

Abstract
Polycystic ovary syndrome (PCOS) is a highly prevalent endocrine-metabolic disorder that implies various severe consequences to female health, including alarming rates of infertility. Although its exact etiology remains elusive, it is known to feature several hormonal disturbances, including hyperandrogenemia, insulin resistance (IR), and hyperinsulinemia. Insulin appears to disrupt all components of the hypothalamus-hypophysis-ovary axis, and ovarian tissue insulin resistance results in impaired metabolic signaling but intact mitogenic and steroidogenic activity, favoring hyperandrogenemia, which appears to be the main culprit of the clinical picture in PCOS. In turn, androgens may lead back to IR by increasing levels of free fatty acids and modifying muscle tissue composition and functionality, perpetuating this IR-hyperinsulinemia-hyperandrogenemia cycle. Nonobese women with PCOS showcase several differential features, with unique biochemical and hormonal profiles. Nevertheless, lean and obese patients have chronic inflammation mediating the long term cardiometabolic complications and comorbidities observed in women with PCOS, including dyslipidemia, metabolic syndrome, type 2 diabetes mellitus, and cardiovascular disease. Given these severe implications, it is important to thoroughly understand the pathophysiologic interconnections underlying PCOS, in order to provide superior therapeutic strategies and warrant improved quality of life to women with this syndrome.
1. Introduction
Polycystic ovary syndrome (PCOS) is an endocrine-metabolic disorder characterized by multiple hormonal imbalances, reflecting on a clinical presentation dominated by manifestations of hyperandrogenism, which generate short and long term consequences on female health [1]. Among these, infertility is one of the most alarming associated morbidities, as it currently affects approximately 48.5 million women aged 20–44 years [2], with PCOS accounting for 6–15% of these cases [3], although up to 70% of women with PCOS may be undiagnosed [4]. Indeed, its optimal diagnosis is often hindered due to its apparent similarities with several other pathologies remarkably, obesity as well as Cushing's syndrome, ovarian and adrenal neoplasms, and congenital adrenal hyperplasia [5].
The manifestations of PCOS are not confined to the gynecological sphere; women afflicted by this disease show an increased prevalence of several comorbidities, including obesity, dyslipidemia, hypertension, metabolic syndrome (MS), and type 2 diabetes mellitus (DM2) in comparison with women without PCOS. These features, along with other alterations such as endothelial dysfunction and a chronic low-grade inflammatory state, underlie the greater risk of developing cardiovascular disease and increased all-cause mortality observed in these subjects [6].
Amongst the complications previously mentioned, obesity stands out as it has reached epidemic proportions [7], with a worldwide prevalence of 35% in females and up to 55% in South America and the Caribbean [8]. Furthermore, in our locality, 32.4% of women are obese [9]. Indeed, both PCOS and obesity boast highly concerning prevalence figures [10]. Several studies have pinpointed insulin resistance (IR) as the fundamental link associating these conditions [11], although IR may be present in PCOS independently of obesity [12]. IR, defined as a metabolic state characterized by a decrease in cellular ability to respond to insulin signaling, appears to be an essential pathophysiologic mechanism in the development of all metabolic complications of PCOS [13]. As a consequence, outstanding proportions of women with PCOS are also diagnosed with DM2 or MS, as well as isolated criteria from the latter [14]. Compensatory hyperinsulinemia appears to mediate many of these deleterious effects. This phenomenon stems as a response of pancreatic β cells in order to preserve lipid and carbohydrate homeostasis in face of diminished insulin sensitivity [15]. This compensation leads to β cell exhaustion and the genesis of not only DM2, but also a series of collateral effects originated by hyperinsulinemia, including the aforementioned frequent comorbidities of PCOS [16].
Notwithstanding the importance of IR in the development of PCOS, both obese and nonobese patients have specific mechanisms leading to ovarian dysfunction independent of IR, reflecting the complexity of this syndrome [17]. Therefore, considering the severe consequences PCOS exerts on the health and lifestyle of the affected women, it is of utmost importance to unravel the intricate pathophysiologic cross-talk among PCOS, IR, and obesity.
2. The Ovarian Cycle in Polycystic Ovary Syndrome: When It All Goes Wrong
Because no specific sole cause for PCOS has been determined, the most accepted premise is a multifactorial model, where interactions between environmental cues and factors intrinsic to each individual act in consonance toward a common result, which is the development of hyperandrogenemia, a biochemical hallmark of this pathology. This alteration is the main culprit behind most clinical manifestations of PCOS [18].
In PCOS, several of the physiological events within the ovarian cycle and folliculogenesis are disrupted. The very beginning of folliculogenesis is compromised due to high levels of Anti-Müllerian Hormone (AMH) [19]. AMH is a 560 amino acid peptide of the TGF-β family, which is secreted by granulosa cells (GC) and displays its greatest expression in small antral follicles and exerts powerful inhibition of primordial follicle initiation and follicle sensitivity to follicle-stimulating hormone (FSH). AMH levels progressively decrease as follicles increase in size, and low levels of this hormone appear to be a requirement for transition from the primordial to the primary stage, dominant follicle selection, and progression to ovulation [20]. In women with PCOS, elevated levels of AMH appear to play an important role in long term disruption of ovarian physiology [21], with greater AMH concentrations being linked to worse fertility outcomes [22].
Feedback disturbances in the hypothalamus-hypophysis-ovary axis (HHOA) are another typical feature of PCOS [23], with increased frequency and amplitude of gonadotropin-releasing hormone (GnRH) and luteinizing hormone (LH) pulsatile secretion. Higher levels of this hormone induce greater androgen synthesis in ovarian theca cells (TC) [24]. In turn, hyperandrogenemia induces a decrease in feedback sensitivity to both estradiol and progesterone in gonadotropic hypothalamic cells, reinforcing GnRH and LH hypersecretion [25]. This represents the first of many self-perpetuating pathophysiologic cycles in which hyperandrogenemia plays a pivotal role in the development and progression of PCOS, while simultaneously warranting the presence of the clinical manifestations. The constant growth of follicles, along with nonselection of a dominant unit, leads to the hyperstimulation of several of these structures, hence the alternative proposed denomination of “polyfollicular ovary syndrome” [26], which maintains all the characteristic hormonal imbalances.
Genetic factors are also considered to play an important role in the development of this syndrome, by setting the stage for abnormally high androgen synthesis in ovarian tissue. The most accepted model proposes a probable Mendelian pattern of inheritance, where key genetic defects would be transmitted to offspring in a dominant autosomal fashion, albeit with widely variable penetrance, dependent on several environmental and epigenetic factors, such as in utero exposure to elevated levels of androgens [27]. Notoriously, mutations in the genes of the androgen receptor, sex hormone binding globulin (SHBG), and steroidogenic enzymes may be especially important in predisposing to the development of hyperandrogenemia [28].
3. Polycystic Ovary Syndrome: The Clinical Picture
In the clinical setting, PCOS presentation is greatly heterogeneous, with a broad clinical manifestation spectrum (Table 1). Several sets of diagnostic criteria for PCOS are presented in Table 2 [29]. Although the presence of oligomenorrhea indicates ovulatory dysfunction, apparent eumenorrhea does not completely rule out anovulation. To this end, progesterone levels <3-4 ng/mL in days 20–24 of the menstrual cycle are sufficient to diagnose the cycle in question as oligo/anovulatory. In contrast, a patient can be diagnosed as anovulatory after ascertaining anovulation in at least 2 subsequent cycles, in the presence of hypoprogesteronemia [30].
Table 1.
Clinical manifestations and conditions associated with polycystic ovary syndrome.
(a).
| Manifestation | Reference | Sample | Prevalence |
|---|---|---|---|
| Manifestations of hyperandrogenism | |||
|
| |||
| Hirsutism | Marcondes et al. [34] | 73 | 83.8% |
| Baldani et al. [35] | 365 | 73.2% | |
| Jedel et al. [36] | 30 | 73% | |
|
| |||
| Acne | Jedel et al. [36] | 30 | 63% |
| Özdemir et al. [37] | 115 | 53% | |
| Baldani et al. [35] | 365 | 49.6% | |
|
| |||
| Alopecia | Özdemir et al. [37] | 115 | 34.8% |
| Sivayoganathan et al. [38] | 70 | 16% | |
|
| |||
| Seborrhea | Özdemir et al. [37] | 115 | 34.8% |
|
| |||
| Manifestations of ovarian dysfunction | |||
|
| |||
| Oligomenorrhea | Valkenburg et al. [39] | 412 | 74% |
| Baldani et al. [35] | 365 | 69.2% | |
| Jedel et al. [36] | 30 | 20% | |
|
| |||
| Amenorrhea | Valkenburg et al. [39] | 412 | 26% |
| Baldani et al. [35] | 365 | 21.5% | |
| Jedel et al. [36] | 30 | 43% | |
|
| |||
| Ultrasound polycystic ovaries | Baldani et al. [35] | 365 | 97.3% |
| Valkenburg et al. [39] | 412 | 89% | |
| Hahn et al. [40] | 200 | 83% | |
(b).
| Condition | Reference | Sample | Prevalence |
|---|---|---|---|
| Associated conditions | |||
|
| |||
| Obesity | Ehrmann et al. [41] | 394 | 80% |
| Hahn et al. [40] | 200 | 52% | |
| Azziz et al. [42] | 267 | 42% | |
|
| |||
| Insulin resistance | Carmina and Lobo [43] | 267 | 77% |
| Hahn et al. [40] | 200 | 71% | |
|
| |||
| Impaired fasting glucose | Ehrmann et al. [41] | 122 | 35% |
| Legro et al. [44] | 254 | 31.1% | |
|
| |||
| Type 2 diabetes mellitus | Legro et al. [44] | 254 | 7.5% |
| Ehrmann et al. [41] | 394 | 6.6% | |
|
| |||
| Arterial hypertension | Ehrmann et al. [41] | 394 | 21% |
| Elting et al. [45] | 346 | 9% | |
|
| |||
| Dyslipidemia | Hahn et al. [40] | 200 | 46.3% |
| Ehrmann et al. [41] | 394 | 32% | |
|
| |||
| Metabolic syndrome | Dokras et al. [46] | 129 | 47.3% |
| Apridonidze et al. [13] | 106 | 43% | |
|
| |||
| Mood disorders | Jedel et al. [36] | 30 | 53% |
| Hollinrake et al. [47] | 103 | 21% | |
All studies cited were carried out in adults.
Table 2.
Diagnostic criteria for polycystic ovary syndrome.
| Clinical or biochemical hyperandrogenism | Oligo/anovulation | US finding of polycystic ovaries∗ | |
|---|---|---|---|
| NIH, 1990 Both of the following: |
+ | + | |
|
| |||
| ESHRE/ASRM, 2003 Only 2 of the following: |
+ | + | + |
|
| |||
| AES, 2006 All 3 of the following: |
+ | + | + |
NIH: National Institute of Health of the United States; ESHRE: European Society of Human Reproduction and Embryology; ASRM: American Society of Reproductive Medicine; AES: Androgen Excess and PCOS Society.
All sets of criteria require the exclusion of other etiologies such as congenital adrenal hyperplasia, androgen-secreting neoplasms, and Cushing's síndrome.
*Ultrasound polycystic ovaries can be defined as the presence of ≥12 follicles of 2–9 mm width or an increase in ovarian volume (>10 mL) in at least one ovary, in women not consuming oral contraceptives.
Although IR is not traditionally considered a diagnostic criterion of PCOS, the presence of this alteration or acanthosis nigricans accompanied by hyperandrogenic signs is highly suggestive of this syndrome [31]. In women with PCOS, obesity is also a very prevalent condition, along with IR. Nevertheless, these may not necessarily be concomitant in all cases, and they may be present independently, translating into distinct metabolic profiles. Each of these phenotypes displays specific biochemical features that render differential profiles, both for cardiovascular risk and fertility outcomes [32].
