

Abstract
Gene expressionis controlled by epigenetic mechanisms including histone methylation. Osteoclasts are bone-resorptive cells that differentiate from hematopoietic-precursor cells by receptor activator of nuclear factor-KB ligand (RANKL) stimulation. Although BIX01294, a specific inhibitor of G9a, which works as a histone H3 lysine 9 (H3K9) methyltransferase, reportedly changes cellular differentiational stage, its effect on osteoclast differentiation is unclear. In this study, the effects of BIX01294 on osteoclast differentiation were examined. Here, we showed that BIX01294 dose-dependently reduced RANKL-induced tartrate-resistant acid phosphatase positive multinuclear osteoclast-like cell differentiation from murine macrophage-like Raw264.7 cells. During differentiation, growth rates reduced only less than 14% of those of cells stimulated with RANKL alone by BIX01294 treatment. Moreover, western blot analysis showed that BIX01294 reduced RANKL-induced carbonic anhydrase II and cathepsin K production and decreased RANKL-induced nuclear factor of activated T-cell c1, a master regulatory transcription factor, production during osteoclast differentiation. These results suggest that BIX01294 suppresses RANKL-induced osteoclast differentiation. This is the first report about the effect of BIX01294 on osteoclast differentiation.
KEY WORDS: osteoclast differentiation, BIX01294, histone methyltransferase, Raw264.7 cell
INTRODUCTION
Bone remodelling is a dynamic process performed by osteoblast and osteoclast. The balance between the amount of bone matrix produced by osteoblasts and bone resorption by osteoclasts is essential for proper bone metabolism. This process is tightly regulated by various hormones and cyto-kines in the local microenvironments, and its impairment can lead to bone diseases [1]. Multinuclear giant osteoclast cells are formed by the differentiation of hematopoietic precursor cells, which shear a common progenitor with macrophages and dendritic cells [2]. Osteoclasts secrete proton and extracellular matrix-digestive enzymes into the resorptive pit, subsequently resulting in bone resorption [3]. An excessive number or an increased function of osteoclasts is responsible for bone resorption during rheumatoid arthritis [4], bone metastasis in certain types of cancers [5, 6], and periodontitis [7]. Therefore, it is very important to study the underlying mechanism for controlling osteoclast development and maturation. Post-translational modification of histones is implicated to play a role in various processes involving gene expression and silencing [8-10]. One of the most studied post-translational modifications of histone is acetylation of lysine residues on histone tails [11, 12]. Histone acetyltransferases acetylate specific lysine residues of histones and activate gene expressions by weakening the tight interaction between positively charged histones and negatively charged DNA [11, 13, 14]. In contrast, histone deacetylases (HDACs) cause histone hypo-acetylation and subsequently induce the affinity between histones and DNA, thereby resulting in transcriptional silencing [11, 13, 14]. Recent studies have shown that HDAC inhibitors suppress osteoclastgenesis in rodent bone marrow cells [15-17] and human peripheral blood mononuclear cells [15]. These reports support the existence of epigenetic regulation during osteoclast differentiation. Another well-characterized histone modification is the methylation of lysine residues of histone [11]. Lysine methylation predominantly occurs in histones H3 and H4 and elicits specific effects depending on the site and degree of methylation: methylation at H3 lysine 4 (H3K4), H3K36, and H3K79 is generally associated with actively transcribed genes, whereas that at H3K9, H3K27, and H4K20 frequently demarcates silent chromatin [18-20]. In addition, methylation of H3K9 (hereafter referred to H3K9me) has a stronger gene repression effect than that of H3K27me [18-20]. Yasui et al. [21] have reported the involvement of H3K27me in osteoclast differentiation. However, there are no reports on the involvement of H3K9 methylation in osteoclast differentiation. In the present study, we demonstrated the effect of H3K9 methylation status on osteoclast differentiation by using BIX01294, a specific inhibitor of G9a that specifically methylates H3K9 [22].
