

Abstract
Zoonotic viruses represent emerging or re-emerging pathogens that pose significant public health threats throughout the world. It is therefore crucial to advance current surveillance mechanisms for these viruses through outlets such as phylogeography. Despite the abundance of zoonotic viral sequence data in publicly available databases such as GenBank, phylogeographic analysis of these viruses is often limited by the lack of adequate geographic metadata. However, many GenBank records include references to articles with more detailed information and automated systems may help extract this information efficiently and effectively. In this paper, we describe our efforts to determine the proportion of GenBank records with “insufficient” geographic metadata for seven well-studied viruses. We also evaluate the performance of four different Named Entity Recognition (NER) systems for automatically extracting related entities using a manually created gold-standard.
Introduction
Zoonotic viruses, viruses that are transmittable between animals and humans, have become increasingly prevalent in the last century leading to the rise and re-emergence of a variety of diseases1. In order to enhance currently available surveillance systems for these viruses, a better understanding of their origins and transmission patterns is required. This need has led to a greater amount of research in the field of phylogeography, the study of geographical lineages of species2. Population health agencies frequently apply phylogeographic techniques to trace the evolutionary changes within viral lineages that affect their diffusion and transmission among animal and human hosts3, 4, 5.
Phylogeography depends on the utilization of both sequence and location data which are often obtained from online resources such as GenBank (http://www.ncbi.nlm.nih.gov/genbank/). While there is an abundance of sequence data records in GenBank, many of them lack sufficient geographical metadata that would enable specific identification of the isolate’s location of collection. In our previous study6 we found that the geographic information of 80% of the GenBank records associated with single or double stranded RNA viruses within tetrapod hosts was less specific than 1st level administrative boundaries (ADM1) such as state or province. More detailed information concerning the location from which sequences were collected is often available within their corresponding journal articles. However, this manual process does not allow researchers to conveniently retrieve the needed data. We investigated the potential of natural language processing (NLP) to enhance the geographical data available for phylogeography studies by extracting information on the origin of viruses from natural language text6. In order to accurately link each GenBank record to its corresponding location of isolation, NER systems can be used to identify additional entities other than location within the text. Such entities can be used to provide extra information that the system can use to distinguish the correct location from other location mentions. Previously we used BANNER and the Stanford NER tool to automatically tag gene and location mentions in text respectively. Here we build on this work by proposing an automated process through which spatial information, dates, genes and species mentions are extracted from journal articles. We validate these results by using a gold standard developed through manual annotation.
Background
Phylogeography has the potential to inform population health endeavors by studying the spread of viruses in relation to migration and host populations. For instance, Gray et al. explores the spatial distribution of Rift Valley Fever Virus (RVFV) within Africa using information regarding the time and location of sample collection combined with sequence data4. Using the work of Lemey et al. 2009 as a guide, phylogeographic analysis was performed using the BEAST software package in order to reconstruct viral transmission and relate these findings to data for livestock population density7, 8. This work demonstrates the benefits of phylogeographic study by highlighting the need to reassess assumptions regarding the spread of RVFV via sheep and cattle populations, considering the spread of the virus over large low density area4. In another example, Weidmann et al. studied the expansion of tick-borne encephalitis virus (TBEV) within central Europe, as opposed to the observed spread throughout the Eurasian region by examining E gene sequences5. Following Bayesian analysis, it was found that west to east viral migration was indicated for central Europe as opposed to an east to west direction in Eurasia. In addition, it was concluded that the emergence of multiple subclades could be explained by recent evolutionary bottlenecks5.
Previously, there has not been work aimed at the automated extraction of spatial data for the enhancement of phylogeography. However, other applications such as geographical information retrieval (GIR) share similar challenges in processing natural language documents containing geographic mentions despite having different goals. Bordogna et al. addressed this subject by developing a GIR model that represents the uncertainty of geographic context of texts by associating fuzzy descriptors of specific locations which indicate the author’s perception9. Other approaches to bio-surveillance have utilized NLP techniques such as NER and n-grams to scan and classify online articles concerning disease outbreaks of potential interest to public health10,11. While these works share similar endeavors, we address the complex issue of improving geospatial data availability relating to genetic sequence data. Furthermore, the framework we develop here will be applied towards the extraction of geospatial data of interest which will allow the linkage of specific mentions of a GenBank record and a location, thereby aiding the development of future phylogeographic models.
