

Abstract
BACKGROUND
Type 1 long QT syndrome (LQT1) is caused by loss-of-function mutations in the KCNQ1-encoded Kv7.1 channel that conducts the slowly activating component of the delayed rectifier K+ current (IKs). Clinically, the diagnosis of LQT1 is complicated by variable phenotypic expressivity, whereby approximately 25% of genotype-positive individuals present with concealed LQT1 (resting corrected QT [QTc] interval ≤ 460 ms).
OBJECTIVE
To determine whether a specific molecular mechanism contributes to concealed LQT1.
METHODS
We identified a multigenerational LQT1 family whereby 79% of the patients genotype-positive for p.Ile235Asn-KCNQ1 (I235N-Kv7.1) have concealed LQT1. We assessed the effect I235N-Kv7.1 has on IKs and the ventricular action potential (AP) by using in vitro analysis and computational simulations.
RESULTS
Clinical data showed that all 10 patients with I235N-Kv7.1 have normal resting QTc intervals but abnormal QTc interval prolongation during the recovery phase of an electrocardiographic treadmill stress test. Voltage-clamping HEK293 cells coexpressing wild-type Kv7.1 and I235N-Kv7.1 (to mimic the patients’ genotypes) showed that I235N-Kv7.1 generated relatively normal functioning Kv7.1 channels but were insensitive to protein kinase A (PKA) activation. Phosphomimetic and quinidine sensitivity studies suggest that I235N-Kv7.1 limits the conformational changes in Kv7.1 channels, which are necessary to upregulate IKs after PKA phosphorylation. Computational ventricular AP simulations predicted that the PKA insensitivity of I235N-Kv7.1 is primarily responsible for prolonging the AP with β-adrenergic stimulation, especially at slower cycle lengths.
CONCLUSIONS
KCNQ1 mutations that generate relatively normal Kv7.1 channels, but limit the upregulation of IKs by PKA activation, likely contribute to concealed LQT1.
Keywords: Long QT syndrome, KCNQ1, Kv7.1, PKA activation, IKs, Treadmill stress test
Introduction
Congenital long QT syndrome (LQTS) is a condition of abnormal cardiac repolarization that affects approximately 1 of every 2000 live births and is characterized clinically by prolongation of the heart rate–corrected QT (QTc) interval on a resting 12-lead electrocardiogram (ECG). Patients with LQTS are at an increased risk of syncope, seizures, and sudden cardiac death secondary to polymorphic ventricular tachyarrhythmias.1 Type 1 long QT syndrome (LQT1) is caused by loss-of-function missense, nonsense, frameshift, or splice-site altering mutations in the KCNQ1 -encoded Kv7.1 α subunit and accounts for an estimated 40% of all genotype-positive LQTS cases.1-4 Kv7.1 α subunits tetramerize to form the pore of the slowly activating delayed rectifier K+ current (IKs) channel complex, and in the human heart, IKs is upregulated by protein kinase A (PKA) activation to prevent ventricular action potential (AP) prolongation during β-adrenergic stimulation.5,6 Consequently, many patients with LQT1 tend to experience life-threatening symptoms while exercising or swimming.7,8 Unfortunately, rendering a diagnosis of LQT1 on the basis of a 12-lead ECG alone presents a significant challenge as an estimated 25% of genotype-positive individuals with LQT1 fail to display an abnormal QTc interval at rest, commonly referred to as a concealed LQT1 phenotype.9,10
Patients with a concealed LQTS phenotype may remain at risk of cardiac events during exercise owing to inappropriate adaptation of repolarization.11 As such, developing a deeper understanding of the molecular mechanisms underlying a concealed LQT1 phenotype might improve personalized diagnostic and management approaches to lower the risk of life-threatening arrhythmias. In this study, we tested the hypothesis that some mutations contribute to a high incidence of the concealed LQT1 phenotype by a specific molecular mechanism.