MATERIALS AND METHODS
Cell culture
Murine macrophage-like Raw264.7 cells, obtained from Dainippon Pharmaceutical (Osaka, Japan), were maintained in α-minimal essential medium (α-MEM, Wako Pure Chemical Industries, Osaka, Japan) containing 10% (v/v) heat-inactivated fetal bovine serum (FBS, Bio-Fill, Elsternwick, Australia) supplemented with 100 U/ mL penicillin and 100 μg/mL streptomycin (Wako) at 37°C in a humidified and 5% CO2 atmosphere.
Osteoclast formation and tartrate-resistant acid phosphatase (TRAP) staining
For osteoclast differentiation, Raw264.7 cells were cultured in 96-well plates at a density of 2 × 103 cells/well. After overnight incubation, the media was replaced with 10% FBS α-MEM containing 50 ng/mL of recombinant mouse receptor activator of nuclear factor-κB ligand (RANKL, also called ODF, TNSF11, or TRANCE; R&D systems, Minneapolis, MN) in the absence or presence of BIX01294 (Wako), and the cells were cultured for 4 days at 37°C. Cells were fixed with 4% paraformaldehyde, stained for tartrate-resistant acid phosphatase (a marker enzyme of osteoclasts) activity as described elsewhere, and the plates were scanned using GT-X800 (EPSON, Suwa, Japan). TRAP-positive multinuclear cells which have more than 4 or more nuclei were counted. These cells were also microscopically observed using the IX70 microscope (Olympus, Tokyo, Japan). Each experiment have done for more than three times.
SYTOX green cell growth assay
Cell-growth rates were measured as an increased amount of cellular DNA using SYTOX green dye. The SYTOX green dye is not cell permeable but is penetrate through cellular membrane and intercalated into genomic DNA when cellular membrane integrity is broken. The intercalated dye emits strong green fluorescent when it is excited. Two thousands of Raw264.7 cells were seeded into each well of 96-well black plate which has transparent bottom (NUNC, Roskilde, Denmark). After overnight incubation, cells were treated with or without 50 mg/mL mouse recombinant RANKL and several concentrations of BIX01294. The plates were frozen at -80°C for more than 24 h to break cellular membrane and 100 μL of 400 nM SYTOX Green dye (BioVision, Mountain View, CA, USA) was applied to each well. The intensity of fluorescence was measured using a Wallac ArvoSX1420 spectrofluorometer (Perkin Elmer, Waltham, MA, USA) with an excitation/emission wavelength of 485/535 nm as the amount of cells in the each well.
Western blot analysis
The Raw264.7 cells (1.3 × 105 cells) were seeded in 6-cm dishes incubated overnight at 37°C, and treated with mouse recombinant RANKL (50 ng/mL) for 72 h in the absence or presence of the indicated concentrations of BIX01294. The cells were lysed in ice-cold lysis buffer; 10 μg of total protein was applied to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked using an Ez Block blocking reagent (ATTO, Tokyo, Japan) and then incubated with rabbit anti-carbonic anhydrase II polyclonal antibody (Santa Cruz, Santa Cruz Biotechnology, CA), mouse anti-cathepsin K (Santa Cruz Biotechnology), or anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Santa Cruz Biotechnology) monoclonal antibody at a dilution of 1:1000 for 1 h. The membranes were washed 3 times in tris-buffered saline containing 0.1% Tween 20 (TBS-T, pH7.5) and then incubated with biotin-conjugated goat anti-rabbit or anti-mouse immunoglobulin G (Invitrogen, Carlsbad, CA) diluted at a ratio of 1:10,000 for 1 h. After the membranes were repeatedly washed with TBS-T, they were treated with streptavidin-peroxidase (KPL, Gaithersburg, MD) for 30 min. Protein bands were visualized by chemiluminescence using an ECL luminescence kit (GE Healthcare, Buckinghamshire, UK); the luminescence emitted from the bands was detected using ChemiDoc XRS Plus (Bio-Rad Laboratories, Hercules, CA). For nuclear factor (NF) of activated T-cell c1 (NFATc1) detection, Raw264.7 cells (1.3 × 105 cells) were seeded in 6-cm dishes and treated with mouse recombinant RANKL (50 ng/mL) for 24 h in the absence or presence of indicated concentrations of BIX01294. The cells were lysed in ice-cold lysis buffer, and 4 μg of total protein was applied for western blot analysis, as described above. Mouse anti-NFATc1 (Santa Cruz Biotechnology) or GAPDH monoclonal antibody was used as the primary antibody.
