

Abstract
Background
Caroli's disease (CD) is a rare congenital disorder. The early diagnosis of the disease and differentiation of types I and II are of extreme importance to patient survival. This study was designed to review and discuss observations in 30 patients with CD and to clarify the clinical characteristics of the disease.
Methods
The demographic and clinical features, laboratory indicators, imaging findings and pathology results for 30 patients with CD were reviewed retrospectively.
Results
Caroli's disease can occur at any age. The average age of onset in the study cohort was 24 years. Patients who presented with symptoms before the age of 40 years were more likely to develop type II CD. Approximately one-third of patients presented without positive signs at original diagnosis and most of these patients were found to have type I CD on pathology. Anaemia, leucopoenia and thrombocytopoenia were more frequent in patients with type II than type I CD. Magnetic resonance cholangiopancreatography (MRCP) and computed tomography (CT) examinations were most useful in diagnosing CD.
Conclusions
No typical symptoms, signs or laboratory indicators are able to distinguish CD from other conditions. Both MRCP and CT were most valuable in diagnosis. The two types of CD may be differentiated by age of onset and routine blood tests.
Introduction
Caroli's disease (CD), also known as communicating cavernous ectasia or congenital saccular dilatation of the intrahepatic bile ducts, is a rare congenital disorder first specifically described in 1958.1 It is characterized by multifocal segmental dilatation of the intrahepatic bile ducts, involving the entire liver, a lobe or a single segment, while these ducts remain connected with the main duct system.2–5 The disease is also characterized by the formation of intraductal calculi and susceptibility to infection.1
Two types of CD were later recognized. Type I, or simple CD, consists of pure cystic dilatations of the intrahepatic bile ducts, whereas type II, or complex CD, also known as Caroli's syndrome (i.e. CD with congenital hepatic fibrosis), is associated with hepatic fibrosis, or even cirrhosis, portal hypertension and oesophageal varices.6 Type II CD may be accompanied by cholangiocarcinoma, calculi of the intrahepatic duct, cholangitis, pancreatic cyst, renal cystic disease or medullary sponge kidney.7 It has even been regarded as precancerous and patients with type II CD are reported to have a 2.5–17.5% likelihood of developing cholangiocarcinoma,8 which is 100-fold higher than in subjects with normal hepatobiliary ducts and 10-fold higher than in those with calculi.9 However, CD has no specific symptoms or signs, which makes diagnosis difficult. Its incidence is extremely low at one case per 1 000 000 people.10 The present study was conducted to clarify the clinical characteristics of this disease through a retrospective review of symptoms, signs, laboratory indicators, imaging results and pathologic findings in 30 patients with CD treated at the study institution.
Materials and methods
Between January 2009 and May 2013, 30 patients with CD were admitted to the 302 Hospital, Beijing. Patient data were collected from the patients' medical records and covered: demographic characteristics, including age, sex, age at onset of CD, and family history; clinical symptoms and signs; laboratory indicators, including results of routine blood tests and biochemical markers; findings on imaging modalities, including ultrasonography, computed tomography (CT), magnetic resonance cholangiopancreatography (MRCP), magnetic resonance imaging (MRI) and endoscopic retrograde cholangiopancreatography (ERCP); pathology results, and details of any associated conditions. The diagnosis of CD was based on imaging results obtained in the study hospital and was confirmed by histopathology. All patients were evaluated by the same pathologist.
All statistical analyses were performed using spss for Windows Version 13.0 (SPSS, Inc., Chicago, IL, USA). Categorical variables were analysed using the chi-squared test or logistic regression, and continuous variables were reported as the mean or median, as appropriate. A P-value of <0.05 was considered to indicate statistical significance.
Results
Demographic features
The 30 patients with CD included 11 males and 19 females (male : female ratio: 1 : 1.7) with a mean age of 24 years (range: 3 months to 82 years). Of these patients, eight were aged 0–6 years, seven were aged 7–17 years, nine were aged 18–40 years, five were aged 41–65 years, and one was aged > 66 years. Patients who presented with symptoms before the age of 40 years were significantly more likely to have type II than type I CD (P = 0.028) (Table 1). Of these 30 patients, only three (10.0%) had a family history of liver cirrhosis or variceal haemorrhage.
Table 1.