Methods
Identification of a concealed LQT1 multigenerational pedigree
In this institutional review board–approved study, a retrospective review of more than 1000 individuals with a referral diagnosis of LQTS evaluated at Mayo Clinic between 1998 and 2008 was used to identify 76 of 249 (30.5%) individuals with LQT1 who featured a concealed LQTS electrocardiographic phenotype at rest (defined as a QTc interval of ≤ 460 ms for both men and women).12 To identify the mutation(s) most likely to confer a mutation-specific risk of concealed LQTS, we further limited this list to LQT1 pedigrees in which ≥ 2 genotype-positive family members featured a concealed LQTS phenotype. This process led to the identification of 3 moderate-to-large LQT1 pedigrees harboring I235N-Kv7.1, Y315C-Kv7.1, and T322A-Kv7.1.12 The concealed LQT1 phenotype was observed in 15 of 19 (79%) individuals who are genotype positive for I235N-Kv7.1, 4 of 7 (57%) individuals who are genotype positive for Y315C-Kv7.1, and 4 of 9 (44%) individuals who are genotype positive for T322A-Kv7.1. Given the electro-physiological defects associated with Y315C-Kv7.113 and T322A-Kv7.114 have been described in detail previously and these mutations showed a lower incidence of concealed LQT1, we focused our investigation on the large LQT1 family harboring the I235N-Kv7.1 mutation. Resting 12-lead and exercise stress test ECGs were analyzed manually, and the QT interval (lead II or V5) was corrected for heart rate by using Bazett’s formula (QTc interval = QT/√RR) as described previously.12, 15
Genetic analysis, mutagenesis, electrophysiology, computational modeling, and statistics
These methods were performed similarly as described previously and are explained in detail in the Online Supplement.16-19
Results
Patients with LQT1 and the I235N-Kv7.1 mutation show marked QTc prolongation during the recovery phase of the ECG treadmill stress test
Ten members of the large I235N-Kv7.1 LQT1 pedigree who underwent a treadmill stress test are depicted in Figure 1A. Each of these patients had a bona fide LQT1 phenotype as evident by the normal QTc interval at rest (Table 1) and ΔQTc > 30 ms at 3 minutes during the recovery phase as compared to baseline (Table 1 and Figure 1B).12 Moreover, marked QTc interval prolongation was seen at all recovery time points tested (Figure 1C), which suggested that I235N-Kv7.1 confers a mutation-specific risk of concealed LQT1. Importantly, genotype-positive patients are at risk of life-threatening cardiac events, as illustrated by the near drowning of the index case at age 15 (IV.8) and the suspicious sudden death of her genotype-positive father at age 41 (III.5).
Figure 1.

I235N-KV7.1 causes concealed LQT1. A: A pedigree of a LQT1 family with I235N-Kv7.1 who underwent an ECG treadmill stress test. Patients with the I235N-KV7.1 mutation are denoted by filled gray symbols. Individual males and females are denoted by squares and circles, respectively; each generation is denoted by a Roman numeral; OC denotes an obligate carrier; and asterisks signify individuals who underwent an ECG treadmill stress test. B: The QTc interval values recorded at baseline and after 3 minutes of recovery used to calculate ΔQTc of the 10 patients positive for I235N-Kv7.1 who underwent an ECG treadmill stress test are plotted. C: The mean QTc interval values recorded during the exercise treadmill stress test were plotted at baseline, peak exercise, and 1, 2, 3, 4, or 5 minutes during the recovery phase (*P < .05 vs baseline). ECG = electrocardiogram; LQT1 = type 1 long QT syndrome; QTc = corrected QT; ΔQTc = difference in QTc interval between baseline recording and 3 minutes after the ECG treadmill stress test.
Table 1.
Clinical characteristics of the genotype-positive I235N-Kv7.1 family
| Genotype-positive patients, n (female, n) | 15 (6) |
| ECG treadmill stress test, n (female, n) | 10 (3) |
| Mean age ± SD (y) | 38 ± 14 |
| Female mean age ± SD (y) | 46 ± 19 |
| Male mean age ± SD (y) | 35 ± 11 |
| Mean resting QTc ± SD (ms) | 445 ± 16 |
| Female mean resting QTc ± SD (ms) | 441 ± 24 |
| Male mean resting QTc ± SD (ms) | 446 ± 14 |
| Mean resting HR ± SD (ms) | 76 ± 17 |
| Female mean resting HR ± SD (ms) | 74 ± 8 |
| Male mean resting HR ± SD (ms) | 76 ± 19 |
| Mean ΔQTc ± SD (ms) | 76 ± 29 |
| Female mean ΔQTc ± SD (ms) | 88 ± 40 |
| Male mean ΔQTc ± SD (ms) | 71 ± 25 |
| Near-drowning episode, n (female, n) | 1 (1) |
| Mean 3 min of recovery HR ± SD (ms) | 117 ± 16 |
| Female mean 3 min of recovery HR ± SD (ms) | 115 ± 7 |
| Male mean 3 min of recovery HR ± SD (ms) | 118 ± 18 |
ECG = electrocardiogram; HR = heart rate; QTc = corrected QT; ΔQTc = difference between the baseline QTc interval and the QTc interval measured at 3 minutes during the recovery phase after the ECG treadmill stress test.