RESULTS
To examine the effect of BIX01294 on RANKL-induced osteoclast differentiation, we first treated murine macro-phage-like Raw264.7 cells with several concentrations of BIX01294 in the presence of 50 ng/mL of mouse recombinant RANKL. As shown in Figure 1A and B, RANKL induced the formation of TRAP-positive multinuclear giant osteoclast-like cells and BIX01294 significantly reduced the number of osteoclast-like cells in a dose-dependent manner. Higher magnified observation also showed that BIX01294 decreased both number and cell size of the os-teoclast-like cells in a dose-dependent manner (Figure 1C). We next examined the effects of BIX01294 on cell-growth rates during RANKL stimulation. As Figure 2 shows, RANKL treatment significantly induced growth rate of Raw 264.7 cell. Although 0.5 μM BIX01294 did not reduce RANKL-induced cell growth, both 1.0 and 1.5 μM of BIX01294 significantly diminished RANKL-induced growth rate of Raw264.7 cells (Figure 2). However, the reduction rate of growth on the cells treated by 1.0 mM or 1.5 mM BIX01294 at 4 days was only 4.8% or 13.2%, respectively. During bone resorption by osteoclast, carbonic anhydrase II, which is localized in the cytoplasm of osteoclasts, produces protons from H2CO3; these protons are secreted into the resorptive cavity through apical H+/ATPase [3].
FIGURE 1.
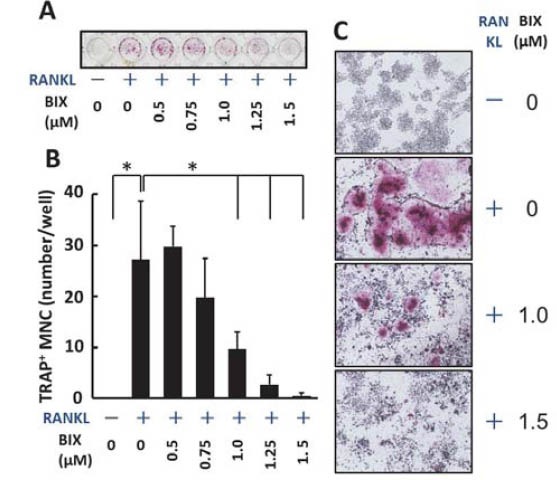
BIX01294 reduced RANKL-induced osteoclast differentiation. (A) Raw264.7 cells in a 98-well plate were treated with recombinant mouse RANKL (50 ng/mL) in the absence or presence of indicated concentrations of BIX01294 for 4 days, and TRAP staining was performed. The plate was scanned using a scanner. (B) TRAP positive multinuclear cells in each well were counted.*, p < 0.01 [student’s t-test] versus RANKL alone treatment. (C) Cells in the several wells were microscopically observed (original magnification x40). BIX, treated with BIX01294; MNC, multinuclear cells.
FIGURE 2.
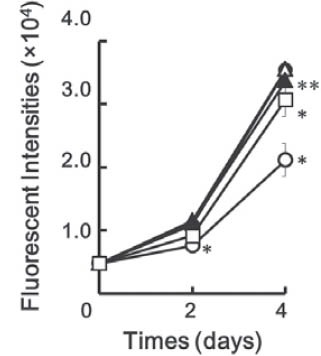
BIX01294 did not so much reduce RANKL-induced growth rate of Raw264.7 cells during differentiation. Raw264.7 cells were cultured in the absence (O) or presence of 50 ng/ mL mouse recombinant RANKL and several concentrations of BIX01294 (•, 0 μM; △, 0.5 μM; ▴, 1.0 μM; □, 1.5 μM) for indicated periods. The plates were frozen at -80°C to break cellular membranes and SYTOX-green dye was applied to each well. Fluorescence emitted from cells in each well was measured by a microplate fluorometer as the amount of cells in the each well. Symbols: *, differences from 0 μL BIX01294 (p < 0.01 [student’s t-test]); **, differences from 0 μL BIX01294 (p < 0.05 [student’s t-test]).