Because skin is a major target for androgen activity, several hyperandrogenemia-triggered dermatologic alterations can be seen in PCOS, most commonly hirsutism, androgenic alopecia, and acne and also seborrhea, onycholysis, and onychorrhexis [33].
3.1. Hirsutism
Hair development is thoroughly affected by hyperandrogenemia. Normally, in females past pubarche the major androgenic molecules are dehydroepiandrosterone sulfate (DHEAS), androstenedione, dehydroepiandrostenedione, testosterone, and dihydrotestosterone (DHT), in descending order of serum concentration. Only the latter two can bind to the androgen receptor and promote hair follicle changes [48, 49]. Hirsutism is defined as the presence of excessive terminal hair in areas of the body that are androgen-dependent and usually hairless or with limited hair growth, such as the face, chest, areolas, abdomen, and upper thighs [50]. Terminal hair should be differentiated from vellus hair, whereas the latter is a longer version of lanugo (the hair that covers fetuses and is shed postnatally) and covers all body surfaces but the lips, palms, and soles; terminal hair is the pigmented, longer, coarser hair that covers the pubic and axillary areas, scalp, eyelashes, eyebrows, and male body and facial hair [48]. Terminal hair development requires androgen stimulation—as seen in pubarche, where androgens trigger vellus to mature into terminal hair [51]—and thus, hirsutism can be seen as the result of the interaction hyperandrogenemia and its influence in the hair follicle unit [52]. It should be differentiated from hypertrichosis [53], which is the overgrowth of vellus in a nonsexual pattern distribution, usually related to persistence of the highly mitotic anagen phase of the hair cycle [54].
Although hyperandrogenemia is the trigger for hirsutism, the rate of hair growth is not proportional to the degree of hyperandrogenism [52], supporting a parallel role for androgen receptor localization (keratinocytes, sebaceous glands, and hair dermal papilla cells) and sensitivity in the development of hair patterns and other skin manifestations, such as acne, alopecia, or seborrhea. Hirsutism can be assessed through the Ferriman-Gallwey Score [55], which evaluates the presence of terminal hair in the upper lip, chin, chest, upper and lower back, upper and lower abdomen, thighs, and arms. Integrated scores from all body areas beyond 15 points are related to a hirsutism diagnosis, although current recommendations suggest the use of the 95th percentile of the score in specific populations, adapting to autochthonous ethnic, hair pattern, and age-related features, in order to diagnose hirsutism properly [52]. Due to the high prevalence of PCOS, all cases of hirsutism during puberty or in women in reproductive age merit determination of sex hormone serum concentrations, as well as pelvic ultrasonography, in order to rule out this syndrome [30].
3.2. Acne and Seborrhea
Sebaceous glands are also androgen-dependent structures, with sebocytes being highly sensitive to androgen signaling, which is exacerbated in PCOS, leading to the development of acne and seborrhea [56]. Androgens stimulate sebocyte proliferation—especially in the mid-back, forehead, and chin—and secretion of sebum, a mixture of lipids including glycerides, squalene, free fatty acids (FFA), and cholesterol [57]. Local bacteria further complicate the process by secreting lypolytic enzymes which break down triglycerides produced in the sebocyte. The resulting FFA that are released into sebaceous ducts by apocrine glands are responsible for the characteristic odor observed in these patients [58].
3.3. Androgenic Alopecia
On the other hand, androgenic alopecia is a disorder in which hair is miniaturized, due to an increased telogen : anagen ratio—with telogen hair being at mitotical rest and anagen hair being mitotically active—and associated to genetic susceptibility related to increased 5α-reductase activity in the hair follicle. This increased enzymatic activity would favor the local conversion of testosterone into DHT, a more powerful androgen [59]. As reported by Cela et al. [60] 67% of the women with alopecia areata have PCOS and elevated levels of testosterone and androstenedione. The balding pattern is dominated by the frontal and parietal scalp zones, leaving the occipital area with great hair density, as opposed to thinner and scarcer hair in the crown area. A variation of this disorder features neither bald spots nor hair loss per se, but only shortening in its length and width, manifesting as wispy distal ends [59].
3.4. Onycholysis and Onychorrhexis
Finally, nails are also subject to possible alterations in PCOS, in the form of onycholysis—separation of the nail plate from the nail bed due to disruption of the onychocorneal band [61]—and onychorrhexis, splitting of nails in lengthway bridges [62]. The association of these nail conditions with excess androgen is not fully understood, but their presence has been observed to be exacerbated when coexisting with hypothyroidism or dysglycemia [63].
4. Insulin Resistance, Hyperinsulinemia, and Hyperandrogenemia: A Vicious Cycle
The role of IR and hyperinsulinemia in the development of PCOS has been thoroughly explored, and it is generally accepted to play an important role in the molecular mechanisms implicated in the androgenic hypersecretion typical of this pathology [64]. This has been evidenced by the decrease in fasting insulin levels observed in women with PCOS that undergo insulin-sensitizing pharmacotherapy, which appears to concurrently lower androgenemia and improve ovarian functionalism [65].
On the other hand, although this association is usually conceived as a one-way relationship from IR to hyperandrogenemia, pathways through which hyperandrogenemia may perpetuate IR and hyperinsulinemia are currently being proposed. Indeed, in the context of PCOS, IR and hyperandrogenemia may assemble a vicious cycle, continuously stimulating each other in a reciprocal fashion. Moreover, this conjunction of endocrine-metabolic alterations sets the stage for the progressive development of additional comorbidities, both metabolic and cardiovascular, further complicating the management of these patients [66]. The main mechanisms suggested within this complex network of interactions are presented in the following paragraphs (Figure 1).
Figure 1.
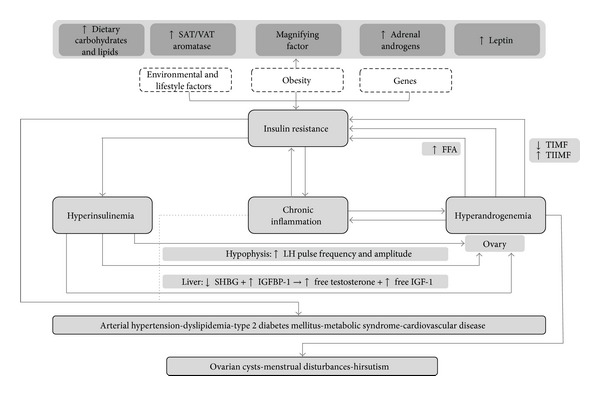
Interactions among insulin resistance, hyperinsulinemia, and hyperandrogenemia in the etiopathogenesis and progression of polycystic ovary syndrome and related comorbidities. SAT = subcutaneous adipose tissue; VAT = visceral adipose tissue LH = luteinizing hormone; SHBG = sex hormone binding globulin; IGFBP-1 = insulin-like growth factor binding protein-1; IGF-1 = insulin-like growth factor-1; FFA = free fatty acids; TIMF = type I muscle fibers; TIIMF = type II muscle fibers. Although details of the exact etiopathogenesis of PCOS are not clear, it appears to be a combination of environmental and genetic factors, which in consonance favor the development of IR. In turn, IR leads to compensatory Hyperinsulinemia, which substantially augments ovarian androgen synthesis by increasing LH pulse frequency at the hypophysis by stimulating GnRH gene transcription in hypothalamic cells. Insulin also triggers hyperandrogenemia by directly activating mitogenic pathways in ovarian cells and increasing transcription of StAR and several key steroidogenic enzymes. Hyperandrogenemia is the key disruption underlying the typical clinical features of PCOS. In addition, increased ovarian production of androgens may in turn worsen IR, thus perpetuating a vicious cycle of IR-hyperinsulinemia-hyperandrogenemia. Indeed, androgens may not only interfere with insulin signaling directly, but also trigger lipolysis, increasing FFA in circulation, favoring IR. Moreover, androgens seem to diminish highly oxidative and insulin-sensitive TIMF and increase glycolytic and less insulin-sensitive TIIMF, further favoring the development of IR. Additionally, obesity appears to magnify all events in this cycle, by increasing androgen synthesis not only in ovaries, but also in SAT and adrenal glands. Leptin also participates by disrupting ovarian physiology and favoring a chronic systemic inflammatory state. Ultimately, both IR and chronic inflammation thrive on all endocrine-metabolic disturbances pertaining PCOS, predisposing patients to the development of comorbidities, such as type 2 diabetes mellitus and cardiovascular disease.
5. From Hyperinsulinemia to Hyperandrogenemia: Systematically Disrupting Ovarian Physiology
5.1. Insulin and Dysregulation of Hypothalamus-Hypophysis-Ovary and Adrenal Signaling
Insulin may play a part in the development of the typical increased amplitude and frequency of GnRH and LH pulse secretion seen in PCOS. Indeed, elevation of LH and GnRH secretion in response to insulin infusion has been observed in vitro, both in dose-dependent and time-dependent fashions [67, 68]. This effect may be mediated by insulin in GnRH-secreting cells of the hypothalamus, by potentiating GnRH gene transcription through the MAPK pathway. As a result, increased GnRH synthesis and secretion lead to a subsequent elevation of LH levels. This continuous stimulation would translate into augmented synthesis of ovarian steroid hormones, particularly androgens [69].
On the other hand, insulin also reinforces adrenal glands as an alternate androgen source parallel to ovaries, by potentiating hypothalamus-hypophysis-adrenal axis (HHAA) activity at several key sites. The hippocampus is an important mediator of HHAA negative feedback, by inhibiting hippocampal activity; insulin indirectly enhances hypothalamic CRH secretion [70] although it may also play a direct role in both the hypothalamus [71] and the hypophysis [72]. Lastly, although the mechanisms remain unclear, insulin appears to augment adrenal cortex sensitivity to ACTH stimulation, with increased androgen secretion [73].
5.2. Insulin and Sex Hormone Binding Globulin
Elevated insulin concentrations have been associated with lower levels of SHBG, leading to enhanced bioavailability of androgens [74]. Although insulin and the insulin-like growth factor 1 (IGF-1) have been demonstrated unable to directly repress shbg [75], they may be key indirect mediators, as they have been associated with decreased total protein secretion in human hepatic cells [76]. Inhibition of SHBG by elevated concentrations of glucose and fructose is also an important component, mediated by downregulation of hepatocyte nuclear factor 4-α (HNF-4α) activity. Nonetheless, in the context of IR, this is yet another indirect effect, as it relies on high concentrations of these monosaccharides due to dysfunctional insulin signaling and not on insulin activity per se [75]. On the other hand, insulin has been shown to repress insulin-like growth factor-1 binding protein (IGFBP-1) synthesis in a direct, rapid, and complete way in both the liver and the ovaries, allowing for greater IGF-1 availability, which in turn boosts insulin activity not only in the liver—further contributing to lower SHBG levels—but also in the ovaries, reinforcing PCOS pathophysiology [77]. This suppression is mediated by intranuclear thymine-rich insulin response elements (TIRE). Although not all components of the signaling cascade linking the insulin receptor (INSR) with TIRE are currently known, inhibition of GSK-3 through the PI3K pathway appears to be essential in this process [78].
5.3. Insulin Signaling in Ovarian Tissue and Selective Insulin Resistance
Pleiotropy is a distinguishing feature of insulin signaling, being involved in a wide catalogue of physiologic and pathophysiologic roles through distinct, yet interconnected, second-messenger pathways (Figure 2). For example, phosphorylation of IRS allows it to act as a docking site for other signaling proteins, such as Grb2, NcK, and PI3K, which are crucial for translocation of GLUT-4. Likewise, further downstream in the PI3K pathway is Akt, which mediates activation of GSK, essential for glycogenesis, as well as mTOR, an important step for insulin-induced protein synthesis. A mitogenic pathway is also activated by insulin, through the binding of phosphorylated IRS or Shc with Grb-2/SOS complexes, leading to MAPK activation through p21Ras and Raf-1 [79].