Materials and Methods
The process undertaken to complete this study can be divided into five distinct stages: selection of the viruses, extraction of relevant GenBank data related to each virus, computation of “sufficiency” statistics on the extracted data, development of an integrated NER system, and evaluation of this system on a manually annotated corpus of full-text PubMed articles. Figure 1 in Appendix A provides a brief overview of each of these steps. A detailed description of each phase is given below.
Virus Selection and GenBank Data Extraction
The domain of this study was limited to zoonotic viruses that are most consistently documented and tracked by public health, agriculture and wildlife state departments within the United States. These viruses include influenza, rabies, hantavirus, western equine encephalitis (WEE), eastern equine encephalitis (EEE), St. Louis encephalitis (SLE), and West Nile virus (WNV). The Entrez Programming Utilities (E-Utilities) was used to download the following fields from 59,595 GenBank records associated with these viruses: GenBank Accession ID, Pubmed Central ID, Strain name, Collection date and Country. This set of records consisted of all records in GenBank related to the selected viruses that contained Pubmed Central (PMC) IDs for referencing articles. We limited our set to records with PMC IDs since the NER system being tested is only relevant for these records. Figure 2 in Appendix A provides a screenshot of the extracted data.
Sufficiency vs. Insufficiency Analysis
The data extracted from Genbank was used to compute the percentage of GenBank records that had insufficient geographic information for each of the selected viruses. In order to perform this computation, we used data from the ISO 3166-1 alpha-2 table12 and the GeoNames13 database. The ISO 3166-1 alpha-2 is the International Standard for representing country names using two-letter codes. The GeoNames database contains a variety of geospatial data for over 10 million locations on earth, including the ISO 3166-1 alpha-2 code for the country of each location and a feature code that can be used to determine the administrative level of each location. To allow for efficient querying, we downloaded the main GeoNames table and the ISO alpha-2 country codes table from their respective websites and stored them in a local SQL database. Prior to adding the ISO data to the database, some commonly used country names and their corresponding country codes were added to the table since it only included a single title for each country. For example, the ISO table included the country name “United States” but not alternate names such as “USA”, “United States of America”, or “US”. Using the created database in conjunction with a parser written in Java, we were able to retrieve most of the geographic information present within the records and classify each of them as sufficient or insufficient.
For the purpose of this project, we considered any geographical boundary more specific than ADM1 to be “sufficient”. Based on this criterion, a feature code in GeoNames was categorized as sufficient only if it was absent from the following list of feature codes: ADM1, ADM1H, ADMD, ADMDH, PCL, PCLD, PCLF, PCLH, PCLI and PCLS. The method for evaluating the geographical sufficiency of a GenBank record was dependent upon whether the record included a country name. A GenBank record with a country mention was called sufficient if the geographic information extracted from that record included another place mention whose feature code fell within the class of sufficient feature codes and whose ISO country code matched that of the retrieved country. Place mentions with matching country codes often had several different feature codes in GeoNames. Such places were only called sufficient if all feature codes corresponding to the given pair of place name and country code were classified as sufficient. In cases where the GenBank record had no country mention, the record was called sufficient only if all matching GeoNames entries for any of the places mentioned in it had sufficient feature codes. The sufficiency criteria were designed to ensure that a geographic location is only called sufficient if its administrative level was found to be more specific than ADM1 without any form of ambiguity. Figure 3a in Appendix A illustrates the pathways of geographical sufficiency for GenBank records in a diagram.
In order to obtain the geographic information for each Genbank record, we used a Java parser which automatically extracted data from the “country” field of each record. Since the “country” field typically contained multiple place mentions divided by a set of delimiters consisting of comma, colon and hyphen, we first split this field using these delimiters. We then checked each string obtained through this process against the ISO country code table to determine whether it was a potential country name for the record’s location. If the query returned no results, then the locally stored GeoNames tables was searched and for each match found, the corresponding ISO country code and feature code were extracted. Figure 3b in Appendix A on the right shows a diagram of this process.
