

Abstract
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) is one of the most common hereditary forms of stroke, and migraine with aura, mood disorders and dementia. CADASIL is caused by mutations of the NOTCH3 gene. This mutation is inherited as an autosomal dominant trait. Most individuals with CADASIL have a parent with the disorder. In extremely rare cases, CADASIL may occur due to a spontaneous genetic mutation that occurs for unknown reasons (de novo mutation). We report a new case of patient with de novo mutation of the NOTCH3 gene and a condition strongly suggestive of CADASIL (migraine, stroke, and white matter abnormalities), except that this patient did not have any first-degree relatives with similar symptoms.
KEY WORDS: CADASIL, NOTCH3 gene, de novo mutation, CT
INTRODUCTION
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an autozomal dominantly inherited form of microangipathy manifested by recurrent strokes, migraine with aura, mood disorders, vascular dementia and premature death [1-4]. CADASIL is caused by mutations within the epidermal growth factor-like repeat domain of the NOTCH3 gene located on chromosome içp13. Most individuals with CA-DASIL have a family history. In extremely rare cases, CA-DASIL may occur due to a spontaneous genetic mutation that occurs for unknown reasons (de novo mutation). There are only two cases of de novo mutation reported in the literature [5,6]. We report a new case of patient with de novo mutation of the NOTCH3 gene and a condition strongly suggestive of CADASIL, except that this patient did not have any first-degree relatives with similar symptoms.
CASE REPORT
Forty seven years old female patient, known with CADASIL, was admitted to the Department of Neurology with altered consciousness near to coma and irregular breathing and increased temperature. Brain CT after the admission showed a characteristic appearance of CADASIL with abnormalities, such as bilateral, supratentorial confluent zones of low attenuations affecting periventricular and sub-cotrical white matter, anterior temporal lobes and external capsulas as well as multiple small infarcts in basal ganglia, thalamus, and pons (Figure 1). The first clinical signs of CADASIL appeared since she was twenty years old with episodes of migraine, depression, hemiparesis and verbal fluency problems. With the development of the disease, symptoms become more intense and more frequent. Past medical history was not remarkable for stroke risk factors such as hypertension, diabetes mellitus, heart disease, or hyperlipidemia. Both parents were alive and healthy, with negative history for stroke, migraine, and dementia. The skin biopsy performed five years ago revealed findings consistent with CADASIL – granular osmiophilic material within the basement membrane surrounding the smooth muscle cells of the small-sized and medium sized vessels on electron microscopy. Afterward, molecular analysis showed a mutation in the NOTCH3 gene confirming CADASIL. The patient died 4 days after the admission of the pneumonia.
FIGURE 1.
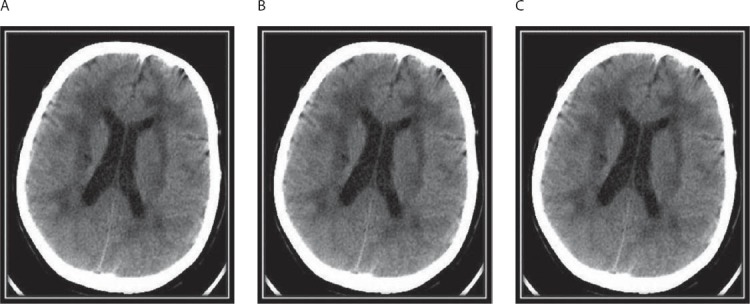
CT scan showing multiple paraventricular and deep white matter hypodensities (A,B), lacunar infarcts in the left nucleus lentiformis (B) and the pons (C), hypodensities in the capsula externa and anterior temporal lobe (C).