Age profile of the 30 patients with Caroli's disease (CD) types I and II
| Age, years | |||||
|---|---|---|---|---|---|
| 0–6 | 7–17 | 18–40 | 41–65 | > 66 | |
| Patients with type I CD, n | 3 | 1 | 1 | 3 | 1 |
| Patients with type II CD, n | 5 | 6 | 8 | 2 | 0 |
Histopathology
Liver biopsy samples were taken from all 30 patients. The type of histopathology performed depended on the presence or absence of hepatic fibrosis and portal hypertension. Type I, or simple CD, is characterized by saccular or cystic dilatation of the segmental intrahepatic bile ducts, with the duct walls lined by hyperplastic or ulcerated biliary epithelium. The colour and texture of the hepatic parenchyma are normal, although fibroplasia is present in the dilated bile ducts, which contain inflammatory cell infiltrates (Fig. 1). Type II CD, or Caroli's syndrome, is characterized by hepatic and dense portal fibrosis, along with dilated bile ducts. The fibrotic regions contain variable numbers of abnormally shaped bile ducts and hypoplastic portal vein branches, and the hepatic parenchyma is subdivided by the overgrowth of portal fibrous tissue, with no evident parenchymal regenerative activity (Fig. 2). In addition, secondary cirrhosis may be found in patients with more severe type II CD.11
Figure 1.
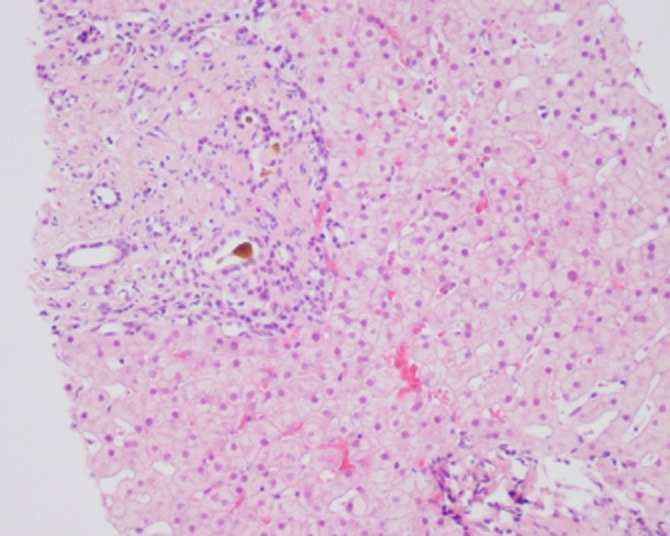
Histopathology of a liver biopsy from a patient with type I Caroli's disease shows cystic dilatation of the segmental intrahepatic bile ducts. (Haematoxylin and eosin stain; original magnification ×10)
Figure 2.
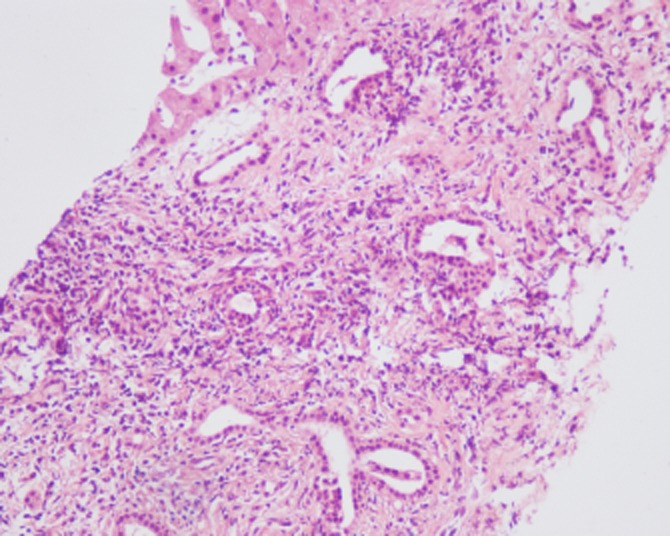
Histopathology of a liver biopsy from a patient with type II Caroli's disease shows hepatic and dense portal fibrosis, along with dilated bile ducts. (Haematoxylin and eosin stain; original magnification ×10)
Of the 30 patients, nine were diagnosed with type I CD, including three who also had chronic cholangitis, and 21 were diagnosed with type II CD, including 14 with cirrhosis and two with biliary cirrhosis.