Figure 2.
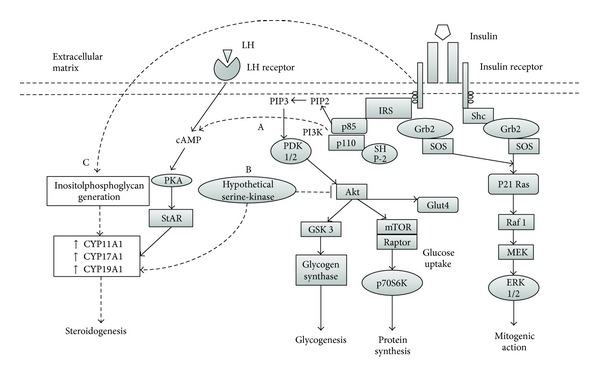
Insulin signaling in ovarian thecal cells and proposed mechanisms for selective insulin resistance. Solid lines represent well-characterized mechanisms. Dashed lines represent incomplete information or hypothetical mechanisms. (A) Insulin may act in synergy with LH to increase intracellular concentration of cAMP, potentiating PKA activation and subsequent phosphorylation of StAR, favoring steroidogenesis. (B) A hypothetical serine-kinase could unify hyperinsulinemia-hyperandrogenemia in PCOS, as it would inhibit Akt activity, negating the metabolic effects of insulin, while activating steroidogenic enzymes. Furthermore, the mitogenic pathway would be left intact and fully functional, favoring thecal proliferation. (C) Inositolphosphoglycan generation appears to be triggered by INSR-prompted mechanisms independent of signaling molecules within metabolic or mitogenic pathways on insulin signaling. Therefore, inositolphosphoglycans may stimulate steroidogenic activity regardless of metabolic and signaling disturbances typical of systemic insulin resistance.
The presence of INSR and IGF-1 receptors in TC, GC, and stromal cells of ovarian tissue unequivocally identifies this organ as a target of insulin activity [80], confirmed by observations of decreased steroidogenesis in TC and GC from both healthy and polycystic ovaries, following in vitro administration of both anti-IGF1R and anti-INSR antibodies [81]. One of the key links in this activity is the acute steroidogenic regulatory protein (StAR), a molecule implicated in transportation of cholesterol to the internal mitochondrial membrane, where the cholesterol side-chain cleavage enzyme (CYP11A1) is anchored, the rate-limiting enzyme in steroid hormone synthesis [82]. Insulin appears to augment not only StAR expression, but also CYP11A1, 17-α-hydroxylase/17,20-lyase (CYP17A1), 3-β-hydroxysteroid dehydrogenase (3-β-HSD), and aromatase (CYP19A1) expression, contributing to an excess in the production of progesterone, 17-α-hydroxyprogesterone, and testosterone in polycystic ovaries in comparison to healthy ovaries [83].
The central paradox in the pathophysiologic association between hyperinsulinemia and hyperandrogenemia in PCOS is that the ovary remains sensitive to insulin activity and subsequent androgen production, despite a systemic state of IR, setting the stage for the “selective insulin resistance” theory [84]. Several mechanisms have been proposed to explain this phenomenon, albeit the true chain of events remains elusive.
5.3.1. cAMP-Dependent Activation of PKA
Insulin appears to act in synergy with LH to elevate intracellular concentration of cAMP, which activates StAR, potentiating steroidogenic activity. Although this effect may be direct through the PI3K pathway, the requirement of cAMP for this activity suggests divergence from the usual insulin cascade; yet the differential molecular interactions are unknown [85]. Similarly, insulin and LH may also act in synergy to increase transcription of LDL-C receptors in GC through the PKA, PI3K, and MAPK pathways. On the other hand, insulin may also augment steroid synthesis through aromatase upregulation in GC, which would serve as substrates for TC for further conversion into androgens [86].
5.3.2. Serine Phosphorylation Theory
This unifying proposal for hyperinsulinemia-hyperandrogenemia in PCOS stems from observations of Dunaif et al., who ascertained a significant decrease in tyrosine-kinase activity, accompanied by considerably higher serine-kinase activity, in fibroblasts of women with PCOS. This differential behavior resulted in attenuated metabolic effects of insulin with normal mitogenesis [87]. Furthermore, in TC, increased serine phosphorylation results in activation of CYP17A1, reflecting into augmented androgen production [88]. Nonetheless, no specific kinase has been evidenced to display these dual effects. However, a key link in the activity of this hypothetical serine-kinase may be the PI3K/Akt pathway, which would explain the coexistence of elevated steroidogenesis with fully functional mitogenic signaling [89].
5.3.3. Inositolphosphoglycan Signaling
Lastly, this pathway stands apart from previous mechanisms as it appears to be independent of all insulin signaling-related molecules except INSR itself [90]. Although the full succession of this cascade is largely unknown, inositolphosphoglycans seem to be able to potentiate steroidogenic activity by stimulating CYP11A1, CYP17A1, and CYP19A1 activity in TC. These observations are noteworthy, as this transduction system may remain intact in the context of IR, in terms of defective tyrosine-kinase activity of INSR substrates and dysfunctional glucose metabolism, warranting ovarian androgen synthesis despite these abnormalities [91].
6. From Hyperandrogenemia Back to Hyperinsulinemia: Completing the Loop
Traditionally, the relationship between IR and PCOS is considered to be a unidirectional pathway toward ovarian disturbances. Nonetheless, recent evidence underpins a complex reciprocal interaction between these phenomena. Indeed, in the context of PCOS, hyperandrogenemia per se may affect insulin sensitivity [66]. This may be mediated by upregulation of β3 adrenergic receptors and hormone-sensitive lipase expression in visceral adipose tissue (VAT) through testosterone or DHEAS signaling [92], modifying lipolytic activity and favoring release of FFA into circulation. This increase in FFA availability causes functional and structural changes in hepatocytes and skeletal myocytes, with the accumulation of metabolites from the long-chain FFA reesterification pathway, including Acyl-CoA and diacylglycerol. In turn, these molecules can activate PKC, a serine/threonine kinase which is widely accepted as pivotal for the mechanisms underlying IR, particularly through serine phosphorylation of IRS-1 [93].
In PCOS, androgens also appear to modify metabolic architecture and functionality in skeletal muscle, by decreasing the amount of type I muscle fibers, which are highly oxidative and insulin-sensitive, and increasing type II fibers, which are glycolytic and less sensitive, as well as decreasing expression of glycogen synthase [94].
Further mechanisms remain poorly characterized, including androgen-driven proinflammatory cytokine secretion from VAT and androgen-induced interference of insulin signaling. In adipocytes, testosterone appears to induce serine phosphorylation of IRS-1 [95], which reflects on inhibition of the metabolic effects of insulin accompanied by normal mitogenic signaling, suggesting that, in PCOS, selective insulin resistance may not only be limited to ovarian tissue, but also be present in adipocytes [96].
7. Obesity: A Key Magnifying Factor of Polycystic Ovary Syndrome
7.1. Obesity As an IR-Independent Pathophysiologic Factor in PCOS
Menstrual irregularities and anovulation appear to be more prevalent and severe in obese women with PCOS than in their nonobese counterparts, and weight loss of at least 5% tends to be associated with improvement of these conditions. Furthermore, obese women with PCOS have greater long term difficulty for conception [97]. Nevertheless, despite the close association between IR and obesity [98], the latter neither requires IR to influence aspects of PCOS pathophysiology, nor is a sine qua non of quality for this entity [99]. Indeed, obesity is a powerful magnifying factor of several aspects of PCOS (Figure 3), which are not limited to favoring the development of IR and hyperinsulinemia.
Figure 3.
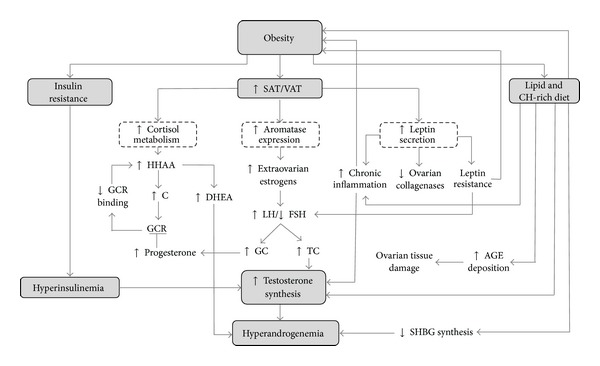
Mechanisms for obesity-mediated potentiation of hyperandrogenemia in polycystic ovary syndrome. SAT = subcutaneous adipose tissue; VAT = visceral adipose tissue; HHAA = hypothalamus-hypophysis-adrenal-axis; C = cortisol; GCR = glucocorticoid receptor; DHEA = dehydroepiandrosterone; LH = luteinizing hormone; FSH = follicle-stimulating hormone; GC = granulosa cells; TC = teca cells; Ch = carbohydrate; AGE = advanced glycation end products; SHBG = sex hormone binding globulin. In PCOS, obesity may potentiate hyperandrogenemia through several mechanisms. Aside from hyperinsulinemia-mediated effects of IR, increased adiposity leads to faster cortisol metabolism, which results in hyperactivation of the HHAA and thus increased DHEA synthesis. Competitive binding to GCR by GC-produced progesterone derives into diminished cortisol binding to GCR, reinforcing HHAA activation. High adiposity also leads to greater aromatase expression in adipocytes, which allows for increased extraovarian estrogen synthesis. In turn, this causes an elevated LH/FSH ratio, prompting hyperplasia of GC and TC, which then synthesizes greater amounts of progesterone and testosterone, respectively. Hyperleptinemia and leptin resistance result in alterations of LH secretion, as well as downregulation of ovarian collagenases, and promotion of a chronic inflammatory state. Carbohydrate-rich diets may induce oxidative stress in circulating mononuclear blood cells and thus chronic inflammation. Lipid-rich diets have been reported to increase testosterone synthesis and downregulate SHBG synthesis, resulting in hyperandrogenemia. These diets also favor endogenous and exogenous AGE deposition, which results in overall ovarian tissue damage.
7.2. Obesogenic Dietary Patterns and Hyperandrogenemia
Both short and long term high-lipid, low-fiber diets have been associated with hyperandrogenemia, possibly acting through intake-induced hyperinsulinemia, which would lower SHBG synthesis, increasing androgen availability [100]. Nevertheless, novel mechanisms suggest a direct effect of diet in ovarian physiology disruption. Advanced glycation end products (AGE) are cytotoxic metabolites derived from disrupted carbohydrate metabolism, which may also be exogenously obtained from a myriad of food typical of Westernized diets [101]. AGE deposition in ovarian tissue induces oxidative stress and aberrant structure modification due to molecule cross-linking, leading to damage of all ovarian cell types and therefore altering all aspects of its functionality. Moreover hyperandrogenemia appears to inhibit glyoxalase-I activity, which is an important enzymatic scavenging system for 2-oxoaldehydes, including major precursors of AGE. Thus, in PCOS, the deleterious effects of AGE deposition may be exacerbated [102].
7.3. Effects of Obesity on Steroid Hormone Physiology and Metabolism
Aside from disturbances in insulin physiology, obesity implies thorough alterations in steroid hormone metabolism, essentially summarized as increased concentrations of nearly all of these messengers. To this end, hyperestrogenemia is a paramount alteration, stemming from extraovarian estrogen production in VAT and subcutaneous adipose tissue (SAT), due to expression of aromatase in these adipocytes [103]. Estrogens stimulate LH and inhibit FSH secretion, contributing to GC and TC hyperplasia. In turn, this would increase androgen synthesis, which not only cause the typical manifestations of PCOS, but also serve as substrates for extraovarian aromatization, reinforcing this cycle of hyperestrogenemia-hyperandrogenemia in obese women with this pathology [104].