I235N-Kv7.1 disrupts a highly conserved amino add in S4 and causes a large reduction in IKs that is largely normalized by the coexpression of wild-type Kv7.1
The Kv7.1 α subunit is composed of cytosolic amino and carboxy termini and 6 transmembrane segments (S1-S6; Figure 2A). 1235 is located in S4 between highly conserved positively charged amino acids critical for the voltage dependence of Kv channel gating (Figure 2B).20 Sequence alignments show that 1235 is also conserved in other Kv α subunits (Figure 2B).20 We tested whether I235N-Kv7.1 generates a unique functional phenotype that accounts for the high incident of the concealed LQT1 clinical phenotype. For all voltage-clamping experiments, we coexpressed the K+ channel β subunit, KCNE1, which is obligatory for Kv7.1 to generate native-like IKs currents.2,3 IKs was recorded from cells expressing wild-type Kv7.1 (WT), I235N-Kv7.1, or coexpressing WT and I235N-Kv7.1 (to mimic the patients’ genotypes) by applying steplike pulses from −80 to 70 mV in 10-mV increments for 5 seconds, immediately followed by applying a “tail” pulse to −50 mV for 5 seconds (Figure 2C). The peak IKs amplitude recorded during the step pulse or at the start of the tail pulse was plotted as a function of the step pulse potential (Figures 2D–2E). These data show that cells expressing WT conducted IKs-like currents; cells expressing I235N-Kv7.1 generated small IKs that activated at positive potentials; and cells coexpressing WT and I235N-Kv7.1 conducted IKs that was slightly smaller than cells expressing WT. We calculated changes in the maximally activated IKs (IMAX) and tested for changes in gating by fitting the individual tail IKs-V relations with a Boltzmann equation (Table 2A and Figure 2E). IKs measured from cells expressing I235N-Kv7.1 without WT were too small to be reliably described by a Boltzmann equation and were excluded from analyses. The mean Boltzmann data showed that compared with cells expressing WT, cells coexpressing WT and I235N-Kv7.1 caused a ~ 30% reduction in IMAX with modestly different gating properties (a slightly more positive midpoint potential for half-maximal activation of IKs [V½] and shallower slope factor [k]). Together, these data demonstrate that the coexpression of WT largely corrected the loss of function caused by I235N-Kv7.1.
Figure 2.

I235N-Kv7.1 disrupts a conserved amino acid residue in S4 and coexpression of WT mostly corrects the loss of function caused by I235N-Kv7.1. A: Membrane topology of a single Kv7.1 pore-forming α subunit embedded in the plasma membrane. B: Amino acid residue sequence alignments for the S4 of Kv7.1 (NP_055194.1), Kv7.2 (NP_742105), Kv7.3 (NP_004510), Kv7.4 (NP_004691.2), Kv7.5 (NP_001153606), Kv1.1 (NP_000208.2), Kv2.1 (NP_004966), Kv3.1 (NP_001106212), Kv4.1 (NP_004970), Kv5.1 (NP_002227), Kv6.1 (NP_002228), Kv9.1 (NP_002242.2), and Kv10.1 (NP_002229.1). Amino acid residues conserved with Kv7.1 are shaded gray. C: Representative traces of whole-cell IKs measured from HEK293 cells transfected WT, I235N-Kv7.1, or WT and I235N-Kv7.1 complementary DNA. D and E: The mean peak (panel D) step or (panel E) tail IKs is plotted as a function of the step voltage for cells expressing WT (n = 11; black squares), I235N-Kv7.1 (n = 8; gray circles), or WT and I235N-Kv7.1 (n = 9; gray triangles). IKs = slowly activating delayed rectifier K+ current; WT = wild-type Kv7.1.
Table 2.
Electrophysiological properties of I235N-Kv7.1 described by using the Boltzmann equation
| Kv7.1 (n) | IMAX (pA/pF) | V½(mV) | k (mV/e-fold Δ) |
|---|---|---|---|
| A. Basal | |||
| WT (11) | 83.53 ± 5.82 | 20.00 ± 3.68 | 13.61 ± 1.61 |
| WT and I235N-Kv7.1 (12) | 57.03 ± 8.26* | 36.36 ± 5.57* | 19.70 ± 2.20* |
| B. Before (-) or after (+) forskolin & IBMX | |||
| WT (-) (6) | 73.99 ± 6.67 | 13.21 ± 3.82 | 9.55 ± 1.37 |
| WT (+) | 112.70 ± 12.46* | 3.62 ± 6.13 | 12.72 ± 1.29 |
| WT and I235N-Kv7.1 (-) (6) | 48.55 ± 7.47† | 28.83 ± 3.10*† | 17.32 ± 1.87 |
| WT and I235N-Kv7.1 (+) | 58.11 ± 10.75† | 30.25 ± 4.79*† | 20.95 ± 1.65 |
| C. Phosphomimetic | |||
| S27A (5) | 83.42 ± 7.79 | 20.73 ± 4.55 | 11.12 ± 1.52 |
| S27D (5) | 140.8 ± 7.86* | 22.68 ± 3.40 | 13.33 ± 0.96 |
| S27A and S27A/I235N (7) | 69.18 ± 10.97† | 39.00 ± 2.81 | 15.74 ± 0.91 |
| S27D and S27D/I235N (9) | 92.83 ± 9.58† | 38.12 ± 5.84 | 16.76 ± 1.88 |
A: To determine IMAX, V½, and k, the peak tail IKs-V relations were described by using the Boltzmann equation for cells expressing WT or WT and I235N-Kv7.1 (gray line; Figure 2E; *P < .05 vs WT). B: Cells expressing WT or WT and I235N-Kv7.1 (gray line; Figures 3B and 3C) before (−) or after (+) perfusion of forskolin and IBMX (*P < .05 vs WT before perfusion; †P < .05 vs WT after perfusion). C: Cells expressing S27A-Kv7.1, S27D-Kv7.1, S27A- and S27A/I235N-Kv7.1, or S27D- and S27D/I235N-Kv7.1 (gray line; Figures 4B and 4C;
P < .05 vs S27A;
P < .05 vs S27D).
