plk3 Phenotypic Cell Assay
The Plk3 gene was fused to the C terminus of EGFP in pEGFP-Bst to form the pEGFP-Bst-Plk3 plasmid. The pEGFP-Bst-Plk3 plasmid was constructed by amplifying Plk3 from a Drosophila Plk3 cDNA (ATCC) with a Forward primer: 5′ CGTAAGCAATTGGACTTCTTTACC and Reverse Primer: 5′ CCTACGACTAGTCTAGGCTGGGCT. The pEGFP-BstNB plasmid was PCR amplified: Forward primer: 5′ GGAACTAGTCAGCGGCCGCGACTCT. Reverse primer: 5′ CCTACGCAATTGCTTGTACAGCTCGTC. Both PCR products were digested with SpeI and MfeI and ligated together with Quick ligase (New England Biolabs). The constructed plasmid pEGFP-Bst-Plk3 was confirmed by sequencing using the following sequencing primer: 5′ CTGCTGCCCGACAACCAC. The caged pEGFP-Bst-Plk3 plasmid was constructed using the same protocol as described above. HeLa cells were passaged into 4 well chamber slides and grown to 70% confluency. The cells were transfected with noncaged and caged pEGFP-Bst-Plk3 plasmids (150 ng) using linear polyethylene imine (LPEI) in an overnight experiment. The cells were irradiated with a UV transilluminator (365 nm, 5 min, 25 W) and were incubated at 37 °C, 5% CO2 for 48 h. The cells were fixed and stained with DAPI (blue) and rhodamine phalloidin (red) then imaged on a Zeiss Z1 Observer microscope (63× magnification objective, filter sets 43 HE DsRed and 38 HE eGFP).
Plk3 Caspase 3 Activity Assay
HeLa cells were grown at 37 °C, 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone), supplemented with 10% fetal bovine serum (Hyclone) and 10% streptomycin/penicillin (MP Biomedicals). The cells were passaged into 24-well plates (1 mL per well, ∼4 × 104 cells per well) and grown to ∼70% confluency within 24 h. The cells were transfected with noncaged and caged pEGFP-Bst-Plk3 plasmids (150 ng) using LPEI in an overnight experiment. The cells were irradiated with UV light (365 nm, 5 min, 25 W UV transilluminator) and were incubated at 37 °C, 5% CO2 for 48 h. The cells were lysed with Mammalian Cell Culture Protein Extraction buffer (GE Healthcare). Total protein was quantified with a Nanodrop spectrometer. HeLa cell protein extract (100 μg) was incubated with 50 μM Caspase-3 substrate (Ac-DEVD-AFC, Calbiochem) in activity buffer (50 mM HEPES, 150 mM NaCl, 50 mM MgCl2, 250 μM EDTA, 10% sucrose, 0.1% CHAPS, pH 7.2) at 37 °C for 20 h. The fluorescence was measured on a BioTek Synergy 4 plate reader (400/505 nm). For each of the triplicates, the data were averaged, normalized to the T0-noncaged control, and standard deviations were calculated. P values were calculated from unpaired t tests.
Zebrafish Maintenance and Injections
All zebrafish experiments were performed with the University of Pittsburgh Institutional Animal Care and Use Committee approval. The Oregon AB* strain was maintained under standard conditions at the University of Pittsburgh School of Medicine in accordance with Institutional and Federal guidelines. Embryos from natural matings were obtained and microinjected with 50 pg of the plasmid constructs using a World Precision Instruments Pneumatic PicoPump injector (100 ng/μL diluted 1:1 in phenol red; 1 nL injection = 50 pg plasmid). Increased plasmid injections were performed with the same dilution (50 ng/μL) but with increased injection amount; for example, the 200 pg injections were performed using 4 nL. Embryos were then irradiated following injection (typically at the 4- or 8-cell stage) for 2 min with a 365 nm UV transilluminator and incubated in the dark at 28 °C for 24 h. Late-stage irradiation experiments were performed at 8 h post fertilization and injection. Manual dechorionation was performed with forceps and zebrafish were treated with 1× Tricaine (MS-222, Sigma). Imaging was performed on a Leica MZ16FA stereo fluorescence microscope with a 1× objective (N.A. 0.14) at 60× (dechorionated 24 h) and 35× (dechorionated 48 h) zooms. Fluorescent (EGFP) and brightfield (BF) images were collected with a QImaging Retiga-EXi Fast 1394 digital camera. EGFP scores were calculated with embryo counts of [(EGFP positive/alive)·100]. For each of the replicates, the data were averaged, and standard deviations were calculated. P values were calculated from unpaired t tests.