Another common finding in women with PCOS is elevated serum concentrations of adrenal androgens, which suggest dysregulation of the HHAA. Parallel to the effects of insulin in this axis [73], adrenal hyperandrogenemia may be reinforced by increased amounts of VAT, which displays a high traffic and catabolism of cortisol, triggering a compensatory activation of the HHAA, which results in elevation of adrenal androgen levels. Additionally, progesterone may interact with the glucocorticoid receptor, impairing activity of these hormones. As a consequence, hyperestrogenemia, such as that found in PCOS and the luteal phase of the menstrual cycle, may exacerbate this HHAA compensation, leading to higher adrenal androgen production [105], which can also be converted back to estrone in adipose tissue and then into estradiol by 17-β-HSD in extraovarian tissues, restarting the hyperestrogenemia-hyperandrogenemia cycle [106]. Moreover, glucocorticoids, which are elevated due to HHAA hyperactivity, induce expression of aromatase, further fueling this positive feedback circuit [103].
7.4. The Role of Leptin in PCOS with Obesity
Leptin, known as the prototypical adipokine, is a 167 amino acid peptide secreted primarily from white adipose tissue, although it is present at several other sites, including the ovary [107]. Leptin secretion occurs predominantly in SAT over VAT in women and appears to be inversely related to adipocyte size [108]. Leptin can be found circulating freely—its metabolically active form—or bound to soluble leptin receptor (sOB-R), a carrier protein derived from alternative splicing of leptin receptor (OB-R) mRNA or ectodomain shedding of OB-R transmembrane structures [109]. sOB-R modulates leptin activity by lengthening clearance and half-life, yet limiting its availability to membrane OB-R [110]. Leptin participates in regulation of energy homeostasis and multiple neuroendocrine, immune, and reproductive functions [111].
Indeed, leptin appears to play a permissive role for adequate functioning of the HHOA, as observed in subjects with congenital leptin deficiency, who are typically infertile [112]. Leptin stimulates LH secretion, as ascertained by correction of LH pulses following leptin administration in women with fasting-induced HHOA dysfunction [113]. Because hypothalamic GnRH-secreting cells do not express leptin receptors, stimulation of LH secretion appears to be an indirect effect, possibly through AgRP/NPY and POMC neurons, which do express the receptor and are anatomically associated with GnRH neurons [114]. Another pathway for leptin-induced GnRH secretion involves kisspeptin; a neuropeptide expressed in the arcuate and periventricular nuclei, which binds to its receptor in hypothalamic GnRH-expressing cells, inducing its secretion [115]. Leptin also exerts direct effects in all ovarian cells and seems to have a physiologic regulatory effect in folliculogenesis [111].
In the context of PCOS, the role of leptin has been subject to profound controversy, with opposing views regarding its true participation. Because leptin concentrations are consistently found to be strongly correlated with weight, some reports consider the hyperleptinemia seen in PCOS as only a byproduct of this condition [116]. On the other hand, findings linking leptin levels to estradiol [117], testosterone, and insulin [118] in women with PCOS advocate for a more complex role of leptin in its pathophysiology. Moreover, reports of elevated leptin in nonobese PCOS patients further question quantitative adiposity as the sole origin of hyperleptinemia in this scenario [119].
Regardless of its origin, in PCOS hyperleptinemia exerts direct effects on ovarian physiology by arresting follicle development [111]. Moreover, ovarian paracrine and autocrine leptin signaling may also be disrupted, parallel to alterations in the HHOA [120]. Likewise, in vitro studies show a decrease in collagenase expression in ovarian tissue after exposure to high concentrations of leptin, adding to the ovulatory disturbances in this scenario [121].
In obese women with PCOS, the consequences of hyperleptinemia may be excacerbated by lower levels of sOB-R, which increase leptin ligand availability. Indeed, sOB-R expression appears to be inverse to both adiposity and DHEAS levels [122]. Finally, matters may be further complicated with the development of leptin resistance, a state of diminished response to the neuroendocrine effects of this hormone, namely, regarding attenuation of appetite [123]. Several factors may contribute to this phenomenon, including saturation of blood brain barrier transporters of leptin, saturation and downregulation of its receptors in kisspeptin-expressing neurons, obesity-induced endoplasmic reticulum stress in target neurons, and a negative feedback loop engrained within its own signaling cascade, which activates in the face of leptin overactivity [124]. In PCOS, leptin resistance results not only in insufficient permissive signaling to achieve optimal LH pulse secretion [125], but also in defective suppression of appetite, which may perpetuate obesity [126].
8. Polycystic Ovary Syndrome in Nonobese Women: Key Differential Features
Given that obesity is only present in roughly half of all cases of PCOS, efforts have been directed to the description of pathophysiologic mechanisms in nonobese women. In this aspect, differential patterns of insulin physiology are paramount in characterizing the metabolic profiles of obese and nonobese women with PCOS [127]. Although nonobese women exhibit lower insulinemia and IR [128] associated with higher SHBG levels [129], hyperinsulinemia is still a common finding in this population [127]. Indeed, several studies have suggested a primary alteration in β cell function as a pathophysiological component independent of weight [130, 131]. Insulin hypersecretion has been suggested as the probable underlying mechanism in this scenario, in light of reports underpinning increased β cell activity—and not IR—as a predictor of hyperandrogenemia in PCOS women [132], being present in both lean and obese subjects [133]. This intrinsic disruption in β cell function may be linked to in utero exposure to elevated androgen levels [134], as supported by animal models wherein this kind of fetal stimulation resulted in altered expression of genes associated with β cell function and mass as well as altered insulin secretion response in vitro and altered β cell quantity [135]. Nevertheless, despite strong evidence for candidacy, variations of the insulin gene VNTR (variable number tandem repeat) minisatellite have been demonstrated not to play a relevant role in the development of PCOS [136], raising the need for further research on the subject.
Another element contributing to hyperinsulinemia in this scenario is increased endogenous opioid signaling by virtue of elevated levels of β endorphins in peripheral circulation. These messengers favor the development of hyperinsulinemia by stimulating its pancreatic secretion and inhibiting its hepatic clearance, as well as regulating IGFBP-I serum levels, therefore modulating IGF-I bioavailability, although the origin of these greater levels of endogenous opioids remains unclear [137].
Nonobese individuals also show greater LH/FSH ratios, which seem to play a major role in this subset of subjects [138]. In this scenario, endogenous opioids also appear to play an important role, albeit in this case, within the central nervous system rather than peripherally. Indeed, an increased opioid signaling tone is observed during the late follicular and luteal phases, associated with slower LH pulse secretion, in physiologic conditions [139]. However, a decreased opioid tone is observed in PCOS, accompanied by increased sensitivity to GnRH, resulting in reinforced LH secretion [137].
Lean women with PCOS have been found to display diminished sensitivity to catecholamine-mediated lipolysis in SAT [140], resulting in preservation of this tissue. Because leptin is primarily secreted from SAT [108], this may partially explain hyperleptinemia found in normal-weight women with PCOS, and although intrinsic dysregulation of its secretion mechanisms may also be involved, the precise chain of events remains unelucidated [119].
Likewise, while obese women with PCOS show higher levels of testosterone than adrenal androgens, this relationship is inversed in nonobese subjects, suggesting that adrenal steroidogenesis may be more severely altered in lean individuals [17]. In addition, although both obese and nonobese women with PCOS have increased adrenal sensitivity to ACTH for cortisol and androgen secretion [141], the increased ratio of the 11-oxo metabolites of cortisol and corticosterone to their 11-hydroxy metabolites in urine of nonobese PCOS women indicates enhanced oxidation by 11-β–hydroxysteroid dehydrogenase (11-β-HSD) in these subjects. In turn, this effect would result in increased cortisol clearance, allowing for greater adrenal androgen synthesis. This 11-β-HSD upregulation has been proposed to be due to hyperandrogenemia or hyperinsulinemia, although further research is required [142].
Lastly, oxidative stress and low-grade inflammation are two features equally shared by obese and nonobese PCOS subjects [143]. Because the genesis of this component does not seem to be attributable to body composition in nonobese women with PCOS, leading premises are genetic in nature, with the proposal of overexpression of several proinflammatory genes as the cause underlying this phenomenon in this subset of females [144].
9. Chronic Inflammation: From Insulin Resistance to Polycystic Ovaries and Beyond
Recent evidence describes a central role for certain proinflammatory mediators in the pathophysiology of PCOS, posing a new focus on the etiological considerations for PCOS, which is currently considered a chronic, low-grade inflammatory disorder, independently of the presence of obesity [145], although these phenomena are indeed exacerbated by adiposity [146]. Reports show that women with PCOS, both with obesity and normal weight, exhibit elevated serum TNF, C-reactive protein (CRP), monocyte and lymphocyte circulating levels, and inflammatory infiltration in ovarian tissue [147].
Several mechanisms have been proposed to explain these features. Among these, the existence of proinflammatory genotypes has been suggested, including polymorphisms of sequences codifying TNF, TNF receptor, IL-6, and IL-10 [148]. Remarkably, greater expression of the CD11c gene is associated with greater proinflammatory macrophage infiltration is SAT and VAT, favoring a transition to decreased secretion of adiponectin and increased TNF and leptin secretion from adipocytes [149].
The role of TNF is especially important in the setting of IR and PCOS. In addition to the diminishing effect it bears over insulin sensitivity, through serine phosphorylation of IRS-1 by PKC [16]. This cytokine also stimulates steroidogenesis and proliferation in TC, as well as follicular atresia, contributing to hyperandrogenemia, posing yet another level of complexity and self-perpetuation for PCOS [150].
Furthermore, hyperglycemia may contribute to inflammation in PCOS, possibly explaining the greater magnitude of manifestations in PCOS (Figure 3). Circulating mononuclear cells utilize glucose as their main redox substrate, with part of its metabolites going into the pentose-phosphate pathway to yield NADPH [151]. Oxidation of this molecule leads to the production of reactive oxygen species (ROS), which induce oxidative stress, with the subsequent activation of NF-kB, a transcription factor involved in the expression of proinflammatory mediators such as TNF and IL-6 [66]. Hence, hyperglycemia may result in increased ROS production. Additionally, oxidative stress appears to also induce key steroidogenic molecules in TC, namely, CYP11A1, CYP17A1, 3-β-HSD, and StAR, favoring hyperandrogenemia [152].
Additionally, hyperleptinemia boosts its effects in immune function regulation, including production of proinflammatory cytokines, such as TNF, IL-6, and IL-12, stimulation of polymorphonuclear cell chemotaxis, inhibition of lymphocyte apoptosis by suppression of Fas-mediated signaling, and induction of Th1 differentiation, both in obese and nonobese females with PCOS [153].
Beyond these implications in PCOS, the reinforcing cross-talk between IR and chronic inflammation generates a welcoming environment for the development of further cardiometabolic disturbances. Oxidative stress caused by ROS production in immune cells plays a fundamental role in the genesis and progression of endothelial dysfunction, which leads to the development of arterial hypertension, and cardiovascular disease [154]. Furthermore, chronic inflammation and IR are two essential elements in the etiopathogenesis of MS and DM2, which in turn open the door for further complications for the overall health of women with PCOS [155].
10. Concluding Remarks
Given the pivotal role IR and obesity play in the etiopathogenesis and progression of PCOS and its potential subsequent metabolic and cardiovascular complications, both should be considered essential therapeutic targets [156]. Although traditionally metformin is thought of as a hallmark of PCOS treatment as the mainstay insulin sensitizer, the advent of the distinct phenotypes for this syndrome and the broader acceptance of this categorization bring into question its indication in all cases of PCOS [157]. Uncertainties are even more commonplace surrounding other pharmacologic alternatives habitually considered for PCOS management, such as thiazolidinediones and statins [158, 159].