Presentations
Symptoms
The most frequent initial symptoms of the disease in the present patients included right upper quadrant pain (n = 14, 46.7%), fever (n = 11, 36.7%), anorexia (n = 8, 26.7%), variceal bleeding (haematemesis, melaena or haematochezia; n = 8, 26.7%), fatigue (n = 6, 20.0%), and jaundice (n = 5, 16.7%). Of the 11 patients with fever, only three (27.3%) had low-grade fever (<38 °C), whereas the other eight (72.7%) had high fever (39–40 °C). Less frequent symptoms included diarrhoea in one patient, and nausea and vomiting, skin itch, and oedema of the lower limbs in two patients each. Four patients were completely asymptomatic and were diagnosed during a health examination conducted for other reasons (Table 2). There were no differences in the frequency of symptoms between patients with types I and II CD (P > 0.05).
Table 2.
Clinical symptoms and signs in the 30 patients with Caroli's disease
| n (%) | |
|---|---|
| Symptoms | |
| Right upper quadrant pain | 14 (46.7%) |
| Fever | 11 (36.7%) |
| Anorexia | 8 (26.7%) |
| Variceal bleeding | 8 (26.7%) |
| Fatigue | 6 (20.0%) |
| Jaundice | 5 (16.7%) |
| None | 4 (13.3%) |
| Nausea and vomiting | 2 (6.7%) |
| Itch of skin | 2 (6.7%) |
| Oedema of lower limbs | 2 (6.7%) |
| Diarrhoea | 1 (3.3%) |
| Signs | |
| None | 10 (33.3%) |
| Hepatosplenomegaly | 8 (26.7%) |
| Ascites | 5 (16.7%) |
| Jaundice | 5 (16.7%) |
| Hepatomegaly | 4 (13.3%) |
| Splenomegaly | 4 (13.3%) |
| Liver palm | 2 (6.7%) |
| Oedema of lower limbs | 1 (3.3%) |
| Lymphadenectasis | 1 (3.3%) |
Signs
On physical examination, 10 patients (33.3%) presented with no signs of CD at the time of first diagnosis; this was more common in type I patients (P = 0.011). Common signs in the present series included hepatosplenomegaly (n = 8, 26.7%), shifting dullness or jaundice (n = 5, 16.7%), and hepatomegaly or splenomegaly (n = 4, 13.3%) (Table 2). There were no differences in the frequency of signs between patients with types I and II CD (P > 0.05).
Laboratory indicators
Findings showed that laboratory indicators were able to distinguish between the two types of CD. A review of the complete blood counts (CBCs) of the 30 patients, including their white blood cell (WBC), red blood cell (RBC) and platelet counts, showed that 13 (43.3%) patients had pancytopoenia, five (16.7%) had anaemia, and one (3.3%) had both leucopoenia and thrombocytopoenia, whereas 11 (36.7%) had normal blood cell counts. Pancytopoenia (P = 0.020), anaemia (P = 0.002) and combined leucopoenia and thrombocytopoenia (P = 0.011) were significantly more frequent in patients with type II than type I CD.
Liver function and electrolytes were also reviewed, including concentrations of albumin, total bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), total bile acids (TBA), γ-glutamyl transpeptidase (GGT), cholinesterase, and potassium (K+), sodium (Na+) and chloride (Cl−) ions. Decreased albumin, cholinesterase and K+ were observed in 12 (40.0%), 15 (50.0%) and seven (23.3%) patients, respectively, and mildly elevated total bilirubin, ALT, AST, ALP, GGT and TBA in 13 (43.3%), 14 (46.7%), 14 (46.7%), 12 (40.0%), 11 (36.7%) and five (16.7%) patients, respectively. The frequency of marker abnormalities, however, did not differ between patients with type I and those with type II CD (P > 0.05) (Table 3).
Table 3.