The released protons demineralize the bone matrix. In addition, mature osteoclasts secrete cathepsin K for the digestion of extracellular matrix proteins [3]. We subsequently examined the effect of BIX01294 on the production of carbonic anhydrase II and cathepsin K using western blotanalysis. The treatment of Raw264.7 cells with RANKL for 72 h augmented the production of carbonic anhydrase II and cathepsin K (Figure 3). In contrast, the RANKL treatment did not induce GAPDH (Figure 3). The induction of carbonic anhydrase II and cathepsin K was inhibited by BIX01294 in a dose-dependent manner (Figure 3). RANKL is one of the most important cytokines that stimulates the differentiation of osteoclast precursor cells into osteoclasts [3]. The binding of RANKL to RANK induces the NF-κ light-chain enhancer of activated B cells (NF-κB) and mitogen-associated protein kinase (MAPK) pathways through adaptor molecules such as TNF receptor-associated factor 6 (TRAF6) [23]. The MAPK family of proteins activate the proteins belonging to the activator protein-1 (AP-1) family (such as c-Jun and c-Fos); then, the AP-1 family proteins induce NFATc1 production, which is a master regulator of osteoclast differentiation, in cooperation with activated NF-κB [23]. Therefore, we examined the effect of BIX01294 on RANKL-induced NFATc1 using western blot analysis. RANKL induced NFATc1 production at 24 h, and BIX01294 reduced the induction in a dose-dependent manner (Figure 4).
FIGURE 3.
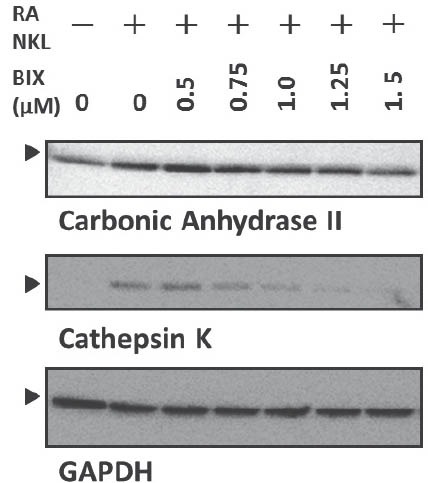
BIX01294 repressed the production of RANKL-induced osteoclast differentiation markers. Raw264.7 cells were treated with mouse recombinant RANKL (50 ng/mL) for 72 h in the absence or presence of indicated concentrations of BIX01294. The cells were lysed in lysis buffer, and 10 μg of total protein was analyzed by western blotting. Glyceraldehyde-3-phospate dehydrogenase (GAPDH) was used as the internal control. BIX, treated with BIX01294. The arrowheads indicate desired bands.
FIGURE 4.
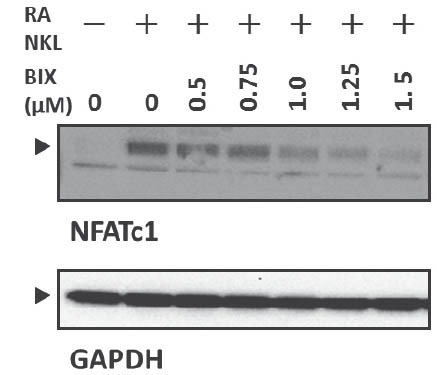
BIX01294 decreased RANKL-induced NFATcl production. Raw264.7 cells were treated with mouse recombinant RANKL (50 ng/mL) for 24 h in the absence or presence of the indicated concentrations of BIX01294. The cells were lysed in lysis buffer, and 4 μg of total protein was analyzed by western blotting. GAPDH was used as the internal control. BIX, treated with BIX01294. The arrowheads indicate desired bands.