Lifestyle modifications in the treatment of PCOS do not escape criticism and controversy despite being widely accepted recommendations. Physical activity (PA) has been reported to ameliorate anovulation, IR, blood pressure, and lipid profiles in women with PCOS, sometimes independently of weight loss [160]; yet PA alone does not seem to be able to equal these parameters to non-PCOS subjects [161]. Therefore, it should be accompanied by a complementary diet plan in order to fully potentiate the effects of a lifestyle-modification therapeutic program. Standard weight loss programs may be sufficient for ameliorating features of PCOS, in the form of a nutritionally adequate, fiber-rich, low fat, moderate protein, and high carbohydrate intake diet with a 500–1,000 kcal/day reduction [162]. On the other hand, more specialized low-carbohydrate ketogenic diets have been reported to significantly reduce weight, LH/FSH ratio, testosterone and fasting insulin ratio, and IR in women with PCOS [163], although concerns on their safety on lipid profiles [164] and cardiovascular risk [165] call for careful consideration. Ultimately, a wide range of dietary programs akin to these general concepts are accepted to similarly improve weight, reproductive and metabolic variables in PCOS so long as they boast nutritional adequacy and long term sustainability [166]. In addition, novel perspectives into the pathophysiology of PCOS, such as the role of ovarian AGE deposition, suggest avoidance of glycotoxin-rich diets as an added recommendation [167].
In light of the controversial considerations regarding treatment of this syndrome, further studies are required to elucidate the intricacies within the pathophysiology of PCOS and the true relationships between IR, hyperinsulinemia, and hyperandrogenemia, as well as other important hormonal disturbances, particularly alterations within steroid hormone metabolism, hyperleptinemia, and leptin resistance. This would allow for the formulation of improved approaches to the management of obesity and IR as therapeutic targets in women with PCOS, in order to improve their quality of life and especially their fertility outcomes, which are often the main concern of both patients and physicians in the management of this pathology.
Acknowledgments
This work was supported by Research Grant no. CC-0437-10-21-09-10 from the Technological, Humanistic, and Scientific Development Council, University of Zulia, and Research Grant no. FZ-0058-2007 from Fundacite-Zulia.
Abbreviations
- 11-β-HSD:
11-β-hydroxysteroid dehydrogenase
- 3-β-HSD:
3-β-hydroxysteroid dehydrogenase
- ACTH:
Adrenocorticotropic hormone
- AGE:
Advanced glycation end products
- AgRP:
Agouti-related protein
- AMH:
Anti-Müllerian hormone
- cAMP:
Cyclic adenosine monophosphate
- CRP:
C-reactive protein
- CRH:
Corticotropin-releasing hormone
- CYP11A1:
Cholesterol side-chain cleavage enzyme
- CYP17A1:
17-α-Hydroxylase/17,20-lyase
- CYP19A1:
Aromatase
- DHEAS:
Dehydroepiandrosterone sulfate
- DHT:
Dihydrotestosterone
- DM2:
Type 2 diabetes mellitus
- FFA:
Free fatty acids
- FSH:
Follicle-stimulating hormone
- GC:
Granulosa cells
- GLUT4:
Glucose transporter type 4
- GnRH:
Gonadotropin-releasing hormone
- GSK-3:
Glycogen synthase kinase 3
- HHAA:
Hypothalamus-hypophysis-adrenal axis
- HHOA:
Hypothalamus-hypophysis-ovary axis
- HNF4α:
Hepatocyte nuclear factor 4-α
- IGF-1:
Insulin-like growth factor-1
- IGFBP-1:
Insulin-like growth factor-1 binding protein
- INSR:
Insulin receptor
- IR:
Insulin resistance
- LDL-C:
Low-density lipoprotein cholesterol
- LH:
Luteinizing hormone
- MAPK:
Mitogen-activated protein kinase
- MS:
Metabolic syndrome
- mTOR:
Mammalian target of rapamycin
- mRNA:
Messenger ribonucleic acid
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NPY:
Neuropeptide Y
- OB-R:
Leptin receptor
- PA:
Physical activity
- PCOS:
Polycystic ovary syndrome
- PI3K:
Phosphoinositide 3-kinase
- PKA:
Protein kinase A
- PKC:
Protein kinase C
- POMC:
Proopiomelanocortin
- ROS:
Reactive oxygen species
- SAT:
Subcutaneous adipose tissue
- SHBG:
Sex hormone binding globulin
- sOB-R:
Soluble leptin receptor
- StAR:
Acute steroidogenic regulatory protein
- TC:
Theca cells
- TGF-β:
Transforming growth factor β
- TIRE:
Thymine-rich insulin response elements
- TNF:
Tumor necrosis factor
- VAT:
Visceral adipose tissue.
Conflict of Interests
The authors have no conflict of interests to disclose.
References
- 1.Teede H, Deeks A, Moran L. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Medicine. 2010;8, article 41 doi: 10.1186/1741-7015-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mascarenhas MN, Flaxman SR, Boerma T, Vanderpoel S, Stevens GA. National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Medicine. 2012;9(12) doi: 10.1371/journal.pmed.1001356.e1001356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fauser BCJM, Tarlatzis BC, Rebar RW, et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-sponsored 3rd PCOS consensus workshop group. Fertility and Sterility. 2012;97(1):28.e25–38.e25. doi: 10.1016/j.fertnstert.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 4.Boyle J, Teede HJ. Polycystic ovary syndrome—an update. Australian Family Physician. 2012;41(10):752–756. [PubMed] [Google Scholar]
- 5.Lane DE. Polycystic ovary syndrome and its differential diagnosis. Obstetrical and Gynecological Survey. 2006;61(2):125–135. doi: 10.1097/01.ogx.0000197817.93201.04. [DOI] [PubMed] [Google Scholar]
- 6.Wild RA. Long-term health consequences of PCOS. Human Reproduction Update. 2002;8(3):231–241. doi: 10.1093/humupd/8.3.231. [DOI] [PubMed] [Google Scholar]
- 7.Legro R. Obesity and PCOS: implications for diagnosis and treatment. Seminars in Reproductive Medicine. 2012;30(6):496–506. doi: 10.1055/s-0032-1328878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miranda J, Herrero V, Chirinos J, et al. Major cardiovascular risk factors in Latin America: a comparison with the United States: the Latin American consortium of studies in obesity (LASO) PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0054056.e54056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bermúdez V, Pacheco M, Rojas J, et al. Epidemiologic behavior of obesity in the Maracaibo City metabolic syndrome prevalence study. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0035392.e35392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gambineri A, Pelusi C, Vicennati V, Pagotto U, Pasquali R. Obesity and the polycystic ovary syndrome. International Journal of Obesity. 2002;26(7):883–896. doi: 10.1038/sj.ijo.0801994. [DOI] [PubMed] [Google Scholar]
- 11.Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Medical Clinics of North America. 2011;95(5):875–892. doi: 10.1016/j.mcna.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Toprak S, Yönem A, Cakir B, et al. Insulin resistance in nonobese patients with polycystic ovary syndrome. Hormone Research. 2001;55(2):65–70. doi: 10.1159/000049972. [DOI] [PubMed] [Google Scholar]
- 13.Apridonidze T, Essah PA, Iuorno MJ, Nestler JE. Prevalence and characteristics of the metabolic syndrome in women with polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism. 2005;90(4):1929–1935. doi: 10.1210/jc.2004-1045. [DOI] [PubMed] [Google Scholar]
- 14.Reaven G. Insulin resistance and compensatory hyperinsulinemia: the linchpin between obesity and cardiovascular disease. Official Journal of the Chair on Cardiometabolic Risk. 2008;1(2):3–10. [Google Scholar]
- 15.Reaven GM. Compensatory hyperinsulinemia and the development of an atherogenic lipoprotein profile: the price paid to maintain glucose homeostasis in insulin-resistant individuals. Endocrinology and Metabolism Clinics of North America. 2005;34(1):49–62. doi: 10.1016/j.ecl.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Rojas J, Bermúdez V, Leal E, et al. Insulinorresistencia e hiperinsulinemia como factores de riesgo para enfermedad cardiovascular. Archivos Venezolanos de Farmacología y Terapéutica. 2008;27(1):29–39. [Google Scholar]
- 17.Moran C, Arriaga M, Rodriguez G, Moran S. Obesity differentially affects phenotypes of polycystic ovary syndrome. International Journal of Endocrinology. 2012;2012:7 pages. doi: 10.1155/2012/317241.317241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diamanti-Kandarakis E, Papailiou J, Palimeri S. Hyperandrogenemia: pathophysiology and its role in ovulatory dysfunction in PCOS. Pediatric Endocrinology Reviews. 2006;3(1):198–204. [PubMed] [Google Scholar]
- 19.Pellatt L, Rice S, Mason HD. Anti-Müllerian hormone and polycystic ovary syndrome: a mountain too high? Reproduction. 2010;139(5):825–833. doi: 10.1530/REP-09-0415. [DOI] [PubMed] [Google Scholar]
- 20.Sánchez F, Smitz J. Molecular control of oogenesis. Biochimica et Biophysica Acta. 2012;1822(12):1896–1912. doi: 10.1016/j.bbadis.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Karkanaki A, Vosnakis C, Panidis D. The clinical significance of anti-müllerian hormone evaluation in gynecological endocrinology. Hormones. 2011;10(2):95–103. doi: 10.14310/horm.2002.1299. [DOI] [PubMed] [Google Scholar]
- 22.Pierre A, Peigné M, Grynberg M, et al. Loss of LH-induced down-regulation of anti-Mullerian hormone receptor expression may contribute to anovulation in women with polycystic ovary syndrome. Human Reproduction. 2013;28(3):762–769. doi: 10.1093/humrep/des460. [DOI] [PubMed] [Google Scholar]
- 23.Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Human Reproduction Update. 2006;12(4):351–361. doi: 10.1093/humupd/dml017. [DOI] [PubMed] [Google Scholar]
- 24.Blank SK, McCartney CR, Chhabra S, et al. Modulation of gonadotropin-releasing hormone pulse generator sensitivity to progesterone inhibition in hyperandrogenic adolescent girls—implications for regulation of pubertal maturation. Journal of Clinical Endocrinology and Metabolism. 2009;94(7):2360–2366. doi: 10.1210/jc.2008-2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burt Solorzano CM, Beller JP, Abshire MY, Collins JS, McCartney CR, Marshall JC. Neuroendocrine dysfunction in polycystic ovary syndrome. Steroids. 2012;77(4):332–337. doi: 10.1016/j.steroids.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karoshi M, Okolo SO. Commentary: polycystic ovarian disease (PCOD): a misnomer, looking for a new name. International Journal of Fertility and Women’s Medicine. 2004;49(4):191–192. [PubMed] [Google Scholar]
- 27.Urbanek M. The genetics of the polycystic ovary syndrome. Nature Clinical Practice Endocrinology and Metabolism. 2007;3(2):103–111. doi: 10.1038/ncpendmet0400. [DOI] [PubMed] [Google Scholar]
- 28.Abbott DH, Dumesic DA, Franks S. Developmental origin of polycystic ovary syndrome—a hypothesis. Journal of Endocrinology. 2002;174(1):1–5. doi: 10.1677/joe.0.1740001. [DOI] [PubMed] [Google Scholar]