Laboratory indicators in the 30 patients with Caroli's disease (CD)
| Laboratory indicators | Patients with type I CD (n = 9), n | Patients with type II CD (n = 21), n |
|---|---|---|
| Complete blood counts | ||
| Pancytopoenia | 1 | 12 |
| Anaemia | 1 | 4 |
| Leucopoenia and thrombocytopoenia | 0 | 1 |
| Normal blood cells | 7 | 4 |
| Liver function | ||
| Decreased albumin | 3 | 9 |
| Elevated alanine aminotransferase | 6 | 8 |
| Elevated aspartate aminotransferase | 5 | 9 |
| Elevated total bilirubin | 5 | 8 |
| Elevated alkaline phosphatase | 4 | 8 |
| Elevated γ-glutamyl transpeptidase | 4 | 7 |
| Elevated total bile acids | 2 | 3 |
| Decreased cholinesterase | 5 | 10 |
| Electrolytes | ||
| Decreased potassium (K+) | 4 | 3 |
| Abnormal sodium (Na+) | 0 | 0 |
| Abnormal chloride (Cl−) ions | 0 | 0 |
Imaging
Of the 30 patients, 22 were examined by ultrasound, 14 by CT, 13 by MRCP, and nine by MRI. The accuracies of these imaging modalities in diagnosing CD were 27.3%, 71.4%, 84.6% and 77.8%, respectively (Table 4), and showed no differences between the two types of CD. Imaging diagnosed cirrhosis in 18 patients, most of whom had type II CD (P = 0.006), which suggests its value in differentiating between the two types.
Table 4.
Results of imaging examinations in the 30 patients with Caroli's disease (CD)
| Imaging modality | Patients examined, n | Patients diagnosed with CD by this modality, n | Accuracy, % |
|---|---|---|---|
| Ultrasound | 22 | 6 | 27.3% |
| Computed tomography | 14 | 10 | 71.4% |
| Magnetic resonance cholangiopancreatography | 13 | 11 | 84.6% |
| Magnetic resonance imaging | 9 | 7 | 77.8% |
Associated conditions
Various conditions were associated with CD. For example, 14 (46.7%) patients had cirrhosis, 11 (36.7%) had renal cysts, 10 (33.3%) had hepatic cysts and seven (23.3%) had cholangiolithiasis. In addition, three (10.0%) patients had cholangitis and three (10.0%) had polycystic kidney, two (6.7%) had polycystic liver, one (3.3%) had pancreatitis, one (3.3%) had septicaemia and one (3.3%) had medullary sponge kidney.
Discussion
Caroli's disease is a rare, autosomally recessive inherited congenital disorder associated with an incomplete and faulty remodelling of the embryonic ductal plate.12–15 It is usually diagnosed during childhood or adolescence,16 but may be diagnosed in adulthood.17,18 Patients who present with symptoms of CD before the age of 40 years are more likely to have type II than type I CD. No gender predominance has been observed,19 including in the present study.
Clinically, CD has no distinguishing symptoms or signs. Patients may remain asymptomatic or may experience symptoms infrequently throughout life.9 Intrahepatic ductal ectasia predisposes to stagnation of the bile, which may lead to the formation of stones and predispose to infections such as cholangitis.10,16 The most frequent symptoms are right upper quadrant pain, fever and jaundice. In the present series 72.7% of patients had a high fever of 39–40 °C. Patients may also present with anorexia and fatigue, often accompanied by severe infections such as abscesses or septicaemia. Repeated cholangitis predisposes to calculi of the intrahepatic ducts, which may aggravate bile obstruction and lead to biliary cirrhosis, as observed in two of the present patients. Variceal bleeding and ascites have also been observed in patients with type II CD. Although no differences in signs or symptoms between patients with types I and II CD were observed in the present study, patients with type I were more likely to lack any positive signs.
The diagnosis of CD and differentiation between the two types depend on laboratory, imaging and pathology examinations. Although differences between laboratory indicators were observed, they were relatively non-specific. Pancytopoenia, anaemia, and leucopoenia and thrombocytopoenia were more likely in type II than in type I patients, thereby differentiating between these two types of CD. Histopathologic examination can yield a definitive diagnosis, but liver biopsy is invasive and can cause injuries. Thus, the diagnosis of CD relies mostly on imaging modalities, such as ultrasound, CT, MRI and MRCP. In addition, the present authors found that imaging was able to differentiate type II CD successfully when the patient developed cirrhosis.