In cases where no sufficient location data was found from the “country” field of a GenBank record, the Java parser searched through its “strain” field. This was done because some viral strains such as influenza include their location of origin integrated into their names. For example, the influenza strain “A/duck/Alberta/35/76” indicates that the geographic origin of the strain is Alberta. The different sections of a strain field are separated by either forward slash, parenthesis, comma, colon, hyphen or underscore and so we used a set of delimiters consisting of these characters to split this field. Each string thus retrieved was queried as before on the ISO country code table and the GeoNames table. GeoNames often returned matches for strings like ‘raccoons’ and ‘chicken’ which were actually meant to be names of host species within the “strain” field and so a list of some of the most frequently seen host name mentions in these records was manually created and filtered out before querying GeoNames.
Some of the place mentions contained very specific location information which resulted in GeoNames not finding a match for them. A list was created for strings like ‘north’, ‘south-east’, ‘governorate’ etc. which when removed from a place mention may produce a match. In cases of potential place mentions which contained any one of these strings and for which GeoNames returned no matching result, a second query was performed after removal of the string.
Development and Evaluation of Integrated NER System
An NER system for identifying species, gene, date and location mentions in text was developed by integrating LINNEAUS14, BANNER15, GeoNamer and Stanford SUTime16. LINNEAUS, BANNER and Stanford SUTime are widely-used, state-of-the-art open source NER systems for recognition of species, gene and temporal expressions respectively. A detailed description of each of these tools is provided in the Appendix. GeoNamer is a dictionary-based location tagging system that we built using GeoNames. The dictionary used by GeoNamer was created by retrieving distinct place names from the GeoNames table and filtering out commonly used words from the retrieved set. Words filtered out include stop words, generic place names such as ‘cave’ and ‘hill’, numbers like ‘one’, domain specific words such as ‘biology’ and ‘DNA’, most commonly used surnames like ‘Garcia’, commonly used animal names such as ‘chicken’ and ‘fox’ and other miscellaneous words such as ‘central’. This was a crucial step since the GeoNames databases contains a wide array of commonly used English used words which may cause a large volume of false positives if not removed. The final dictionary consisted of 5,396,503 entries. To recognize place mentions in a given set of text files, GeoNamer first builds a Lucene index on the contents of the files. It then constructs a phrase query for every entry in the Geonames dictionary and runs each query on the Lucene index. The document id, query text, start offset and end offset for every match found is written to an output file.
The developed system was tested on a set of twenty-seven manually-annotated full-text PubMed Central articles downloaded manually in the pdf version and converted to text using Adobe Acrobat. The number of papers selected for each virus was influenced by the number of GenBank records with PMC ids that were available for the given virus. Ten papers were selected for rabies, nine for influenza, two for hantavirus, WEE and WNV and one for SLE and EEE. The articles for each virus were chosen by using Excel’s RAND function on the subset of extracted GenBank records related to that virus which had insufficient geographic information.
Three annotators tagged the following five entities in each article using the freely available annotation tool, BRAT17: gene names, locations, dates, organisms and viruses. A detailed description of the annotation guidelines is given in the appendix. Before creating the guidelines, each annotator individually annotated six common articles and compared and discussed their results to devise a reasonable set of rules for annotating each entity. After discussion, the annotators re-annotated the common articles based on the guidelines and divided the remaining articles amongst themselves. The inter-annotator agreement was calculated for each pair of annotators and the entity taggers were evaluated on the annotated corpus.
Results
Sufficiency vs. Insufficiency Analysis
The results of the sufficiency vs. insufficiency analysis are given in Table 1. 64% of all GenBank records extracted for this project contained insufficient geographic information. Amongst the seven studied viruses, WEE had the highest and EEE had the lowest percentage of insufficient records.
Table 1.
Percentage of GenBank records with insufficient geographic information for each virus.
| Submission Type | Number of Entries | % Insufficient |
|---|---|---|
| WEE | 67 | 90 |
| Rabies | 4450 | 85 |
| WNV | 1084 | 79 |
| SLE | 141 | 74 |
| Hanta | 1745 | 66 |
| Influenza | 51734 | 62 |
| EEE | 374 | 51 |
| All | 59595 | 64 |
Inter-rater Agreement
The results for the comparison of the annotations performed by our three annotators on 6 common papers can be found in Table 3 of Appendix B. We used both the Jaccard Similarity and the harmonic mean of the intersection between two annotators divided by the total number annotated by each as a measure of inter-rater agreement and had over 90% agreement with overlap matching and over 86% agreement with exact matching in all cases.