DISCUSSION
CADASIL is an autosomal dominant vascular disorder clinically characterized by a variety of symptoms including migraine with aura, mood disorder, vascular dementia, ischemic stroke and premature death [1-4]. The vascular lesions underlying CADASIL are a non-arteriosclerotic, non-amyloid arteriopathy affecting small perforating cerebral arteries with accumulating of granular and osmiophilic materials within the vascular smooth muscles cell (VSMC) basement membrane. This initial thickening and expansion of the extracellular matrix can be found to a lesser extent in extra cerebral arteries such as skin arterioles. VSMC gradualy undego degeneration with mural fibrosis resulting in stenosis of the distal segment of medullaty arteries. Cerebral vessels loosing the ability to autoregulate and functionally becoming blood pressure dependant [2]. These changes leading to small infarcts in the white matter, deep gray matter and the pons. CADASIL is caused by mutations within the epidermal growth factor-like repeat domain of the NOTCH3 gene located on chromosome 19p13. NOTCH3 gene is essential for the normal maturation of blood vessels in both fetal and adult brains in mammals. There is a large spectrum of CADASIL mutations. More than 150 different mutation in 18 exon have been already described [2, 8, 9]. In European population exon 4 is the most frequently involved site of NOTCH3 gene mutation followed by exon 8 and 3 [10]. Genetic analysis remains the diagnostic gold standard of the diseases but it is expensive and may be false negative if only a cluster of most probably exons is examined [11].
Regarding neuroimaging findings in CADASIL, CT hypodense and T2 MR hyperintense lesions in subcortical white matter apear regularly in CADASIL patients, but the list of differential diagnoses is large comprising other causes of vascular involvement and inflammatory diseases [11]. Typical findings are T2 MR hyperintensities and CT hypodensities in the temporopolar white matter and the capsula externa. Lesions of the anterior temporal lobe seem to be very specific for CADASIL, but their occurrence is not the rule, especially in the Asian CADASIL population. These lesions best relate to the numerous fluid-filled perivascular spaces or increased number of perivascular spaces rather than the presence of lacnar infarcts [7]. We found specific hypodense lesions in the anterior temporal lobe and the external capsule (Figure 1). Currently, the clinical diagnosis of CADASIL is based on the satisfaction of the following conditions: (1) development of the condition at a relatively young age (40-50 years), (2) no risk for stroke, (3) repeated attacks of lacunar infarction with gradual progression to pseudobulbar paralysis and dementia (migraine, emotional disturbance, cerebral infarction, and dementia in 30%, 20%, 85%, and 30-90% of patients, respectively), and (4) family members with similar symptoms (autosomal dominant inheritance) [3]. The diagnosis is also established on the basis of the following findings of imaging and laboratory studies: the presence of (1) leukoaraiosis and multiple small infarcts in the bilateral deep white matter, basal ganglia, thalamus, and pons revealed by MRI; (2) granular and osmiophilic materials around the vascular smooth muscles in the brain, skeletal muscle, peripheral nerves, and skin demonstrated by electron microscopy; and (3) NOTCH3 mutations revealed by DNA analysis [3]. Our patient fulfilled all diagnostic criteria except of early beginning of the disease in her twenties and she did not have any first-degree relatives with similar symptoms. None of the family members had a history of typical risk factors for microangiopathy such as hypertension or diabetesthat migth impact clinical manifestation. The absence of prior manifestation of disease in family refers to de novo mutation, which is extremely rare. The history of describing a CADASIL began in 1955 and until now were registrated only two patients with de novo mutations. The exact prevalence of CADASIL is not known [4]. The estimated prevalence of CADASIL in Scotland is approximately 1 in 50,000 adults [12]; however, this estimate cannot be generalized worldwide. Patients with NOTCH3 gene mutation have been reported in different ethnic groups [13, 14]. However, because the genetic test for CADASIL was not available before 2000, many cases were misdiagnosed as multiple sclerosis, Alzheimer’s disease, or other neurodegenerative diseases. As de novo mutations of the NOTCH3 gene are very rare, reports of their occurrence help in finding common characteristics that may give some light to the etiology and pathogenesis of CADASIL. Our opinion coincides with opinion from other authors, which consider that the absence of positive family history does not deny the diagnosis of CADASIL, which means that this disorder can be more oftener than is actual known, bearing in mind probability that many patients with existing symptomatology but without positive family history, are not subjects of further, particularly, genetic diagnostic methods. Lethal result usually comes 12-23 years after symptomatology starts. Pneumonia is the most frequent cause of death, followed by sudden unexpected death and asphyxia.