Cells coexpressing WT and I235N-Kv7.1 generate PKA-insensitive IKs
An important functional role of IKs is to shorten the ventricular AP in response to β-adrenergic stimulation, β-adrenergic stimulation activates PKA, which increases IKs by phosphorylating Kv7.1 at S27.5,21 We previously showed that the KCNQ1 mutation R231H-Kv7.1, which is 4 amino acid residues upstream of I235, is insensitive to PKA activation.16 Therefore, we tested whether cells coexpressing WT and I235N-Kv7.1 might also generate PKA-insensitive IKs. We used a similar voltage protocol as in Figure 2C and measured IKs before and after the perfusion of forskolin and IBMX (Figure 3A). All these experiments were performed with the coexpression of KCNE1 and A-kinase anchoring protein 9 (Yotiao; AKAP9), which is the A-kinase anchoring protein important for coupling PKA to the IKs channel complex.5,21,22 The peak IKs amplitude recorded at the start of the tail pulse was plotted as a function of the step pulse potential and described with a Boltzmann equation (Table 2B and Figures 3B–3C). These data show that PKA activation in cells expressing WT increased IMAX However, it did not increase IMAX in cells coexpressing WT and I235N-Kv7.1. I235N-Kv7.1 appears to prevent the upregulation of IKs through PKA activation.
Figure 3.
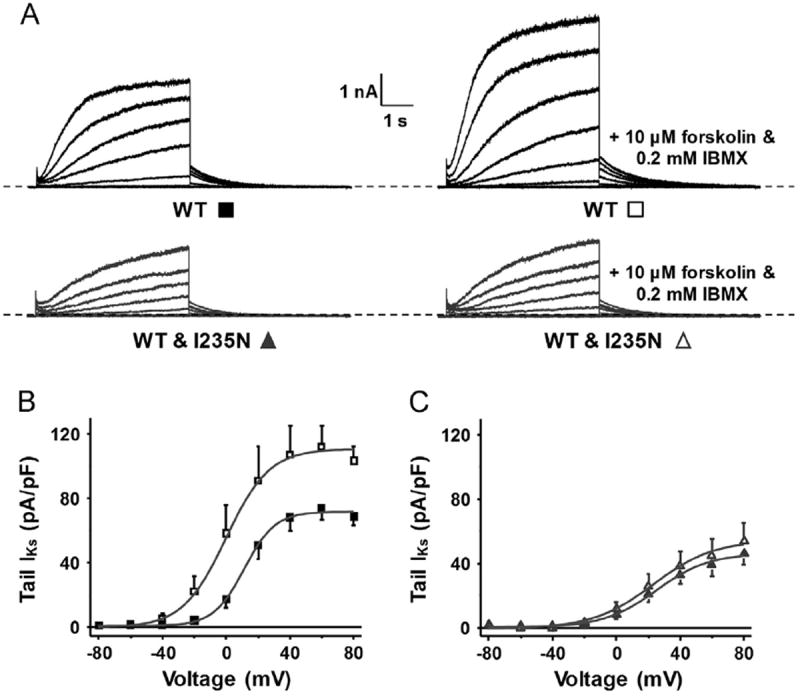
I235N-Kv7.1 generates PKA-insensitive Kv7.1 channels. (A) Representative traces of whole-cell IKs measured from cells transfected with WT or WT and I235N-Kv7.1 complementary DNA before and after extracellular perfusion of 10 μM of forskolin and 0.2 mM of IBMX. The mean peak tail IKs-V relations are plotted for cells expressing (B) WT (n = 6) before (black squares) and after (black open squares) perfusion or (C) WT and I235N-Kv7.1 (n = 6) before (triangles) and after (open triangles) perfusion. IKs = slowly activating delayed rectifier K+ current; PKA = protein kinase A; WT = wild-type Kv7.1.
I235N-Kv7.1 blunts the increase in IKs caused by the phosphomimetic substitution of S27D
I235N-Kv7.1 might prevent PKA stimulation of IKs by inhibiting phosphorylation at S27. To test this, we introduced amino acid residue substitutions at S27 in WT or I235N-Kv7.1 that prevent or mimic S27 phosphorylation (S27A or S27D, respectively).5,22 We recorded IKs from cells expressing S27A-Kv7.1 or S27D-Kv7.1 and from cells coexpressing S27A- and S27A/I235N-Kv7.1 or S27D- and S27D/I235N-Kv7.1 by using the same voltage protocol as in Figure 2C (Figure 4A). Boltzmann analyses of the tail IKs-V relations showed that as compared with cells expressing S27A-Kv7.1, cells expressing S27D-Kv7.1 had a larger IMAX but did not alter V½ or k (Table 2C and Figures 4B–4C). In contrast, cells coexpressing S27D-Kv7.1 and S27D/I235N-Kv7.1 blunted the increase in IMAX as compared with cells coexpressing S27A- and S27A/I235N-Kv7.1. These data suggest that S27D does not upregulate IKs in channels with the I235N-Kv7.1 mutation.