Acknowledgments
This work was supported in part by the NIH (R01GM079114 to AD), North Carolina State University, and the University of Pittsburgh. The authors thank Ezenwa Obi Onuoha for assistance with zebrafish breeding, injections, and analysis.
Supporting Information Available
Sequence tables, melting temperatures, plasmid gel analysis, UV irradiation cell viability assay, cellular micrographs for control and caged plasmids, FACS dot plots and raw values, Tmut and T0-noncaged controls for PLk3 overexpression, T0-noncaged controls for plasmid-based zebrafish EGFP expression, additional images for the UV activation of T3-caged EGFP expression in zebrafish, UV irradiation optimization in zebrafish. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mayer G.; Heckel A. Angew. Chem., Int. Ed. Engl. 2006, 45, 4900. [DOI] [PubMed] [Google Scholar]
- Deiters A. Curr. Opin Chem. Biol. 2009, 13, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. D.; Deiters A. Org. Biomol. Chem. 2007, 5, 999. [DOI] [PubMed] [Google Scholar]
- Deiters A. ChemBioChem 2010, 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence D. S. Curr. Opin Chem. Biol. 2005, 9, 570. [DOI] [PubMed] [Google Scholar]
- Lee H. M.; Larson D. R.; Lawrence D. S. ACS Chem. Biol. 2009, 4, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Angew. Chem., Int. Ed. 2012, 51, 8446. [DOI] [PubMed] [Google Scholar]
- Fenno L.; Yizhar O.; Deisseroth K. Annu. Rev. Neurosci. 2011, 34, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher J. E.; Voigt C. A.; Weiner O. D.; Lim W. A. Nat. Methods 2011, 8, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker C. L. Prog. Brain Res. 2012, 196, 95. [DOI] [PubMed] [Google Scholar]
- Arenkiel B.; Peca J.; Davison I.; Feliciano C.; Deisseroth K.; Augustine G.; Ehlers M.; Feng G. Neuron 2007, 54, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge M.; Schneider F.; Tsunoda S.; Shilyansky C.; Wietek J.; Deisseroth K.; Hegemann P. J. Biol. Chem. 2012, 287, 31804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.; Young D. D.; Deiters A. ChemBioChem 2010, 11, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill J.; Chou C.; Chin J. W.; Deiters A. J. Am. Chem. Soc. 2013, 135, 13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröck L.; Heckel A. Angew. Chem., Int. Ed. 2005, 44, 471. [DOI] [PubMed] [Google Scholar]
- Polstein L. R.; Gersbach C. A. J. Am. Chem. Soc. 2012, 134, 16480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teraoka A.; Murakoshi K.; Fukamauchi K.; Suzuki A. Z.; Watanabe S.; Furuta T. Chem. Commun. (Cambridge) 2014, 50, 664. [DOI] [PubMed] [Google Scholar]
- Karginov A.; Zou Y.; Shirvanyants D.; Kota P.; Dokholyan N.; Young D.; Hahn K.; Deiters A. J. Am. Chem. Soc. 2011, 133, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. I.; Frey D.; Lungu O. I.; Jaehrig A.; Schlichting I.; Kuhlman B.; Hahn K. M. Nature 2009, 461, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A.; Deiters A.; Chin J. W. J. Am. Chem. Soc. 2011, 133, 2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher J. E.; Weiner O. D.; Lim W. A. Cell 2013, 155, 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A.; Weiner O.; Lim W.; Voigt C. Nature 2009, 461, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M. J.; Hughes R. M.; Peteya L. A.; Schwartz J. W.; Ehlers M. D.; Tucker C. L. Nat. Methods 2010, 7, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A.; Nguyen D. P.; Lusic H.; An W.; Deiters A.; Chin J. W. J. Am. Chem. Soc. 2010, 132, 4086. [DOI] [PubMed] [Google Scholar]
- Kamensek U.; Sersa G.; Vidic S.; Tevz G.; Kranjc S.; Cemazar M. Mol. Imaging Biol. 2011, 13, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura H.; Stinski M. F.; Kudoh A.; Nakayama S.; Murata T.; Sato Y.; Iwahori S.; Tsurumi T. J. Virol. 2008, 82, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini C. M. Physiol. Rev. 2009, 89, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Deiters A. Acc. Chem. Res. 2014, 47, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J. R.; Sarasin A.; Marrot L. Photochem. Photobiol. 2002, 75, 437. [DOI] [PubMed] [Google Scholar]