Performance Analysis of NER Systems
The performance metrics for the NER systems at tagging the desired entities in the test set are listed in Table 2. The highest performance was achieved by Stanford SUTime for date tagging. Tagging of genes had the lowest performance.
Table 2.
Performance statistics of the integrated NER system
| Entity | Precision (Exact;Overlap) |
Recall (Exact;Overlap) |
F-measure (Exact;Overlap) |
|---|---|---|---|
| GeneName | 0.070; 0.239 | 0.114; 0.395 | 0.087; 0.297 |
| Location | 0.452; 0.626 | 0.658; 0.783 | 0.536; 0.696 |
| Species | 0.853; 0.962 | 0.563; 0.658 | 0.678; 0.781 |
| Date | 0.800; 0853 | 0.681; 0.727 | 0.736; 0.785 |
Discussion
Based on our analysis, at least half of the GenBank records for each virus lack sufficient geographic information and the proportion of insufficient records can be as high as 90%. The virus with the highest level of sufficiency, WEE, had a large number of records with county level information in the “country” field. However, the insufficient records for this virus typically contained no place mention, not even at the country level. A key reason for our calculated percentage of sufficient GenBank records being higher for these seven viruses than what we previously computed in Scotch et al.6 was the inclusion of the “strain” field. The “strain” field often contained specific location information which, when combined with place mentions present within the “country” field, made the record geographically sufficient. The virus for which the inclusion of “strain” field had the greatest impact on boosting the sufficiency percentage was influenza. Most of the GenBank records associated with this virus had structured “strain” fields from which the parser could easily separate place mentions using GeoNames.
Although the sufficiency classifications produced by our system were correct most of the time, there were a few cases where a record got incorrectly labeled as insufficient even when it contained detailed geographic information. This typically happened because GeoNames failed to return matching results for these places. For instance, the country field “India: Majiara, WB” was not found to be sufficient even though Majiara is a city in India because GeoNames has no entry for it. In some cases the lack of matching result was due to spelling variations of the place name. For instance the country field “Indonesia: Yogjakarta” was called insufficient since “Yogjakarta” is spelled as “Yogyakarta” in GeoNames. Sometimes the database simply did not contain the exact string present in the GenBank record. For instance, it does not have any entry for the place “south Kalimantan” but it contains the place name “kalimantan”. Errors due to inexact matching were greatly mitigated by removing strings such as “south” from the place mention, as described in the “Methods” section.
Most of the NER systems performed significantly better with overlap measures than with exact-match measures. This is because our annotation guidelines typically involved tagging the longest possible match for each entity and the automated systems frequently missed portions of each annotation. Stanford SUTime had the best overlap f-measure of 0.785, closely followed by LINNEAUS with an overlap f-measure of 0.781. Although Stanford SUTime was fairly effective at finding date mentions in text, it tagged all four-digit-numbers such as “1012” and “2339” as years, leading to a number of false positives. The poor recall of LINNEAUS was mostly caused because the dictionary used by LINNEAUS tagged only species mentions in text while we tagged genus and family mentions as well. It also missed a lot of commonly used animal names such as monkey, bat, badger and wolf. GeoNamer was the third best performer with the highest recall but second lowest precision. This is because the GeoNames dictionary contains an extensively large list of location names, many of which are commonly used words such as “central”. Even though we filtered out a vast majority of these words, it still produced false positives such as “wizard”. However, its performance was considerably better than that of the Stanford location tagger we used in our last paper. BANNER performed the worst amongst all the entity taggers. This is primarily because of the differences between the data set used to train the BANNER model and the annotation corpus used to test our system. The journal articles we selected had a large number of tables and BANNER was not able to identify the gene mentions in them. Instead, it tagged several entries within the table as a single gene name. It also incorrectly tagged strings in all capital letters such as VEEV and H1N1 as gene names. As a result, BANNER had both poor recall and precision.