DISCUSSION
In the present study, we examine the effect of BIX01294 on osteoclast differentiation of Raw264.7 cells. BIX01294 suppressed RANKL-induced formation of TRAP-positive multinulear giant cells (Figure 1A, B, and C). Production of other osteoclast markers, such as carbonic anhydrase II and cathepsin K, were also dose-dependently decreased by BIX01294 treatment (Figure 3). Although BIX01294 treatment reduced RANKL-induced cell growth rate at the concentration of more than 1.0 mM, the reduction rate was only less than 14% (Figure 2). These data suggested that BIX01294 suppresses osteoclast differentiation. Moreover, BIX01294 treatment dose-dependently depressed NFATc1 induction, which was induced by RANKL stimulation (Figure 4). Thus, it was suggested that BIX01294 inhibited the upstream signalling pathways of NFATc1. At this time, we do not have data showing what molecule or pathway was disturbed by BIX01294 and how BIX01294 was associated with the reduction of RANKL-induced NFATc1 induction. Therefore, further studies are needed to elucidate these aspects. G9a histone methyltransferase catalyzes H3K9 methylation in mammals because disruption of the Gça gene resulted in a drastic decrease in H3K9 methylation [24-27]. As H3K9 is primarily found in the silenced region within the euchromatin, G9a has been implicated to play a role in the silencing of gene expression [28-30]. Consequently, BIX01294 suppresses the silencing of gene expression through G9a inhibition. In this study, we demonstrated that BIX01294 inhibited RANKL-induced osteoclast differentiation via suppression of RANKL-induced NFATc1 production. Thus, we can hypothesize that BIX01294 may upregulate some signalling molecules that suppress RANKL-stimulated NFATc1 induction. However, there is no evidence supporting this hypothesis. Yasui et al. [21] demonstrated that RANKL stimulation upregulated the jumonji domain-containing 3 (jmjd3) gene, and short hairpin RNAs of jmjd3 gene diminished RANKL-induced osteoclast differentiation, suggesting that JMJD3, a H3K27 demeth-ylase, plays an important role in osteoclast differentiation. Moreover, as proof that H3K27 methylation status controls osteoclast genesis, our data showed that H3K9 methylation status also plays an important role in osteoclast differentiation. Therefore, H3K9 and H3K27 methylation might be therapeutic targets for the treatment of bone resorptive diseases.
CONCLUSION
In this study, we examined the effects of BIX01294 on osteoclast differentiation of Raw264.7 cells. BIX01294 reduced RANKL-induced formation of TRAP positive multinuclear osteoclast-like cells in a dose dependent manner. In addition, BIX01294 reduced RANKL-induced production of osteoclast differentiation markers, such as carbonic anhydrase II, cathepsin K, and NFATc1. These results suggest that BIX01294 suppresses osteoclast differentiation on mouse macrophage-like Raw264.7 cells.
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Young Scientists (B21792160) and a Grant-in-Aid for Scientific Research (C23592714) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; an UemuraFund, Nihon University School of Dentistry (2008); a grant from the Dental Research Center, Nihon University School of Dentistry (2008); Sato Fund, Nihon University School of Dentistry (2009); a Strategic Research Base Development Program for Private Universities from Ministry of Education, Culture, Sports, Science and Technology of Japan, 2010-2014 (S1001024); and a Nihon University Joint Research Grant (2012).