Figure 4.

The phosphomimetic substitution of S27D does not increase the functional expression of I235N-Kv7.1. A: Representative traces of whole-cell IKs measured from cells transfected with S27A- or S27D-Kv7.1, and S27A- and S27A/I235N-Kv7.1 or S27D- and S27D/I235N-Kv7.1 complementary DNA. B and C: The mean peak tail IKs-V relations are plotted for cells expressing (panel B) S27A-Kv7.1 (n = 5; black squares) or S27D-Kv7.1 (n = 5; open squares) and (panel C) S27A- and S27A/I235N-Kv7.1 (n = 7; gray triangles) or S27D- and S27D/I235N-Kv7.1 (n = 9, open triangles). IKs = slowly activating delayed rectifier K+ current.
Since the phosphomimetic mutation did not rescue the upregulation of IKs, we tested whether I235N-Kv7.1 prevents the necessary conformational changes in Kv7.1 channels required to upregulate IKs after PKA activation. Previous studies by Yang and colleagues23,24 showed that PKA-stimulated Kv7.1 channels or S27D-Kv7.1 reduce quinidine block of IKs compared with nonstimulated Kv7.1 channels. These data suggest that the PKA phosphorylation of S27 or S27D cause conformational changes in Kv7.1 channels that increase IKs and restricts the access of quinidine to its binding site.25 In other words, the relative sensitivity of IKs to quinidine block is a good indicator of the conformational changes associated with S27 phosphorylation and the phosphomimetic substitution of S27D. We compared the % block of IKs caused by quinidine from cells expressing S27A-Kv7.1, S27D-Kv7.1, or S27D- and S27D/I235N-Kv7.1 (Figure 5). Similar to what was shown previously, cells expressing S27D-Kv7.1 conducted IKs that was less sensitive to quinidine block than cells expressing S27A-Kv7.1. However, cells coexpressing S27D- and S27D/I235N-Kv7.1 conducted IKs and demonstrated sensitivity to quinidine block similar to that shown by cells expressing S27A-Kv7.1. Together, these data suggest that I235N-Kv7.1 prevents the PKA upregulation of IKs by limiting the conformational changes in Kv7.1 channels associated with S27 phosphorylation.
Figure 5.

I235N-Kv7.1 preserves the sensitivity of IKs to quinidine block. Representative IKs traces before (black) and after the perfusion of 100 μM quinidine (Quin; red) measured from cells transfected with S27A-Kv7.1, S27D-Kv7.1, or S27D- and S27D/I235N-Kv7.1 complementary DNA. IKs was recorded by applying a step pulse to 50 mV for 5 seconds followed by applying a tail pulse to −50 mV for 5 seconds. The mean % block of the peak tail IKs are plotted (*P < .05). IKs = slowly activating delayed rectifier K+ current.
Computational simulations of ventricular AP with or without β-adrenergic stimulation suggest that IKs insensitivity to PKA is primarily responsible for a ventricular AP prolongation
Next, we wanted to test how I235N-Kv7.1 might alter the duration of a ventricular AP over a wide range of cycle lengths with or without β-adrenergic stimulation by using computational modeling.18 To mimic the effects that I235N-Kv7.1 has on IKs, the IKs component was reduced by 30% and made insensitive to PKA. We compared these simulations with control simulations or simulations that simply reduced the IKs component by 30% (without changing PKA sensitivity). The steady-state action potential duration at 90% repolarization (APD90) at cycle lengths ranging from 300 to 1000 ms was calculated. In the absence of β-adrenergic stimulation, a 30% reduction in IKs resulted in a l%–2% prolongation of the APD90 between 300 and 1000 ms (Figures 6A–6C). The presence of β-adrenergic stimulation indicated that reducing the IKs component by 30% still increased the APD90 by only l%-2%. However, reducing the IKs component by 30% and making it PKA-insensitive increased the APD90 from 6% to 10% at cycle lengths between 300 and 1000 ms. These computational data suggest that the PKA insensitivity of I235N-Kv7.1 is primarily responsible for the concealed LQT1 phenotype.
Figure 6.

Computational simulations suggest that making the IKs component insensitive to PKA selectively increases the ventricular AP duration with β-adrenergic stimulation. The APD90 was plotted as a function of the cycle length. Shown are the APD90 calculated at cycle lengths between 300 and 1000 ms for control simulations (black squares), simulations in which the IKs component was reduced by 30% (blue circles), and simulations in which the IKs component was reduced by 30% and made insensitive to PKA (red triangles) (A) for basal conditions or (B) with β-adrenergic stimulation. (C) Representative AP waveforms and the corresponding IKs for control simulations (black line), simulations in which the IKs component was reduced by 30% (blue line), and simulations in which the IKs component was reduced by 30% and rendered PKA-insensitive (red line) for 300 or 1000 ms. The black and red AP waveforms are not visible for the basal simulations because they are overlaid by the blue AP waveform, and the red IKs is not visible for the basal simulations because it is overlaid by the blue IKs. To mimic dominant-negative IKs suppression, simulations were repeated for control IKs (black squares) or in which the IKs component was reduced by 70% (aqua diamonds) for (D) basal conditions or (E) β-adrenergic stimulation. Also shown are simulations in which IKs is only PKA-insensitive (orange open inverted triangles) or is reduced by 70% and PKA-insensitive (gold triangles). AP = action potential; APD90 = steady-state action potential duration at 90% repolarization; IKs = slowly activating delayed rectifier K+ current; PKA = protein kinase A.