Although this study explores the problem of insufficient geographic information in GenBank more thoroughly than our prior paper6, expanding the number of viruses that we included in our survey and loosening the definition of sufficiency to account for additional metadata present in the “strain” field, the number of papers annotated as the gold standard increased, but is still limited. Thus, the performance of the taggers reported can be construed as a preliminary estimate at best. The set of taggers and their performance seem to be adequate for a large-scale application, with the exception of the gene tagger. However, we did not make any changes to the BANNER system (specifically, re-training). We had used it before and we had already identified limitations, but changes to it are not possible until sufficient data is annotated for retraining.
Conclusion
It can be concluded that the majority of GenBank records for zoonotic viruses do not contain sufficient geographic information concerning their origin. In order to enable phylogeographic analysis of these viruses and thereby monitor their spread, it is essential to develop an efficient mechanism for extracting this information from published articles. Automated NER systems may help accelerate this process significantly. Our results indicate that the NER systems LINNEAUS, Stanford SUTime and GeoNamer produce satisfactory performance in this domain and thus can be used in the future for linking GenBank records with their corresponding geographic information. However, the current version of BANNER is not well-suited for this task. We will need to train BANNER specifically for this purpose before incorporating it within our system.
Acknowledgments
Research reported in this publication was supported by the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R56AI102559 to MS and GG. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Appendix A. Figure
Figure 1.
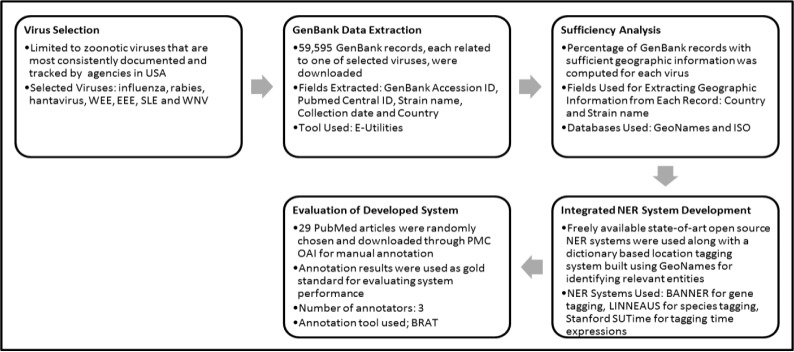
Flowchart of experiment procedure
Figure 2.
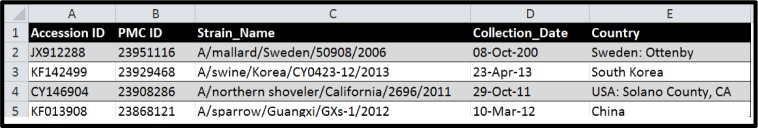
Screenshot of data automatically extracted from GenBank
Figure 3.
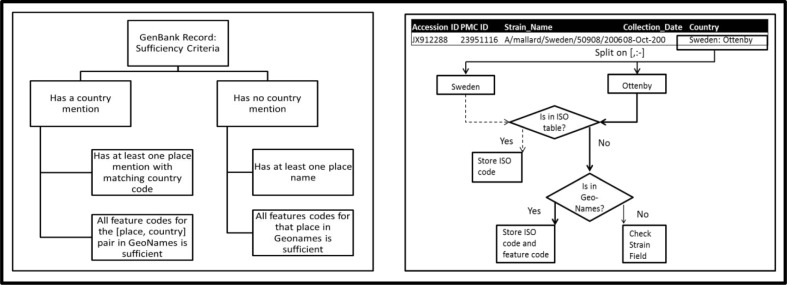
Description of Sufficiency criteria for GenBank record
Appendix B. Annotation Data
Table 1.
Entity type frequency table
| Entity | Annotator A | Annotator B | Annotator C |
|---|---|---|---|
| Date | 386 | 387 | 390 |
| GeneName | 230 | 209 | 208 |
| Location | 846 | 846 | 903 |
| Organism | 916 | 866 | 850 |
| Virus | 1037 | 994 | 1031 |
Table 2.