CONCLUSION
We report a new case of de novo mutation of NOTCH3 gene, as an extremely rare cause of CADASIL. The absence of positive family history does not deny the diagnosis of CADASIL, and these patients should be subjects of further, particularly, genetic diagnostic methods.
DECLARATION OF INTEREST
The authors declared no conflicts of interest.
REFERENCES
- [1].Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. CADASIL. Lancet Neurol. 2009;8:643–653. doi: 10.1016/S1474-4422(09)70127-9. [DOI] [PubMed] [Google Scholar]
- [2].Hervé D, Chabriat H. CADASIL. J Geriatr Psychiatry Neurol. 2010;23:269–276. doi: 10.1177/0891988710383570. [DOI] [PubMed] [Google Scholar]
- [3].Uchino M. The pathomechanism and treatment of CADASIL. Rinsho Shinkeigaku. 2011;51:945–948. doi: 10.5692/clinicalneurol.51.945. [DOI] [PubMed] [Google Scholar]
- [4].Choi JC. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalpoathy: a genetic cause of small vessel disease. J Clin Neurol. 2010;6:1–9. doi: 10.3988/jcn.2010.6.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Joutel A, Dodick DD, Parisi JE, Cecillon M, Tournier-Lasserve E, Bousser MG. De novo mutation in the Notch3 gene causing CA-DASIL. Ann Neurol. 2000;47:388–391. [PubMed] [Google Scholar]
- [6].Coto E, Menéndez M, Navarro R, Garcia-Castro M, Alvarez V. A new de novo Notch3 mutation causing CADASIL. Eur J Neurol. 2006;13:628–631. doi: 10.1111/j.1468-1331.2006.01337.x. [DOI] [PubMed] [Google Scholar]
- [7].Yamamoto Y, Ihara M, Tham C, Low R, Slade J, Moss T, et al. Neuropathological correlates of temporal white matter hyperintensities in CADASIL. Stroke. 2009;40:2004–2011. doi: 10.1161/STROKEAHA.108.528299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee YC, Liu CS, Chang MS. Population-specific spectrum of NOTCH3 mutations, MRI features and founder effect of CADASIL in Chinese. J Neurol. 2009;256:249–255. doi: 10.1007/s00415-009-0091-3. [DOI] [PubMed] [Google Scholar]
- [9].Mignarri A, Martini G, Malandrini A, Bianchi S, Tassi R, Federico A, et al. First report of Iraqi Kurdish CADASIL patient. Neurol Sci. 2011;32:359–360. doi: 10.1007/s10072-010-0399-x. [DOI] [PubMed] [Google Scholar]
- [10].Federico A, Bianchi S, Dotti MT. The spectrum of mutations for CADASIL diagnosis. Neurol Sci. 2005;26:117–124. doi: 10.1007/s10072-005-0444-3. [DOI] [PubMed] [Google Scholar]
- [11].Peisker T, Musil L, Hrebicek M, Vlaskova H, Cihelkova I, Bartos A. Clinical spectrum in CADASIL family with a new mutation. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2013;157:379–382. doi: 10.5507/bp.2013.055. [DOI] [PubMed] [Google Scholar]
- [12].Razvi SS, Davidson R, Bone I, Muir KW. The prevalence of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalpoathy (CADASIL) in the West Scotland. J Neurol Neurosurg Psychiatry. 2005;76:739–741. doi: 10.1136/jnnp.2004.051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shahien R, Bianchi S, Bowirrat A. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy in an Israeli family. Neuropsychiatr Dis Treat. 2011;7:383–390. doi: 10.2147/NDT.S19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vyshka G, Kruja J. Clinical variability of the cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalpoathy phenotype in two simblings of the large family showing the same mutation. International Medical Case Reports Journal. 2013;6:59–63. doi: 10.2147/IMCRJ.S51875. [DOI] [PMC free article] [PubMed] [Google Scholar]