DECLARATION OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- [1].Walsh NC, Crotti TN, Goldring SR, Gravallese EM. Rheumatic diseases: the effects of inflammation on bone. Immunol Rev. 2005;208:228–251. doi: 10.1111/j.0105-2896.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- [2].Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocr Rev. 1992;13(1):66–80. doi: 10.1210/edrv-13-1-66. [DOI] [PubMed] [Google Scholar]
- [3].Väänänen HK, Laitala-Leinonen T. Osteoclast lineage and function. Arch Biochem Biophys. 2008;473(2):132–138. doi: 10.1016/j.abb.2008.03.037. [DOI] [PubMed] [Google Scholar]
- [4].Shiozawa S, Tsumiyama K, Yoshida K, Hashiramoto A. Pathogenesis of joint destruction in rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) 2011;59(2):89–95. doi: 10.1007/s00005-011-0116-3. [DOI] [PubMed] [Google Scholar]
- [5].Clézardin P. Therapeutic targets for bone metastases in breast cancer. Breast Cancer Res. 2011;13(2):207. doi: 10.1186/bcr2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sturge J, Caley MP, Waxman J. Bone metastasis in prostate cancer: emerging therapeutic strategies. Nat Rev Clin Oncol. 2011;8(6):357–368. doi: 10.1038/nrclinonc.2011.67. [DOI] [PubMed] [Google Scholar]
- [7].Bartold PM, Cantley MD, Haynes DR. Mechanisms and control of pathologic bone loss in periodontitis. Periodontol 2000. 2010;53:55–69. doi: 10.1111/j.1600-0757.2010.00347.x. [DOI] [PubMed] [Google Scholar]
- [8].Mimura I, Tanaka T, Wada Y, Kodama T, Nangaku M. Pathophysiological response to hypoxia - from the molecular mechanisms of malady to drug discovery: epigenetic regulation of the hypoxic response via hypoxia-inducible factor and histone modifying enzymes. J Pharmacol Sci. 2011;115(4):453–458. doi: 10.1254/jphs.10r19fm. [DOI] [PubMed] [Google Scholar]
- [9].Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108(4):475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- [10].Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- [11].Hamby ME, Coskun V, Sun YE. Transcriptional regulation of neuronal differentiation: the epigenetic layer of complexity. Biochim Biophys Acta. 2008;1779(8):432–437. doi: 10.1016/j.bbagrm.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93(1):57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- [13].Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- [14].Richon VM, O’Brien JP. Histone deacetylase inhibitors: a new class of potential therapeutic agents for cancer treatment. Clin Cancer Res. 2002;8(3):662–664. [PubMed] [Google Scholar]
- [15].Cantley MD, Fairlie DP, Bartold PM, Rainsford KD, Le GT, Lucke AJ, et al. Inhibitors of histone deacetylases in class I and class II suppress human osteoclasts in vitro. J Cell Physiol. 2011;226(12):3233–3241. doi: 10.1002/jcp.22684. [DOI] [PubMed] [Google Scholar]
- [16].Nakamura T, Kukita T, Shobuike T, Nagata K, Wu Z, Ogawa K, et al. Inhibition of histone deacetylase suppresses osteoclastogenesis and bone destruction by inducing IFN-beta production. J Immunol. 2005;175(9):5809–5816. doi: 10.4049/jimmunol.175.9.5809. [DOI] [PubMed] [Google Scholar]
- [17].Rahman MM, Kukita A, Kukita T, Shobuike T, Nakamura T, Ko-hashi O. Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood. 2003;101(9):3451–3459. doi: 10.1182/blood-2002-08-2622. [DOI] [PubMed] [Google Scholar]
- [18].Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- [19].Jenuwein T. The epigenetic magic of histone lysine methylation. FEBS J. 2006;273(14):3121–3135. doi: 10.1111/j.1742-4658.2006.05343.x. [DOI] [PubMed] [Google Scholar]
- [20].Lee DY, Teyssier C, Strahl BD, Stallcup MR. Role of protein methylation in regulation of transcription. Endocr Rev. 2005;26(2):147–170. doi: 10.1210/er.2004-0008. [DOI] [PubMed] [Google Scholar]
- [21].Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, Tanaka S. Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J Bone Miner Res. 2011;26(11):2665–2671. doi: 10.1002/jbmr.464. [DOI] [PubMed] [Google Scholar]
- [22].Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teo-doro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25(3):473–81. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- [23].Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- [24].Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12(6):1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- [25].Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain- containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001;276(27):25309–25317. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- [26].Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a histone methyltransferase plays a dominant role in euchro- matic histone H3 lysine 9 methylation and is essential for early em- bryogenesis. Genes Dev. 2002;16(14):1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;19(7):815–826. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 2008;27(20):2691–2701. doi: 10.1038/emboj.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Ger-son A, Ueda J, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol. 2008;15(5):1176–1183. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol. 2006;8(2):188–194. doi: 10.1038/ncb1353. [DOI] [PubMed] [Google Scholar]