- Forman J.; Dietrich M.; Monroe W. T. Photochem. Photobiol. Sci. 2007, 6, 649. [DOI] [PubMed] [Google Scholar]
- Dong Q.; Svoboda K.; Tiersch T.; Monroe W. J. Photochem. Photobiol. B—Biol. 2007, 88, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusic H.; Lively M. O.; Deiters A. Mol. Biosyst. 2008, 4, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. D.; Lively M. O.; Deiters A. J. Am. Chem. Soc. 2010, 132, 6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly C. M.; Uprety R.; Hemphill J.; Deiters A. Mol. Biosyst. 2012, 8, 2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blidner R. A.; Svoboda K. R.; Hammer R. P.; Monroe W. T. Mol. Biosyst. 2008, 4, 431. [DOI] [PubMed] [Google Scholar]
- Govan J. M.; Uprety R.; Hemphill J.; Lively M. O.; Deiters A. ACS Chem. Biol. 2012, 7, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govan J. M.; Lively M. O.; Deiters A. J. Am. Chem. Soc. 2011, 133, 13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikat V.; Heckel A. RNA 2007, 13, 2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordoukhanian P.; Taylor J. S. Bioconjugugate Chem. 2000, 11, 94. [DOI] [PubMed] [Google Scholar]
- Tang X.; Maegawa S.; Weinberg E. S.; Dmochowski I. J. J. Am. Chem. Soc. 2007, 129, 11000. [DOI] [PubMed] [Google Scholar]
- Tallafuss A.; Gibson D.; Morcos P.; Li Y.; Seredick S.; Eisen J.; Washbourne P. Development 2012, 139, 1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini A. J.; Schuler A. D.; Zebala J. A.; Mayer A. N. Genesis 2009, 47, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X.; Shestopalov I. A.; Sinha S.; Zheng G.; Pitt C. L.; Li W. H.; Olson A. J.; Chen J. K. J. Am. Chem. Soc. 2009, 131, 13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Wu L.; Wang P.; Lv C.; Yang Z.; Tang X. Nucleic Acids Res. 2012, 40, 11155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazoe S.; Shestopalov I. A.; Provost E.; Leach S. D.; Chen J. K. Angew. Chem., Int. Ed. Engl. 2012, 51, 6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Swaminathan J.; Gewirtz A. M.; Dmochowski I. J. Nucleic Acids Res. 2008, 36, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J. L.; Seward G. K.; Wang Y. H.; Dmochowski I. J. ChemBioChem 2010, 11, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B.; O’Connor T. R.; Wang Y. ACS Chem. Biol. 2010, 5, 1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusic H.; Young D. D.; Lively M. O.; Deiters A. Org. Lett. 2007, 9, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter W. D.; Hüdepohl U.; Zillig W. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. D.; Edwards W. F.; Lusic H.; Lively M. O.; Deiters A. Chem. Commun. (Cambridge) 2008, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. D.; Lusic H.; Lively M. O.; Yoder J. A.; Deiters A. ChemBioChem 2008, 9, 2937. [DOI] [PubMed] [Google Scholar]
- Barr F. A.; Silljé H. H.; Nigg E. A. Nat. Rev. Mol. Cell Biol. 2004, 5, 429. [DOI] [PubMed] [Google Scholar]
- Xu D.; Wang Q.; Jiang Y.; Zhang Y.; Vega-Saenzdemiera E.; Osman I.; Dai W. Exp. Hematol. Oncol. 2012, 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N.; Wang X.; Jhanwar-Uniyal M.; Darzynkiewicz Z.; Dai W. J. Biol. Chem. 2006, 281, 10577. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Xie S.; Chen J.; Fukasawa K.; Naik U.; Traganos F.; Darzynkiewicz Z.; Jhanwar-Uniyal M.; Dai W. Mol. Cell. Biol. 2002, 22, 3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Gao J.; Dai W.; Lu L. J. Biol. Chem. 2008, 283, 25928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder N.; Xu Q. Methods Mol. Biol. 1999, 97, 487. [DOI] [PubMed] [Google Scholar]
- Culp P.; Nüsslein-Volhard C.; Hopkins N. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q. Methods Mol. Biol. 1999, 127, 125. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S.; Chen Y.; Nakajima S.; Furuta T.; Nagamune T. Chem. Commun. (Cambridge) 2010, 46, 2244. [DOI] [PubMed] [Google Scholar]
- Ando H.; Furuta T.; Tsien R. Y.; Okamoto H. Nat. Genet. 2001, 28, 317. [DOI] [PubMed] [Google Scholar]
- Monroe W. T.; McQuain M. M.; Chang M. S.; Alexander J. S.; Haselton F. R. J. Biol. Chem. 1999, 274, 20895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.