The data from the LQTS patient registry indicate that dominant-negative LQT1 mutations, which decrease total IKs by > 50%, are an independent risk factor for life-threatening events.26 We used computational modeling to assess the pathological/clinical relevance of PKA-insensitive vs dominant-negative Kv7.1 mutations. We compared simulations in which IKs was PKA-insensitive, a simulation that mimicked a dominant-negative LQT1 mutation (the IKs component was reduced by 70%), or a simulation that mimicked a PKA-insensitive dominant-negative LQT1 mutation. During simulations in which the IKs component was only PKA-insensitive, the APD90 increased with β-adrenergic stimulation by 6%–8%. A dominant-negative IKs simulation increased the APD90 by 3%–4% in the basal simulations and by 6%–8% in β-adrenergic simulations for cycle lengths 300–1000 ms (Figures 6D–6E). Coupling the PKA insensitivity with a dominant-negative reduction in IKs increased the APD90 by 8%–12% for cycle lengths 300–1000 ms. These data suggest that PKA-insensitive Kv7.1 mutations, which do not reduce basal IKs, can prolong the AP as much as dominant-negative Kv7.1 mutations in conditions of β-adrenergic stimulation. We conclude that assessing the PKA sensitivity of seemingly normal functioning Kv7.1 mutations is critical to determine their pathological significance.
Discussion
The increasing availability of clinical genetic testing for LQTS promises to one day allow for the development of personalized approaches to the prevention of life-threatening arrhythmias in individuals afflicted with this genetic disorder. However, the probabilistic interpretation of LQTS genetic testing results is complicated by the observation that 4%–8% of the ostensibly healthy individuals are expected to harbor rare amino acid–altering genetic variants in 1 of 3 major LQTS-susceptibility genes (KCNQ1, KCNH2, and SCN5A).27 In addition, it is estimated that approximately 25% of the individuals with an LQTS-causative mutation fail to manifest any abnormalities in their QTc interval on resting ECG (ie, concealed LQTS).11 There remain few insights into the genetic and/or electrophysiological mechanisms that underlie concealed LQTS phenotypes.26,28 The fact that individuals with a concealed LQTS phenotype still have a > 10-fold higher relative risk of sudden cardiac arrest/death than their genotype-negative relatives highlights the pressing need to develop a deeper understanding of the mechanisms underlying this phenomenon.27 Therefore, the purpose of this study was to determine whether a specific molecular mechanism contributes to the concealed LQT1 phenotype.
In this study, we identified a large LQT1 multigenerational pedigree whereby the vast majority of individuals who were genotype-positive for I235N-Kv7.1 displayed a concealed LQT1 phenotype. The family was brought to clinical attention owing to a near-drowning episode of the I235N-positive index case and the sudden unexpected death of her genotype-positive father. Therefore, we directly evaluated the functional properties of I235N-Kv7.1 in HEK293 cells. While homomeric I235N-Kv7.1 caused a severe loss of function, the coexpression of WT markedly improved the dysfunctional IKs phenotype. These data contradict a recent report by Henrion et al,29 who previously suggested that I235N-Kv7.1 causes dominant-negative suppression of WT when expressed in Xenopus laevis oocytes. Although we cannot completely account for this discrepancy, we suspect that it might be due in part to the alternative expression system used.30,31
The activation of the β-adrenergic receptor initiates a signaling cascade through Gαs proteins that activate adenylate cyclase to increase intracellular levels of cyclic adenosine monophosphate cAMP. cAMP activates PKA, and PKA phosphorylates several substrate proteins in cardiomyocytes, specifically phosphorylating S27 on the N terminus of Kv7.1, which upregulates IKs.5 These data suggest that patients with LQT1 who have dominant-negative LQT1 mutations (>50% reduction in IKs) that generate Kv7.1 channels insensitive to PKA activation are likely to manifest a severe clinical phenotype. For example, Heijman et al22 and Barsheshet et al32 reported several dominant-negative LQT1-linked mutations that generate PKA-insensitive Kv7.1 channels and associate with severe clinical phenotypes. Most recently, Wu et al33 showed that a mildly dominant-negative LQTl-linked mutation (G269S-Kv7.1) primarily contributes to an adrenergic-induced LQTS phenotype. We now show that I235N-Kv7.1, which does not cause dominant-negative effect on IKs but generates PKA-insensitive Kv7.1 channels, contributes to a high incidence of the concealed LQT1 phenotype. Computational ventricular AP simulations that incorporate the functional effects of I235N-Kv7.1 suggest that the mild dysfunctional phenotype minimally alters the AP duration in basal conditions, but β-adrenergic stimulation, especially at slow cycle lengths, unmasks AP prolongation similar to a dominant-negative LQT1 mutation. The exacerbated prolongation of the ventricular AP duration at slower cycle lengths qualitatively mimics the changes in the QTc interval during the exercise treadmill stress test for patients with I235N-Kv7.1 (QTc prolongation was exacerbated as the heart rate slows during the recovery phase of the ECG stress test).