On ultrasound, CD appears as intrahepatic cystic anechoic areas, which may include fibrovascular bundles, stones and linear bridging or septa. The fibrovascular bundles are composed of portal veins and hepatic arteries, as shown by Doppler ultrasonography.9 Despite the ability of ultrasound to visualize liver cysts and possible intrahepatic lithiasis and to provide information on the common bile duct, it remains difficult to differentiate cysts arising from CD from liver cysts arising from other conditions, such as polycystic liver disease.20 In the present study, ultrasound was found to have an accuracy of 27.3%, which is much lower than those of other imaging modalities. Computed tomography scans may show central dots (Fig. 3a) in CD patients, representing a sign that is specific and essential, and indicates fibrovascular bundles containing the portal vein radical and a branch of the hepatic artery bridging the saccule.20 However, central dots may not always be present as the accuracy of CT was found to be 71.4%. Magnetic resonance imaging was not more advantageous than CT and had an accuracy of 77.8%. The highest level of sensitivity in the diagnosis of CD was associated with ERCP,21 which suggests that this method can evaluate the entire biliary tree while also identifying any intrahepatic masses.22 Imaging by MRCP may show the connections between the secular ectasias and the normal biliary tract (Fig. 3b), thus establishing a diagnosis of CD and ruling out other conditions such as multiple liver abscesses and polycystic liver disease.20 There are also substantial additional therapeutic benefits to be gained by using ERCP to assess preoperative transient recovery from acute episodes of cholangitis.23 However, because it is invasive and carries a risk for infection, ERCP is superfluous and should not be performed in patients with established diagnoses.
Figure 3.
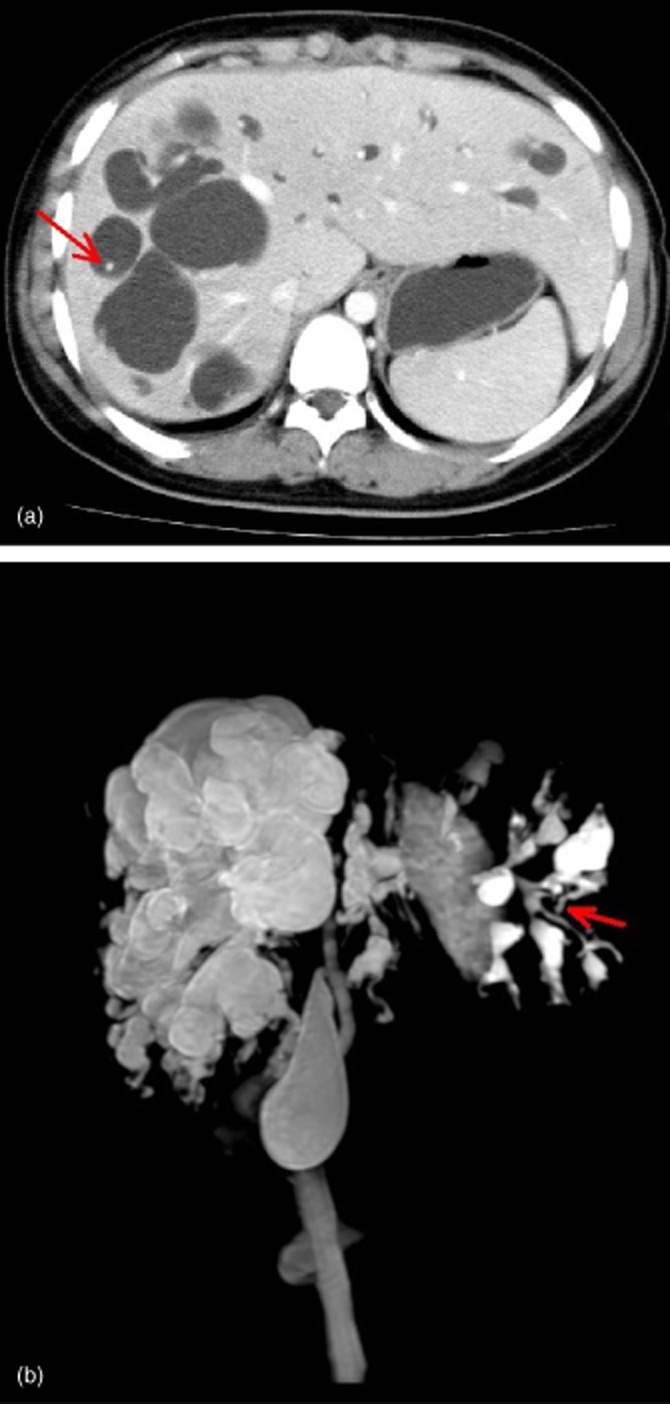
(a) Computed tomography in a 16-year-old girl with Caroli's disease showing central dotting (arrow). (b) Magnetic resonance cholangiopancreatography in the same patient shows the connection between the secular ectasias and the normal biliary tract (arrow)
The treatment of patients with CD depends on the clinical features of the disease and the locations of biliary abnormalities. Localized forms, involving either the left or the right half of the liver, should be treated by hemi-hepatectomy, following obstruction of the biliary tract or cholangitis. Diffuse involvement of both lobes can be treated conservatively, including with endoscopic therapy and an internal biliary bypass procedure,24 but patients with such involvement are often difficult to manage. Liver transplantation may be the optimal management option in these patients, even if they are asymptomatic.20 Survival after liver transplantation was found to be poor in patients with congenital hepatic fibrosis (Caroli's syndrome) and in those with cholangitis at the time of orthotopic liver transplantation.25 Although a recent multicentre study on the longterm results of the surgical management of patients with either Caroli's disease or syndrome showed encouraging results in terms of postoperative mortality and 5-year overall survival, the poor prognosis and the sizeable morbidity and mortality rates after liver transplantation still suggest that the timely recognition of indications for surgical treatment is of major importance.26 Thus, the early diagnosis of the disease and differentiation between types I and II are of extreme importance to patient survival.