Additional inter-rater agreement measurements
| Entity |
(Exact;Overlap) |
(Exact;Overlap) |
(Exact;Overlap) |
(Exact;Overlap) |
(Exact;Overlap) |
(Exact;Overlap) |
|---|---|---|---|---|---|---|
| Date | .977; .979 | .974; .977 | .984; .992 | .974; .982 | .966; .977 | .959; .969 |
| GeneName | .870; .880 | .962; .976 | .870; .887 | .962; .981 | .909; .952 | .913; .957 |
| Location | .943; .961 | .948; .961 | .939; .962 | .879; .901 | .944; .966 | .885; .905 |
| Organism | .885; .930 | .935; .984 | .843; .906 | .902; .976 | .906; .950 | .924; .968 |
| Virus | .932; .938 | .972; .979 | .945; .963 | .951; .969 | .965; .973 | .930; .938 |
Table 3.
Inter-rater agreement measurements (H.M. = Harmonic Mean, J.S. = Jaccard Similarity).
| Entity | H.M.(
,
) (Exact;Overlap) |
H.M. (
,
) (Exact;Overlap) |
H.M. (
,
) (Exact;Overlap) |
J.S. (A,B) (Exact;Overlap) |
J.S. (A,C) (Exact;Overlap) |
J.S.(B, C) (Exact;Overlap) |
|---|---|---|---|---|---|---|
| Date | .975; .978 | .979; .987 | .962; .973 | .952; .957 | .950; .965 | .928; .947 |
| GeneName | .914; .926 | .913; .932 | .911; .954 | .845; .868 | .840; .872 | .837; .913 |
| Location | .945; .961 | .907; .931 | .914; .935 | .897; .925 | .831; .871 | .841; .877 |
| Organism | .909; .956 | .874; .940 | .915; .959 | .833; .916 | .792; .905 | .843; .922 |
| Virus | .952; .958 | .947; .966 | .947; .955 | .907; .920 | .903; .937 | .900; .914 |
| Mean | .939; .956 | .924; .951 | .930; .955 | .887; .917 | .863; .910 | .870; .914 |
Appendix C. Annotation Schema
Entity Labels: Date, Location, Organism, Virus, GeneName
Entity Description and Tagging Guideline:
General
Non-entity specific guidelines for tagging
Instructions for tagging: When we have a group of words that collectively can be tagged as one entity, but individually mean another, then tag the group and the individual words separately. For example, “Eastern equine encephalitis” should be tagged as a virus, and separately “equine” should be tagged as an organism.
The components of a multi-word entity should not be tagged if the resulting tags represent the multi-word entity minus descriptive words. For example “fruit bat” should be tagged just as “fruit bat” rather than “fruit bat” and “bat” and “South Africa” should only be tagged “South Africa” rather than “South Africa” and “Africa”.
Strings that may represent multiple entity types should be tagged as all representative entities if the meaning is unclear in the eyes of the annotator. For example the string “Fort Morgan virus” would be tagged as “Fort Morgan” and “Fort Morgan Virus” and the entity types would be Location and Virus respectively. However, if the text were to simply mention “Fort Morgan” without being followed by the string “virus” it cannot be assumed that this then is a Location entity. If the context does not make it abundantly clear which entity type it represents, then it would be tagged as all reasonable entity types.
Lastly, entity types should be identified based on their meaning within the text. For example, “tree” in “phylogenetic tree” would not be tagged as an organism and “Monte Carlo” in “Markov Chain Monte Carlo” would not be tagged as a location.
Date
Any date that identifies a decade or any time quantification more specific than that. Examples include “1970s”; “Jan 2013”; “June 16th, 1973”.
Instructions for tagging: For a specific date mention, group every adjacent component of the date into one tag. For example, “Jun 16th, 1973” should NOT be tagged in components “Jun”, “16th”, and “1973”. One tag should cover the entire date.
Special cases: In the cases of 10+ years ago, if a term of uncertainty is included before it (eg roughly, about) then do not tag it. Otherwise include “#### years ago” as a tag. If the reference to a date includes confidence intervals relating to the origin of speciation, for example “1,000 to 1,500 years ago,” then related dates should not be tagged.
If a range of dates is given, such as “1942 to 1952”, they should be tagged as separate entities. In the case of “May/June 1986,” this would be tagged as one date, otherwise information would be lost.