Although an increasing number of LQT1-linked mutations are being linked to PKA insensitivity, the molecular mechanisms for most PKA-insensitive mutations have not been fully elucidated. The suppression of IKs upregulation has previously been reported in an LQT1-linked mutation G589D-Kv7.1, which disrupts the binding of AKAP9 to the C terminus.5 Heijman et al22 showed that A341V-Kv7.1 prevents IKs upregulation through PKA activation by inhibiting the phosphorylation at S27. Unlike A341V-Kv7.1, the phosphomimetic substitution of S27 in I235N-Kv7.1 did not increase IKs. Moreover, phosphomimetic I235N-Kv7.1 generated IKs that maintained a high sensitivity to quinidine block. Recently, Yang et al25 performed extensive Kv7.1 mutagenesis and molecular modeling to examine the mechanism for quinidine block. These studies suggest that quinidine allosterically inhibits IKs by binding to an intracellular pocket generated by amino acid residues in the S4–S5 linker and S6, which raises the intriguing possibility that S27 phosphorylation causes a conformational change that upregulates IKs and alters the quinidine binding pocket to decrease quinidine block. The presence of I235N-Kv7.1 prevents both PKA-induced conformational changes and IKs upregulation. These data identify a novel mechanism by which LQT1 mutations generate PKA-insensitive Kv7.1 channels.
Study limitations
This study has several limitations. The data were obtained in a widely used heterologous expression system that might not completely recapitulate in vivo phenotypes. Additional family specific factors, including KCNQ1 polymorphisms in the 3′ allele, might contribute to the concealed LQT1 phenotype in this family.
Conclusions
Our study suggests that the exercise treadmill stress test for patients with concealed LQT1 might help identify patients who harbor PKA-insensitive Kv7.1 channels. To our knowledge, these are the first data that link PKA insensitivity to an abnormal QT response during the ECG exercise treadmill stress test. Future studies incorporating the exercise treadmill stress test combined with mutation-specific risk assessment will likely improve the treatment of each patient with LQT1. We conclude that some PKA-insensitive LQT1 mutations, which generate relatively mild dysfunctional phenotypes in basal conditions, can contribute to a high penetrance of the concealed LQT1 phenotype.
Supplementary Material
Acknowledgments
We thank Dr Robert Kass (Columbia University, New York, NY) for providing the AKAP9 plasmid DNA.
This work was supported by the American Heart Association predoctoral award PRE7370003 (to Dr Bartos) and the National Heart Lung and Blood Institute grants R01 HL087039 (to Dr Delisle) and F30 HL106993 (to Dr Giudicessi). Dr Ackerman is a consultant for Biotronik, Boston Scientific, Medtronic, St Jude Medical, and Transgenomic. In addition, there is a license agreement between Transgenomic and Mayo Clinic Health Solutions and royalties are distributed in accordance with the Mayo Clinic policy.
ABBREVIATIONS
- AKAP9
A-kinase anchoring protein 9 (Yotiao)
- AP
action potential
- APD90
steady-state action potential duration at 90% repolarization
- ECG
electrocardiogram/electrocardiographic
- HEK293
human embryonic kidney 293
- IKs
slowly activating delayed rectifier K+ current
- IMAX
maximally activated IKs
- k
slope factor
- LQT1
type 1 long QT syndrome
- LOTS
long QT syndrome
- PKA
protein kinase A
- QTc
corrected QT
- V½
midpoint potential for half-maximal activation of IKs
- WT
wild-type Kv71
Footnotes
Appendix
Supplementary data
Supplementary data associated with this article can be found in the online version at http://dx.doi.Org/10.1016/j.hrthm.2013.11.021.