Conclusions
Caroli's disease is an inherited disorder that may cause severe, life-threatening cholangitis or hepatobiliary degeneration, or even cancer.27 Despite its rare incidence, CD should not be forgotten in the differential diagnosis of patients with recurrent cholangitis. The onset of CD can occur at any age and in either sex, although patients who present with symptoms before the age of 40 years were found to be more likely to have type II CD, or Caroli's syndrome. There are no specific symptoms or signs with which to distinguish the two types of CD, although positive signs are infrequently encountered in type I patients during physical examinations. Laboratory findings cannot be relied upon for diagnosis, although reduced blood cell counts suggest type II CD. The imaging modalities most useful in the diagnosis of CD are MRCP and CT.
Conflicts of interest
None declared.
References
- Caroli J, Soupault R, Kossakowski J, Plocker L, Paradowska E. Congenital polycystic dilation of the intrahepatic bile ducts, attempt at classification. Sem Hop. 1958;34:488–495. In French. [PubMed] [Google Scholar]
- Guy F, Cognet F, Dranssart M, Cercueil JP, Conciatori L, Krause D. Caroli's disease: magnetic resonance imaging features. Eur Radiol. 2002;12:2730–2736. doi: 10.1007/s00330-002-1471-6. [DOI] [PubMed] [Google Scholar]
- Levy AD, Rohrmann CA, Jr, Murakata LA, Lonergan GJ. Caroli's disease: radiologic spectrum with pathologic correlation. AJR Am J Roentgenol. 2002;179:1053–1057. doi: 10.2214/ajr.179.4.1791053. [DOI] [PubMed] [Google Scholar]
- Miller WJ, Sechtin AG, Campbell WL, Pieters PC. Imaging findings in Caroli's disease. AJR Am J Roentgenol. 1995;165:333–337. doi: 10.2214/ajr.165.2.7618550. [DOI] [PubMed] [Google Scholar]
- Fulcher AS, Turner MA, Sanyal AJ. Case 38: Caroli disease and renal tubular ectasia. Radiology. 2001;220:720–723. doi: 10.1148/radiol.2203000825. [DOI] [PubMed] [Google Scholar]
- Caroli J. Diseases of the intrahepatic biliary tree. Clin Gastroenterol. 1973;2:147–161. [PubMed] [Google Scholar]
- Karim AS. Caroli's disease. Indian Pediatr. 2004;41:848–850. [PubMed] [Google Scholar]
- Taylor AC, Palmer KR. Caroli's disease. Eur J Gastroenterol Hepatol. 1998;10:105–108. doi: 10.1097/00042737-199802000-00001. [DOI] [PubMed] [Google Scholar]
- Wu KL, Changchien CS, Kuo CM, Chuah SK, Chiu YC, Kuo CH. Caroli's disease – a report of two siblings. Eur J Gastroenterol Hepatol. 2002;14:1397–1399. doi: 10.1097/00042737-200212000-00019. [DOI] [PubMed] [Google Scholar]
- Ozlem Y, Yusuf B. Clinical characteristics of Caroli's disease. World J Gastroenterol. 2007;13:1930–1933. doi: 10.3748/wjg.v13.i13.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Ren XS, Nakanuma Y. Caroli's disease: current knowledge of its biliary pathogenesis obtained from an orthologous rat model. Int J Hepatol. 2012;2012:107945. doi: 10.1155/2012/107945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini G, Mandrioli L, Morini M. Bile duct dysplasia and congenital hepatic fibrosis associated with polycystic kidney (Caroli syndrome) in a rat. Vet Pathol. 2003;40:693–694. doi: 10.1354/vp.40-6-693. [DOI] [PubMed] [Google Scholar]