Location
Any named geographic location. Continents, countries, states, provinces, regions, territories, counties, named lakes, named mountain ranges, named deserts, named bodies of water, etc. General terms such as “the river”, “swamplands”, “in mountains” that cannot be used to identify specific locations should not be tagged.
Instructions for tagging: Include all parts of a location (as long as it provides more information) in the tag. For example, “upstate New York” and “South Lebanon” should include “upstate” and “South” in their tags.
Special cases: In cases like “Central and South America” tag the whole string as one entity, unless they could still be reasonably identified separately (Central has no specific meaning without America). Cases where a city and state are listed as [City, State] (or similar case), tag the city and state separately. Zip codes should be tagged as separate locations.
Organism
Any specific non-viral organism mentioned in the text. Broad terms such as “animals” should not be tagged.
Instructions for tagging: Include descriptive words attached to organism mentions if the descriptive word adds value beyond that which is implied. Such words may describe the region it’s from, if it’s domestic, or other types. Examples include “Canadian duck”, or “domestic poultry”. Examples of words that would not add value include “wild lion”.
Genus-species names should also be tagged (separate from its generic name). Abbreviations for organisms such as “gp” for guinea pig should be tagged as separate entities. If there is a mention of a genus or family name in text, this should be tagged as an organism separately from the common name of the organism.
Special cases: Different words addressing humans should be tagged: soldier, girl, boy, he, she, etc. Examples of words we would not tag are “trees” in reference to phylogenetic trees, or “host” without any descriptive information.
Virus
Any entity recognized as a virus by the annotators such as “West Nile virus”, “Flanders virus” or “Western equine encephalitis”. This also includes any abbreviations indicated in text to represent this virus such as “rabies”, (“RV”). All virus families, genus and subspecies should also be tagged as a virus.
Instructions for tagging: Tag all components of the virus name as one tag. This includes the word virus, except for commonly identified viruses such as “rabies” and “influenza”.
Special Cases: Words describing the virus should not be included as part of the tag, with the exception of “avian influenza” and “swine influenza”.
GeneName
Any entity recognized as a gene by the annotators. Some examples include “E1 envelope glycoprotein” gene, “N” gene, and “Matrix” gene.
Instructions for tagging: Include all components of the gene in a single tag, excluding the word gene. If an entity is tagged as a GeneName entity earlier in the text, and the context does not clearly indicate that the entity is no longer referring to a gene, then it should be tagged as a gene as well.
Appendix D. Description of Data Sources and Software
A number of freely available databases and open source systems were used to complete this project. This section provides a concise overview of each of these resources.
ISO 3166-1 alpha-2 Table
ISO (International Organization for Standardization) is the principal designer of voluntary International Standards in the world. The standard developed by them for representing countries is called ISO 3166-1. This standard allows representation of country names by a two-letter code (alpha-2), a three-letter code (alpha-3) and a three-digit numeric code (numeric-3) respectively. The table of alpha-2 country codes is freely available for non-commercial purposes and currently contains codes for 249 distinct countries.
GeoNames Database
The GeoNames database is a freely available, manually curated database that contains geospatial data for over 10 million locations on earth. Some of its most important data sources include National Geospatial-Intelligence Agency’s (NGA) and the U.S. Board on Geographic Names, U.S. Geological Survey Geographic Names Information System and Ordnance Survey OpenData. Users also have the option to manually edit and add places using a wiki interface. Each location in this database is assigned a specific feature code corresponding to one of nine distinct feature classes. The database also contains the ISO country code of the country where each place is located.
BANNER
BANNER is an open-source named-entity recognition system, designed principally for biomedical text. It is implemented using conditional random fields and incorporates a rich feature set consisting primarily of orthographic, morphological and shallow-syntax features. It also provides users with the option of including a dictionary. BANNER was evaluated using 5x2 cross validation on the training corpus for BioCreative 2 Gene Mention Task and found to perform better than two of the existing, freely-available, state-of-the-art systems, ABNER and LingPipe, with a precision, recall and f-measure of 82.39%, 76.21% and 79.18% respectively. It is currently one of the most widely cited NER systems in biomedical literature for gene tagging.