- 29.Sirmans SM, Pate KA. Epidemiology, diagnosis, and management of polycystic ovary syndrome. Clinical Epidemiology. 2014;6:1–13. doi: 10.2147/CLEP.S37559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azziz R, Carmina E, Dewailly D, et al. The androgen excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertility and Sterility. 2009;91(2):456–488. doi: 10.1016/j.fertnstert.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 31.Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatology Online Journal. 2008;14(9, article 2) [PubMed] [Google Scholar]
- 32.Głuszak O, Stopińska-Głuszak U, Glinicki P, et al. Phenotype and metabolic disorders in polycystic ovary syndrome. ISRN Endocrinology. 2012;2012:7 pages. doi: 10.5402/2012/569862.569862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Essah PA, Wickham EP, III, Nunley JR, Nestler JE. Dermatology of androgen-related disorders. Clinics in Dermatology. 2006;24(4):289–298. doi: 10.1016/j.clindermatol.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 34.Marcondes JAM, Hayashida SAY, Barcellos CRG, Rocha MP, Maciel GAR, Baracat EC. Metabolic syndrome in women with polycystic ovary syndrome: prevalence, characteristics and predictors. Arquivos Brasileiros de Endocrinologia e Metabologia. 2007;51(6):972–979. doi: 10.1590/s0004-27302007000600012. [DOI] [PubMed] [Google Scholar]
- 35.Baldani DP, Skrgatić L, Goldstajn MS, et al. Clinical and biochemical characteristics of polycystic ovary syndrome in Croatian population. Collegium Antropologicum. 2012;36(4):1413–1418. [PubMed] [Google Scholar]
- 36.Jedel E, Waern M, Gustafson D, et al. Anxiety and depression symptoms in women with polycystic ovary syndrome compared with controls matched for body mass index. Human Reproduction. 2010;25(2):450–456. doi: 10.1093/humrep/dep384. [DOI] [PubMed] [Google Scholar]
- 37.Özdemir S, Özdemir M, Görkemli H, Kiyici A, Bodur S. Specific dermatologic features of the polycystic ovary syndrome and its association with biochemical markers of the metabolic syndrome and hyperandrogenism. Acta Obstetricia et Gynecologica Scandinavica. 2010;89(2):199–204. doi: 10.3109/00016340903353284. [DOI] [PubMed] [Google Scholar]
- 38.Sivayoganathan D, Maruthini D, Glanville JM, Balen AH. Full investigation of patients with polycystic ovary syndrome (PCOS) presenting to four different clinical specialties reveals significant differences and undiagnosed morbidity. Human Fertility. 2011;14(4):261–265. doi: 10.3109/14647273.2011.632058. [DOI] [PubMed] [Google Scholar]
- 39.Valkenburg O, Steegers-Theunissen RPM, Smedts HPM, et al. A more atherogenic serum lipoprotein profile is present in women with polycystic ovary syndrome: a case-control study. Journal of Clinical Endocrinology and Metabolism. 2008;93(2):470–476. doi: 10.1210/jc.2007-1756. [DOI] [PubMed] [Google Scholar]
- 40.Hahn S, Tan S, Elsenbruch S, et al. Clinical and biochemical characterization of women with polycystic ovary syndrome in North Rhine-Westphalia. Hormone and Metabolic Research. 2005;37(7):438–444. doi: 10.1055/s-2005-870236. [DOI] [PubMed] [Google Scholar]
- 41.Ehrmann DA, Liljenquist DR, Kasza K, et al. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism. 2006;91(1):48–53. doi: 10.1210/jc.2005-1329. [DOI] [PubMed] [Google Scholar]
- 42.Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. Journal of Clinical Endocrinology and Metabolism. 2004;89(6):2745–2749. doi: 10.1210/jc.2003-032046. [DOI] [PubMed] [Google Scholar]
- 43.Carmina E, Lobo RA. Use of fasting blood to assess the prevalence of insulin resistance in women with polycystic ovary syndrome. Fertility and Sterility. 2004;82(3):661–665. doi: 10.1016/j.fertnstert.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 44.Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. Journal of Clinical Endocrinology and Metabolism. 1999;84(1):165–169. doi: 10.1210/jcem.84.1.5393. [DOI] [PubMed] [Google Scholar]
- 45.Elting MW, Korsen TJM, Bezemer PD, Schoemaker J. Prevalence of diabetes mellitus, hypertension and cardiac complaints in a follow-up study of a Dutch PCOS population. Human Reproduction. 2001;16(3):556–560. doi: 10.1093/humrep/16.3.556. [DOI] [PubMed] [Google Scholar]
- 46.Dokras A, Bochner M, Hollinrake E, Markham S, VanVoorhis B, Jagasia DH. Screening women with polycystic ovary syndrome for metabolic syndrome. Obstetrics and Gynecology. 2005;106(1):131–137. doi: 10.1097/01.AOG.0000167408.30893.6b. [DOI] [PubMed] [Google Scholar]
- 47.Hollinrake E, Abreu A, Maifeld M, van Voorhis BJ, Dokras A. Increased risk of depressive disorders in women with polycystic ovary syndrome. Fertility and Sterility. 2007;87(6):1369–1376. doi: 10.1016/j.fertnstert.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 48.Uno H. Biology of hair growth. Seminars in Reproductive Endocrinology. 1986;4(2):131–141. [Google Scholar]
- 49.Burger HG. Androgen production in women. Fertility and Sterility. 2002;77(supplement 4):S3–S5. doi: 10.1016/s0015-0282(02)02985-0. [DOI] [PubMed] [Google Scholar]
- 50.Redmond GP, Bergfeld WF. Diagnostic approach to androgen disorders in women: acne, hirsutism, and alopecia. Cleveland Clinic Journal of Medicine. 1990;57(5):423–427. doi: 10.3949/ccjm.57.5.423. [DOI] [PubMed] [Google Scholar]
- 51.Auchus RJ, Rainey WE. Adrenarche—physiology, biochemistry and human disease. Clinical Endocrinology. 2004;60(3):288–296. doi: 10.1046/j.1365-2265.2003.01858.x. [DOI] [PubMed] [Google Scholar]
- 52.Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the androgen excess and polycystic ovary syndrome society. Human Reproduction Update. 2012;18(2):146–170. doi: 10.1093/humupd/dmr042. [DOI] [PubMed] [Google Scholar]
- 53.Wendelin DS, Pope DN, Mallory SB. Hypertrichosis. Journal of the American Academy of Dermatology. 2003;48(2):161–179. doi: 10.1067/mjd.2003.100. [DOI] [PubMed] [Google Scholar]
- 54.Al-Nuaimi Y, Baier G, Watson REB, Chuong C, Paus R. The cycling hair follicle as an ideal systems biology research model. Experimental Dermatology. 2010;19(8):707–713. doi: 10.1111/j.1600-0625.2010.01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yildiz BO, Bolour S, Woods K, Moore A, Azziz R. Visually scoring hirsutism. Human Reproduction Update. 2009;16(1):51–64. doi: 10.1093/humupd/dmp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zouboulis CC. Acne and sebaceous gland function. Clinics in Dermatology. 2004;22(5):360–366. doi: 10.1016/j.clindermatol.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 57.Zouboulis CC, Degitz K. Androgen action on human skin—from basic research to clinical significance. Experimental Dermatology, Supplement. 2004;13(supplement 4):5–10. doi: 10.1111/j.1600-0625.2004.00255.x. [DOI] [PubMed] [Google Scholar]
- 58.Makrantonaki E, Ganceviciene R, Zouboulis C. An update on the role of the sebaceous gland in the pathogenesis of acne. Dermato-Endocrinology. 2011;3(1):41–49. doi: 10.4161/derm.3.1.13900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Price VH. Androgenetic alopecia in women. Journal of Investigative Dermatology Symposium Proceedings. 2003;8(1):24–27. doi: 10.1046/j.1523-1747.2003.12168.x. [DOI] [PubMed] [Google Scholar]
- 60.Cela E, Robertson C, Rush K, et al. Prevalence of polycystic ovaries in women with androgenic alopecia. European Journal of Endocrinology. 2003;149(5):439–442. doi: 10.1530/eje.0.1490439. [DOI] [PubMed] [Google Scholar]
- 61.Daniel CR, III, Iorizzo M, Piraccini BM, Tosti A. Simple onycholysis. Cutis. 2011;87(5):226–228. [PubMed] [Google Scholar]
- 62.van de Kerkhof PCM, Pasch MC, Scher RK, et al. Brittle nail syndrome: a pathogenesis-based approach with a proposed grading system. Journal of the American Academy of Dermatology. 2005;53(4):644–651. doi: 10.1016/j.jaad.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 63.Ramos-e-Silva M, Chaves Azevedo-e-Silva M, Carneiro SC. Hair, nail, and pigment changes in major systemic disease. Clinics in Dermatology. 2008;26(3):296–305. doi: 10.1016/j.clindermatol.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 64.Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocrine Reviews. 2012;33(6):981–1030. doi: 10.1210/er.2011-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pauli JM, Raja-Khan N, Wu X, Legro RS. Current perspectives of insulin resistance and polycystic ovary syndrome. Diabetic Medicine. 2011;28(12):1445–1454. doi: 10.1111/j.1464-5491.2011.03460.x. [DOI] [PubMed] [Google Scholar]
- 66.Gambineri A, Pelusi C, Vicennati V, Pagotto U, Pasquali R. Obesity and the polycystic ovary syndrome. International Journal of Obesity. 2002;26(7):883–896. doi: 10.1038/sj.ijo.0801994. [DOI] [PubMed] [Google Scholar]
- 67.Sekar N, Garmey JC, Veldhuis JD. Mechanisms underlying the steroidogenic synergy of insulin and luteinizing hormone in porcine granulosa cells: joint amplification of pivotal sterol-regulatory genes encoding the low-density lipoprotein (LDL) receptor, steroidogenic acute regulatory (StAR) protein and cytochrome P450 side-chain cleavage (P450scc) enzyme. Molecular and Cellular Endocrinology. 2000;159(1-2):25–35. doi: 10.1016/s0303-7207(99)00203-8. [DOI] [PubMed] [Google Scholar]
- 68.Morley P, Calaresu FR, Barbe GJ, Armstrong DT. Insulin enhances luteinizing hormone-stimulated steroidogenesis by porcine theca cells. Biology of Reproduction. 1989;40(4):735–743. doi: 10.1095/biolreprod40.4.735. [DOI] [PubMed] [Google Scholar]
- 69.Kim HH, DiVall SA, Deneau RM, Wolfe A. Insulin regulation of GnRH gene expression through MAP kinase signaling pathways. Molecular and Cellular Endocrinology. 2005;242(1-2):42–49. doi: 10.1016/j.mce.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 70.Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocrine Reviews. 1991;12(2):118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- 71.Grunstein HS, James DE, Storlien LH, Smythe GA, Kraegen EW. Hyperinsulinemia suppresses glucose utilization in specific brain regions: in vivo studies using the euglycemic clamp in the rat. Endocrinology. 1985;116(2):604–610. doi: 10.1210/endo-116-2-604. [DOI] [PubMed] [Google Scholar]
- 72.Unger JW, Lange W. Insulin receptors in the pituitary gland: morphological evidence for influence on opioid peptide-synthesizing cells. Cell and Tissue Research. 1997;288(3):471–483. doi: 10.1007/s004410050833. [DOI] [PubMed] [Google Scholar]
- 73.Alesci S, Koch CA, Bornstein SR, Pacak K. Adrenal androgens regulation and adrenopause. Endocrine Regulations. 2001;35(2):95–100. [PubMed] [Google Scholar]
- 74.Wallace IR, McKinley MC, Bell PM, Hunter SJ. Sex hormone binding globulin and insulin resistance. Clinical Endocrinology. 2013;78(3):321–329. doi: 10.1111/cen.12086. [DOI] [PubMed] [Google Scholar]
- 75.Selva DM, Hogeveen KN, Innis SM, Hammond GL. Monosaccharide-induced lipogenesis regulates the human hepatic sex hormone-binding globulin gene. Journal of Clinical Investigation. 2007;117(12):3979–3987. doi: 10.1172/JCI32249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crave JC, Lejeune H, Brebant C, Baret C, Pugeat M. Differential effects of insulin and insulin-like growth factor I on the production of plasma steroid-binding globulins by human hepatoblastoma-derived (Hep G2) cells. Journal of Clinical Endocrinology and Metabolism. 1995;80(4):1283–1289. doi: 10.1210/jcem.80.4.7536204. [DOI] [PubMed] [Google Scholar]