References
- 1.Crotti L, Celano G, Dagradi F, Schwartz PJ. Congenital long QT syndrome. Orphanet J Rare Dis. 2008;3:18. doi: 10.1186/1750-1172-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V) LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of K(V) LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 5.Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 6.Volders PG, Stengl M, van Opstal JM, et al. Probing the contribution of IKs to canine ventricular repolarization: key role for beta-adrenergic receptor stimulation. Circulation. 2003;107:2753–2760. doi: 10.1161/01.CIR.0000068344.54010.B3. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 8.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation. 2004;110:2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 9.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 10.Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res. 2013;161:1–14. doi: 10.1016/j.trsl.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldenberg I, Horr S, Moss AJ, et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51–59. doi: 10.1016/j.jacc.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horner JM, Horner MM, Ackerman MJ. The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm. 2011;8:1698–1704. doi: 10.1016/j.hrthm.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 13.Bianchi L, Priori SG, Napolitano C, et al. Mechanisms of I(Ks) suppression in LQT1 mutants. Am J Physiol Heart Circ Physiol. 2000;279:H3003–H3011. doi: 10.1152/ajpheart.2000.279.6.H3003. [DOI] [PubMed] [Google Scholar]
- 14.Burgess DE, Bartos DC, Reloj AR, et al. High-risk long QT syndrome mutations in the Kv7.1 (KCNQ1) pore disrupt the molecular basis for rapid K(+) permeation. Biochemistry. 2012;51:9076–9085. doi: 10.1021/bi3009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giudicessi JR, Ackerman MJ. Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity. Circ Cardiovasc Genet. 2013;6:193–200. doi: 10.1161/CIRCGENETICS.112.964684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartos DC, Anderson JB, Bastiaenen R, et al. A KCNQ1 mutation causes a high penetrance for familial atrial fibrillation. J Cardiovasc Electrophysiol. 2013;24:562–569. doi: 10.1111/jce.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartos DC, Duchatelet S, Burgess DE, et al. R231C mutation in KCNQ1 causes long QT syndrome type 1 and familial atrial fibrillation. Heart Rhythm. 2011;8:48–55. doi: 10.1016/j.hrthm.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys J. 2010;99:2038–2047. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tester DJ, Will ML, Ackerman MJ. Mutation detection in congenital long QT syndrome: cardiac channel gene screen using PCR, dHPLC, and direct DNA sequencing. Methods Mol Med. 2006;128:181–207. doi: 10.1385/1-59745-159-2:181. [DOI] [PubMed] [Google Scholar]
- 20.Papazian DM, Timpe LC, Jan YN, Jan LY. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 1991;349:305–310. doi: 10.1038/349305a0. [DOI] [PubMed] [Google Scholar]
- 21.Kurokawa J, Motoike HK, Rao J, Kass RS. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc Natl Acad Sci U S A. 2004;101:16374–16378. doi: 10.1073/pnas.0405583101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heijman J, Spatjens RL, Seyen SR, et al. Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circulation Res. 2012;110:211–219. doi: 10.1161/CIRCRESAHA.111.249482. [DOI] [PubMed] [Google Scholar]
- 23.Yang T, Kanki H, Zhang W, Roden DM. Probing the mechanisms underlying modulation of quinidine sensitivity to cardiac I(Ks) block by protein kinase A-mediated I(Ks) phosphorylation. Br J Pharmacol. 2009;157:952–961. doi: 10.1111/j.1476-5381.2009.00293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang T, Kanki H, Roden DM. Phosphorylation of the IKs channel complex inhibits drug block: novel mechanism underlying variable antiarrhythmic drug actions. Circulation. 2003;108:132–134. doi: 10.1161/01.CIR.0000082708.86266.B8. [DOI] [PubMed] [Google Scholar]
- 25.Yang T, Smith JA, Leake BF, Sanders CR, Meiler J, Roden DM. An allosteric mechanism for drug block of the human cardiac potassium channel KCNQ1. Mol Pharmacol. 2013;83:481–489. doi: 10.1124/mol.112.081513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giudicessi JR, Kapplinger JD, Tester DJ, et al. Phylogenetic and physicochemical analyses enhance the classification of rare nonsynonymous single nucleotide variants in type 1 and 2 long-QT syndrome. Circ Cardiovasc Genet. 2012;5:519–528. doi: 10.1161/CIRCGENETICS.112.963785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henrion U, Strutz-Seebohm N, Duszenko M, Lang F, Seebohm G. Long QT syndrome-associated mutations in the voltage sensor of I(Ks) channels. Cell Physiol Biochem. 2009;24:11–16. doi: 10.1159/000227828. [DOI] [PubMed] [Google Scholar]
- 30.Furutani M, Trudeau MC, Hagiwara N, et al. Novel mechanism associated with an inherited cardiac arrhythmia: defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation. 1999;99:2290–2294. doi: 10.1161/01.cir.99.17.2290. [DOI] [PubMed] [Google Scholar]
- 31.Nakajima T, Furukawa T, Hirano Y, et al. Voltage-shift of the current activation in HERG S4 mutation (R534C) in LQT2. Cardiovasc Res. 1999;44:283–293. doi: 10.1016/s0008-6363(99)00195-9. [DOI] [PubMed] [Google Scholar]
- 32.Barsheshet A, Goldenberg I, O-Uchi J, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: implications for mutation-specific response to beta-blocker therapy in type 1 long-QT syndrome. Circulation. 2012;125:1988–1996. doi: 10.1161/CIRCULATIONAHA.111.048041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, Naiki N, Ding W-G, et al. A molecular mechanism for adrenergic-induced long QT syndrome. J Am Coll Cardiol. 2013 doi: 10.1016/j.jacc.2013.08.1648. [DOI] [PubMed] [Google Scholar]
- 34.Amin AS, Giudicessi JR, Tijsen AJ, et al. Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J. 2012;33:714–723. doi: 10.1093/eurheartj/ehr473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.