- Torra R, Badenas C, Darnell A, Brú C, Escorsell A, Estivill X. Autosomal dominant polycystic kidney disease with anticipation and Caroli's disease associated with a PKD1 mutation. Rapid communication. Kidney Int. 1997;52:33–38. doi: 10.1038/ki.1997.300. [DOI] [PubMed] [Google Scholar]
- Parada LA, Hallén M, Hägerstrand I, Tranberg KG, Johansson B. Clonal chromosomal abnormalities in congenital bile duct dilatation. Gut. 1999;45:780–782. doi: 10.1136/gut.45.5.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Harada K, Kizawa K, Sanzen T, Furubo S, Yasoshima M, et al. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli's disease. Am J Pathol. 2005;166:49–60. doi: 10.1016/S0002-9440(10)62231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Tommaso AM, Santos DS, Hessel G. Caroli's disease: 6 case studies. Acta Gastroenterol Latinoam. 2003;33:47–51. [PubMed] [Google Scholar]
- Desmet J. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- Hussman KL, Friedwald JP, Gollub MJ, Melamed J. Caroli's disease associated with infantile polycystic kidney disease. Prenatal sonographic appearance. Ultrasound Med. 1991;10:235–237. doi: 10.7863/jum.1991.10.4.235. [DOI] [PubMed] [Google Scholar]
- Madjov R, Chervenkov P, Madjova V, Balev B. Caroli's disease. Report of 5 cases and review of literature. Hepatogastroenterology. 2005;52:606–609. [PubMed] [Google Scholar]
- Kassahun WT, Kahn T, Wittekind C, Mossner J, Caca K, Hauss J, et al. Caroli's disease: liver resection and liver transplantation. Experience in 33 patients. Surgery. 2005;138:888–898. doi: 10.1016/j.surg.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Rickes S, Neye H, Wirth J, Ortner M, Lochs H, Wermke W. Improved accuracy in the diagnosis of intrahepatic bile duct ectasia in Caroli's disease by combination of ultrasound and endoscopic retrograde cholangiography. Ultraschall Med. 2000;21:223–225. doi: 10.1055/s-2000-7988. [DOI] [PubMed] [Google Scholar]
- Totkas S, Hohenberger P. Cholangiocarcinoma associated with segmental Caroli's disease. Eur J Surg Oncol. 2000;26:520–521. doi: 10.1053/ejso.1999.0936. [DOI] [PubMed] [Google Scholar]
- Chawla YK, Sharma BC, Dilawari JB. Endoscopic nasobiliary drainage in acute suppurative cholangitis. Indian J Gastroenterol. 1993;12:97–98. [PubMed] [Google Scholar]
- Desmet VJ. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol. 2004;19:356–360. (Suppl.): [Google Scholar]
- Habib S, Shakil O, Couto OF, Demetris AJ, Fung JJ, Marcos A, et al. Caroli's disease and orthotopic liver transplantation. Liver Transpl. 2006;12:416–421. doi: 10.1002/lt.20719. [DOI] [PubMed] [Google Scholar]
- Mabrut JY, Kianmanesh R, Nuzzo G, Castaing D, Boudjema K, Létoublon C, et al. Surgical management of congenital intrahepatic bile duct dilatation, Caroli's disease and syndrome: longterm results of the French Association of Surgery Multicentre Study. Ann Surg. 2013;258:713–721. doi: 10.1097/SLA.0000000000000269. [DOI] [PubMed] [Google Scholar]
- De Kerckhove L, De Meyer M, Verbaandert C, Mourad M, Sokal E, Goffette P, et al. The place of liver transplantation in Caroli's disease and syndrome. Transpl Int. 2006;19:381–388. doi: 10.1111/j.1432-2277.2006.00292.x. [DOI] [PubMed] [Google Scholar]