- 77.Mounier C, Dumas V, Posner BI. Regulation of hepatic insulin-like growth factor-binding protein-1 gene expression by insulin: central role for mammalian target of rapamycin independent of forkhead box O proteins. Endocrinology. 2006;147(5):2383–2391. doi: 10.1210/en.2005-0902. [DOI] [PubMed] [Google Scholar]
- 78.Finlay D, Patel S, Dickson LM, et al. Glycogene synthase kinase-3 regulates IGFBP-1 gene transcription through the thymine-rich insulin response element. BMC Molecular Biology. 2004;5, article 15 doi: 10.1186/1471-2199-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fröjdö S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochimica et Biophysica Acta. 2009;1792(2):83–92. doi: 10.1016/j.bbadis.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 80.Dunaif A, Wu X, Lee A, Diamanti-Kandarakis E. Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS) The American Journal of Physiology—Endocrinology and Metabolism. 2001;281(2):E392–E399. doi: 10.1152/ajpendo.2001.281.2.E392. [DOI] [PubMed] [Google Scholar]
- 81.Mukherjee S, Maitra A. Molecular & genetic factors contributing to insulin resistance in polycystic ovary syndrome. Indian Journal of Medical Research. 2010;131(6):743–760. [PubMed] [Google Scholar]
- 82.Zhang G, Garmey JC, Veldhuis JD. Interactive stimulation by luteinizing hormone and insulin of the steroidogenic acute regulatory (StAR) protein and 17α-hydroxylase/17, 20-lyase (CYP17) genes in porcine theca cells. Endocrinology. 2000;141(8):2735–2742. doi: 10.1210/endo.141.8.7595. [DOI] [PubMed] [Google Scholar]
- 83.Jamnongjit M, Hammes SR. Ovarian steroids: the good, the bad, and the signals that raise them. Cell Cycle. 2006;5(11):1178–1183. doi: 10.4161/cc.5.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Book C, Dunaif A. Selective insulin resistance in the polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism. 1999;84(9):3110–3116. doi: 10.1210/jcem.84.9.6010. [DOI] [PubMed] [Google Scholar]
- 85.Méndez E, Montserrat N, Planas JV. Modulation of the steroidogenic activity of luteinizing hormone by insulin and insulin-like growth factor-I through interaction with the cAMP-dependent protein kinase signaling pathway in the trout ovary. Molecular and Cellular Endocrinology. 2005;229(1-2):49–56. doi: 10.1016/j.mce.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 86.Rice S, Christoforidis N, Gadd C, et al. Impaired insulin-dependent glucose metabolism in granulosa-lutein cells from anovulatory women with polycystic ovaries. Human Reproduction. 2005;20(2):373–381. doi: 10.1093/humrep/deh609. [DOI] [PubMed] [Google Scholar]
- 87.Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. Journal of Clinical Investigation. 1995;96(2):801–810. doi: 10.1172/JCI118126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bremer AA, Miller WL. The serine phosphorylation hypothesis of polycystic ovary syndrome: a unifying mechanism for hyperandrogenemia and insulin resistance. Fertility and Sterility. 2008;89(5):1039–1048. doi: 10.1016/j.fertnstert.2008.02.091. [DOI] [PubMed] [Google Scholar]
- 89.Draznin B. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85 α: the two sides of a coin. Diabetes. 2006;55(8):2392–2397. doi: 10.2337/db06-0391. [DOI] [PubMed] [Google Scholar]
- 90.Nestler JE. Inositolphosphoglycans (IPGs) as mediators of insulin’s steroidogenic actions. Journal of Basic and Clinical Physiology and Pharmacology. 1998;9(2–4):197–204. doi: 10.1515/jbcpp.1998.9.2-4.197. [DOI] [PubMed] [Google Scholar]
- 91.Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. Journal of Clinical Endocrinology and Metabolism. 1998;83(6):2001–2005. doi: 10.1210/jcem.83.6.4886. [DOI] [PubMed] [Google Scholar]
- 92.de Pergola G. The adipose tissue metabolism: role of testosterone and dehydroepiandrosterone. International Journal of Obesity. 2000;24(supplement 2):S59–S63. doi: 10.1038/sj.ijo.0801280. [DOI] [PubMed] [Google Scholar]
- 93.Boden G. Obesity, insulin resistance and free fatty acids. Current Opinion in Endocrinology, Diabetes and Obesity. 2011;18(2):139–143. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giallauria F, Palomba S, Vigorito C, et al. Androgens in polycystic ovary syndrome: the role of exercise and diet. Seminars in Reproductive Medicine. 2009;27(4):306–315. doi: 10.1055/s-0029-1225258. [DOI] [PubMed] [Google Scholar]
- 95.Allemand MC, Irving BA, Asmann YW, et al. Effect of testosterone on insulin stimulated IRS1 ser phosphorylation in primary rat myotubes—a potential model for PCOS-related insulin resistance. PLoS ONE. 2009;4(1) doi: 10.1371/journal.pone.0004274.e4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Corbould A. Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. Journal of Endocrinology. 2007;192(3):585–594. doi: 10.1677/joe.1.07070. [DOI] [PubMed] [Google Scholar]
- 97.Pasquali R, Gambineri A, Pagotto U. The impact of obesity on reproduction in women with polycystic ovary syndrome. BJOG: An International Journal of Obstetrics and Gynaecology. 2006;113(10):1148–1159. doi: 10.1111/j.1471-0528.2006.00990.x. [DOI] [PubMed] [Google Scholar]
- 98.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 99.Toprak S, Yönem A, Cakir B, et al. Insulin resistance in nonobese patients with polycystic ovary syndrome. Hormone Research. 2001;55(2):65–70. doi: 10.1159/000049972. [DOI] [PubMed] [Google Scholar]
- 100.Mukherjee S, Maitra A. Molecular & genetic factors contributing to insulin resistance in polycystic ovary syndrome. Indian Journal of Medical Research. 2010;131(6):743–760. [PubMed] [Google Scholar]
- 101.Diamanti-Kandarakis E, Piperi C, Korkolopoulou P, et al. Accumulation of dietary glycotoxins in the reproductive system of normal female rats. Journal of Molecular Medicine. 2007;85(12):1413–1420. doi: 10.1007/s00109-007-0246-6. [DOI] [PubMed] [Google Scholar]
- 102.Kandaraki E, Chatzigeorgiou A, Piperi C, et al. Reduced ovarian glyoxalase-I activity by dietary glycotoxins and androgen excess: a causative link to polycystic ovarian syndrome. Molecular Medicine. 2012;18:1183–1189. doi: 10.2119/molmed.2012.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stocco C. Tissue physiology and pathology of aromatase. Steroids. 2012;77(1-2):27–35. doi: 10.1016/j.steroids.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pasquali R, Casimirri F. The impact of obesity on hyperandrogenism and polycystic ovary syndrome in premenopausal women. Clinical Endocrinology. 1993;39(1):1–16. doi: 10.1111/j.1365-2265.1993.tb01744.x. [DOI] [PubMed] [Google Scholar]
- 105.Pasquali R, Vicennati V. The abdominal obesity phenotype and insulin resistance are associated with abnormalities of the hypothalamic-pituitary-adrenal axis in humans. Hormone and Metabolic Research. 2000;32(11-12):521–525. doi: 10.1055/s-2007-978680. [DOI] [PubMed] [Google Scholar]
- 106.Simpson ER. Aromatase: biologic relevance of tissue-specific expression. Seminars in Reproductive Medicine. 2004;22(1):11–23. doi: 10.1055/s-2004-823023. [DOI] [PubMed] [Google Scholar]
- 107.Dardeno TA, Chou SH, Moon H, Chamberland JP, Fiorenza CG, Mantzoros CS. Leptin in human physiology and therapeutics. Frontiers in Neuroendocrinology. 2010;31(3):377–393. doi: 10.1016/j.yfrne.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van Harmelen V, Reynisdottir S, Eriksson P, et al. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes. 1998;47(6):913–917. doi: 10.2337/diabetes.47.6.913. [DOI] [PubMed] [Google Scholar]
- 109.Yang G, Ge H, Boucher A, Yu X, Li C. Modulation of direct leptin signaling by soluble leptin receptor. Molecular Endocrinology. 2004;18(6):1354–1362. doi: 10.1210/me.2004-0027. [DOI] [PubMed] [Google Scholar]
- 110.Lammert A, Kiess W, Bottner A, Glasow A, Kratzsch J. Soluble leptin receptor represents the main leptin binding activity in human blood. Biochemical and Biophysical Research Communications. 2001;283(4):982–988. doi: 10.1006/bbrc.2001.4885. [DOI] [PubMed] [Google Scholar]
- 111.Brannian JD, Hansen KA. Leptin and ovarian folliculogenesis: implications for ovulation induction and ART outcomes. Seminars in Reproductive Medicine. 2002;20(2):103–112. doi: 10.1055/s-2002-32501. [DOI] [PubMed] [Google Scholar]
- 112.Sadaf Farooqi I, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. Journal of Clinical Investigation. 2002;110(8):1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chan JL, Matarese G, Shetty GK, et al. Differential regulation of metabolic, neuroendocrine, and immune function by leptin in humans. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(22):8481–8486. doi: 10.1073/pnas.0505429103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hill JW, Elmquist JK, Elias CF. Hypothalamic pathways linking energy balance and reproduction. The American Journal of Physiology—Endocrinology and Metabolism. 2008;294(5):E827–E832. doi: 10.1152/ajpendo.00670.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hameed S, Jayasena CN, Dhillo WS. Kisspeptin and fertility. Journal of Endocrinology. 2011;208(2):97–105. doi: 10.1677/JOE-10-0265. [DOI] [PubMed] [Google Scholar]
- 116.Mantzoros CS, Dunaif A, Flier JS. Leptin concentrations in the polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism. 1997;82(6):1687–1691. doi: 10.1210/jcem.82.6.4017. [DOI] [PubMed] [Google Scholar]
- 117.Mendonça HC, Montenegro RM, Jr., Foss MC, Silva de Sá MF, Ferriani RA. Positive correlation of serum leptin with estradiol levels in patients with polycystic ovary syndrome. Brazilian Journal of Medical and Biological Research. 2004;37(5):729–736. doi: 10.1590/s0100-879x2004000500015. [DOI] [PubMed] [Google Scholar]
- 118.Chakrabarti J. Serum leptin level in women with polycystic ovary syndrome: correlation with adiposity, insulin, and circulating testosterone. Annals of Medical and Health Sciences Research. 2013;3(2):191–196. doi: 10.4103/2141-9248.113660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yildizhan R, Ilhan GA, Yildizhan B, Kolusari A, Adali E, Bugdayci G. Serum retinol-binding protein 4, leptin, and plasma asymmetric dimethylarginine levels in obese and nonobese young women with polycystic ovary syndrome. Fertility and Sterility. 2011;96(1):246–250. doi: 10.1016/j.fertnstert.2011.04.073. [DOI] [PubMed] [Google Scholar]
- 120.Löffler S, Aust G, Köhler U, Spanel-Borowski K. Evidence of leptin expression in normal and polycystic human ovaries. Molecular Human Reproduction. 2001;7(12):1143–1149. doi: 10.1093/molehr/7.12.1143. [DOI] [PubMed] [Google Scholar]