LINNEAUS
LINNEAUS is a well-known, open-source species name identification and normalization system which uses a dictionary-based approach. The species dictionary used by this system was constructed using the NCBI taxonomy which contains 386,108 species names along with 116,557 genera and other higher-order taxonomic units. Different heuristics were used for successful disambiguation of overlapping mentions. The system was evaluated on various corpora by comparing both document level tags (such as MESH tags) and mention level tags (tags indicating the exact location of each mention) with those produced by LINNEAUS. On a manually annotated corpus of 100 full-text PubMed articles, LINNEAUS had a recall and precision of 94% and 97% respectively at mention level.
Stanford SUTime
SUTime is a temporal tagger created by the Stanford Natural Language Processing Group. It uses regular expressions to recognize and normalize time expressions and includes TIMEX3 tags in its annotations. TIMEX3 is a part of TimeML, a widely used formal specification language for events and temporal expressions. SUTime was evaluated on the TemEval-2 Task and found to be the best performer in the recognition of temporal expression extents with a token level precision, recall and f-measure of 0.88, 0.96 and 0.92 respectively.
References
- 1.Krauss H. Zoonoses : infectious diseases transmissible from animals to humans. 3rd. Washington, D.C.: ASM Press; 2003. [Google Scholar]
- 2.Avise John C. Phylogeography : the history and formation of species. Cambridge, Mass.: Harvard University Press; 2000. [Google Scholar]
- 3.Ciccozzi M, et al. Epidemiological history and phylogeography of West Nile virus lineage 2. Infection, Genetics and Evolution. 2013;17:46–50. doi: 10.1016/j.meegid.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 4.Gray RR, Salemi M. Integrative molecular phylogeography in the context of infectious diseases on the human-animal interface. Parasitology-Cambridge. 2012;139:1939–1951. doi: 10.1017/S0031182012001102. [DOI] [PubMed] [Google Scholar]
- 5.Weidmann M, et al. Molecular phylogeography of tick-borne encephalitis virus in Central Europe. Journal of General Virology. 2013;94:2129–2139. doi: 10.1099/vir.0.054478-0. [DOI] [PubMed] [Google Scholar]
- 6.Scotch Matthew, et al. Enhancing phylogeography by improving geographical information from GenBank. Journal of biomedical informatics. 2011;44:S44–S47. doi: 10.1016/j.jbi.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian Phylogeography Finds Its Roots. PLoS Comput Biol. 2009;5(9):e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drummond AJ, et al. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012 Aug;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bordogna G, Ghisalberti G, Psaila G. Geographic information retrieval: Modeling uncertainty of user’s context. Fuzzy Sets and Systems. 2012;196:105–124. [Google Scholar]
- 10.Conway M, Doan S, Kawazoe A, Collier N. Classifying disease outbreak reports using n-grams and semantic features. International journal of medical informatics. 2009;78:e47–e58. doi: 10.1016/j.ijmedinf.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Doan S, Vinh NTN, Phuong TM. Classifying Vietnamese disease outbreak reports with important sentences and rich features. Proceedings of the Third Symposium on Information and Communication Technology. 2012 Aug;:23–24. 260–265. [Google Scholar]
- 12.Iso.org. [Internet] Genève: c2013. [cited 2013 Oct 10] Available from http://www.iso.org/iso/home/standards/country_codes.htm. [Google Scholar]
- 13.Geonames.org. [Internet] Egypt: c2013. [updated 2013 Apr 30; cited 2013 Sep 26] Available from http://www.geonamesorg/EG/administrative-division-egypt.html. [Google Scholar]
- 14.Gerner M, Nenadic G, Bergman CM. LINNAEUS: A species name identification system for biomedical literature. BMC Bioinformatics. 2010;11(85) doi: 10.1186/1471-2105-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leaman R, Gonzalez G. BANNER: An executable survey of advances in biomedical named entity recognition. Pacific Symposium on Biocomputing. 2008;13:652–663. [PubMed] [Google Scholar]
- 16.Chang AX, Manning CD. SUTime: A Library for Recognizing and Normalizing Time Expressions.
- 17.Stenetorp P, et al. BRAT: A Web-based Tool for NLP-Assisted Text Annotation. EACL ’12 Proceedings of the Demonstrations at the 13th Conference of the European Chapter of the Association for Computational Linguistics. 2012:102–107. [Google Scholar]