- 121.Yang Z, Kong B, Mosser DM, Zhang X. TLRs, macrophages, and NK cells: our understandings of their functions in uterus and ovary. International Immunopharmacology. 2011;11(10):1442–1450. doi: 10.1016/j.intimp.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 122.Sepilian VP, Crochet JR, Nagamani M. Serum soluble leptin receptor levels and free leptin index in women with polycystic ovary syndrome: relationship to insulin resistance and androgens. Fertility and Sterility. 2006;85(5):1441–1447. doi: 10.1016/j.fertnstert.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 123.Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance. Annual Review of Physiology. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 124.Jung CH, Kim MS. Molecular mechanisms of central leptin resistance in obesity. Archives of Pharmacal Research. 2013;36(2):201–207. doi: 10.1007/s12272-013-0020-y. [DOI] [PubMed] [Google Scholar]
- 125.Spritzer PM, Poy M, Wiltgen D, Mylius LS, Capp E. Leptin concentrations in hirsute women with polycystic ovary syndrome or idiopathic hirsutism: influence on LH and relationship with hormonal, metabolic, and anthropometric measurements. Human Reproduction. 2001;16(7):1340–1346. doi: 10.1093/humrep/16.7.1340. [DOI] [PubMed] [Google Scholar]
- 126.LaZovic G, Radivojevic U, Milicevic S, Spremovic S. Influence of adiposity on leptin, LH and androgen levels in lean, overweight and obese PCOS patients. International Journal of Fertility and Women’s Medicine. 2007;52(2-3):82–88. [PubMed] [Google Scholar]
- 127.Shi YH, Zhao DN, Zhao JL, et al. Characteristics of glucose metabolism in non-obese and obese women with polycystic ovarian syndrome. Zhonghua Fu Chan Ke Za Zhi. 2010;45(8):575–577. [PubMed] [Google Scholar]
- 128.Unluer AN, Findik RB, Sevinc N, Karakaya J. Comparison of HbA1c levels in obese and non-obese polycystic ovarian patients. Clinical and Experimental Obstetrics and Gynecology. 2013;40(1):148–150. [PubMed] [Google Scholar]
- 129.Silfen ME, Denburg MR, Manibo AM, et al. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between nonobese and obese adolescents. Journal of Clinical Endocrinology and Metabolism. 2003;88(10):4682–4688. doi: 10.1210/jc.2003-030617. [DOI] [PubMed] [Google Scholar]
- 130.Diamanti-Kandarakis E, Xyrafis X, Boutzios G, Christakou C. Pancreatic β-cells dysfunction in polycystic ovary syndrome. Panminerva Medica. 2008;50(4):315–325. [PubMed] [Google Scholar]
- 131.Messer C, Boston R, Leroith D, et al. Pancreatic β-cell dysfunction in polycystic ovary syndrome: the role of metformin. Endocrine Practice. 2012;18(5):685–693. doi: 10.4158/EP11375.OR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Goodarzi MO, Erickson S, Port SC, Jennrich RI, Korenman SG. β-cell function: a key pathological determinant in polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism. 2005;90(1):310–315. doi: 10.1210/jc.2004-1006. [DOI] [PubMed] [Google Scholar]
- 133.Vrbíková J, Bendlová B, Hill M, Vanková M, Vondra K, Stárka L. Insulin sensitivity and beta-cell function in women with polycystic ovary syndrome. Diabetes Care. 2002;25(7):1217–1222. doi: 10.2337/diacare.25.7.1217. [DOI] [PubMed] [Google Scholar]
- 134.Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Reviews in Endocrine and Metabolic Disorders. 2007;8(2):127–141. doi: 10.1007/s11154-007-9046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rae M, Grace C, Hogg K, et al. The pancreas is altered by in utero androgen exposure: implications for clinical conditions such as polycystic ovary syndrome (PCOS) PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0056263.e56263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Powell BL, Haddad L, Bennett A, et al. Analysis of multiple data sets reveals no association between the insulin gene variable number tandem repeat element and polycystic ovary syndrome or related traits. Journal of Clinical Endocrinology and Metabolism. 2005;90(5):2988–2993. doi: 10.1210/jc.2004-2485. [DOI] [PubMed] [Google Scholar]
- 137.Eyvazzadeh AD, Pennington KP, Pop-Busui R, Sowers M, Zubieta J, Smith YR. The role of the endogenous opioid system in polycystic ovary syndrome. Fertility and Sterility. 2009;92(1):1–12. doi: 10.1016/j.fertnstert.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 138.Morales AJ, Laughlin GA, Bützow T, Maheshwari H, Baumann G, Yen SSC. Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: common and distinct features. Journal of Clinical Endocrinology and Metabolism. 1996;81(8):2854–2864. doi: 10.1210/jcem.81.8.8768842. [DOI] [PubMed] [Google Scholar]
- 139.Smith YR, Zubieta J, del Carmen MG, et al. Brain opioid receptor measurements by positron emission tomography in normal cycling women: relationship to luteinizing hormone pulsatility and gonadal steroid hormones. Journal of Clinical Endocrinology and Metabolism. 1998;83(12):4498–4505. doi: 10.1210/jcem.83.12.5351. [DOI] [PubMed] [Google Scholar]
- 140.Ek I, Arner P, Bergqvist A, Carlström K, Wahrenberg H. Impaired adipocyte lipolysis in nonobese women with the polycystic ovary syndrome: a possible link to insulin resistance? Journal of Clinical Endocrinology and Metabolism. 1997;82(4):1147–1153. doi: 10.1210/jcem.82.4.3899. [DOI] [PubMed] [Google Scholar]
- 141.Invitti C, de Martin M, Delitala G, Veldhuis JD, Cavagnini F. Altered morning and nighttime pulsatile corticotropin and cortisol release in polycystic ovary syndrome. Metabolism. 1998;47(2):143–148. doi: 10.1016/s0026-0495(98)90210-4. [DOI] [PubMed] [Google Scholar]
- 142.Rodin A, Thakkar H, Taylor N, Clayton R. Hyperandrogenism in polycystic ovary syndrome: evidence of dysregulation of 11β-hydroxysteroid dehydrogenase. New England Journal of Medicine. 1994;330(7):460–465. doi: 10.1056/NEJM199402173300703. [DOI] [PubMed] [Google Scholar]
- 143.Dhindsa G, Bhatia R, Dhindsa M, Bhatia V. Insulin resistance, insulin sensitization and inflammation in polycystic ovarian syndrome. Journal of Postgraduate Medicine. 2004;50(2):140–144. [PubMed] [Google Scholar]
- 144.Chazenbalk G, Chen YH, Heneidi S, et al. Abnormal expression of genes involved in inflammation, lipid metabolism, and Wnt signaling in the adipose tissue of polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism. 2012;97(5):E765–E770. doi: 10.1210/jc.2011-2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Escobar-Morreale HF, Luque-Ramírez M, González F. Circulating inflammatory markers in polycystic ovary syndrome: a systematic review and metaanalysis. Fertility and Sterility. 2011;95(3):1048.e2–1058.e2. doi: 10.1016/j.fertnstert.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.González F. Inflammation in polycystic ovary syndrome: underpinning of insulin resistance and ovarian dysfunction. Steroids. 2012;77(4):300–305. doi: 10.1016/j.steroids.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Xiong YL, Liang XY, Yang X, Li Y, Wei LN. Low-grade chronic inflammation in the peripheral blood and ovaries of women with polycystic ovarian syndrome. European Journal of Obstetrics Gynecology and Reproductive Biology. 2011;159(1):148–150. doi: 10.1016/j.ejogrb.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 148.Vural P, Değirmencioğlu S, Saral NY, Akgül C. Tumor necrosis factor α (-308), interleukin-6 (-174) and interleukin-10 (-1082) gene polymorphisms in polycystic ovary syndrome. European Journal of Obstetrics and Gynecology and Reproductive Biology. 2010;150(1):61–65. doi: 10.1016/j.ejogrb.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 149.Tao T, Li S, Zhao A, Zhang Y, Liu W. Expression of the CD11c gene in subcutaneous adipose tissue is associated with cytokine level and insulin resistance in women with polycystic ovary syndrome. European Journal of Endocrinology. 2012;167(5):705–713. doi: 10.1530/EJE-12-0340. [DOI] [PubMed] [Google Scholar]
- 150.Spaczynski RZ, Arici A, Duleba AJ. Tumor necrosis factor-α stimulates proliferation of rat ovarian theca-interstitial cells. Biology of Reproduction. 1999;61(4):993–998. doi: 10.1095/biolreprod61.4.993. [DOI] [PubMed] [Google Scholar]
- 151.Piotrowski PC, Rzepczynska IJ, Kwintkiewicz J, Duleba AJ. Oxidative stress induces expression of CYP11A, CYP17, STAR and 3bHSD in rat theca-interstitial cells. Journal of the Society for Gynecologic Investigation. 2005;12(2, supplement, article 319A) [Google Scholar]
- 152.Duleba AJ, Dokras A. Is PCOS an inflammatory process? Fertility and Sterility. 2012;97(1):7–12. doi: 10.1016/j.fertnstert.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Martin SS, Qasim A, Reilly MP. Leptin resistance. A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. Journal of the American College of Cardiology. 2008;52(15):1201–1210. doi: 10.1016/j.jacc.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. Journal of Cellular and Molecular Medicine. 2010;14(1-2):70–78. doi: 10.1111/j.1582-4934.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Moran LJ, Misso ML, Wild RA, Norman RJ. Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Human Reproduction Update. 2010;16(4):347–363. doi: 10.1093/humupd/dmq001. [DOI] [PubMed] [Google Scholar]
- 156.Wild RA, Carmina E, Diamanti-Kandarakis E, et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the androgen excess and polycystic ovary syndrome (AE-PCOS) society. Journal of Clinical Endocrinology and Metabolism. 2010;95(5):2038–2049. doi: 10.1210/jc.2009-2724. [DOI] [PubMed] [Google Scholar]
- 157.Marshall JC, Dunaif A. Should all women with PCOS be treated for insulin resistance? Fertility and Sterility. 2012;97(1):18–22. doi: 10.1016/j.fertnstert.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li XJ, Yu YX, Liu CQ, et al. Metformin vs thiazolidinediones for treatment of clinical, hormonal and metabolic characteristics of polycystic ovary syndrome: a meta-analysis. Clinical Endocrinology. 2011;74(3):332–339. doi: 10.1111/j.1365-2265.2010.03917.x. [DOI] [PubMed] [Google Scholar]
- 159.Hoeger KM. Polycystic ovary syndrome, inflammation, and statins: do we have the right target? Journal of Clinical Endocrinology and Metabolism. 2009;94(1):35–37. doi: 10.1210/jc.2008-2376. [DOI] [PubMed] [Google Scholar]
- 160.Harrison CL, Lombard CB, Moran LJ, Teede HJ. Exercise therapy in polycystic ovary syndrome: a systematic review. Human Reproduction Update. 2011;17(2):171–183. doi: 10.1093/humupd/dmq045. [DOI] [PubMed] [Google Scholar]
- 161.Harrison CL, Stepto NK, Hutchison SK, Teede HJ. The impact of intensified exercise training on insulin resistance and fitness in overweight and obese women with and without polycystic ovary syndrome. Clinical Endocrinology. 2012;76(3):351–357. doi: 10.1111/j.1365-2265.2011.04160.x. [DOI] [PubMed] [Google Scholar]
- 162.Poehlman ET, Dvorak RV, DeNino WF, Brochu M, Ades PA. Effects of resistance training and endurance training on insulin sensitivity in nonobese, young women: a controlled randomized trial. Journal of Clinical Endocrinology and Metabolism. 2000;85(7):2463–2468. doi: 10.1210/jcem.85.7.6692. [DOI] [PubMed] [Google Scholar]
- 163.Mavropoulos JC, Yancy WS, Hepburn J, Westman EC. The effects of a low-carbohydrate, ketogenic diet on the polycystic ovary syndrome: a pilot study. Nutrition and Metabolism. 2005;2, article 35 doi: 10.1186/1743-7075-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sumithran P, Proietto J. Ketogenic diets for weight loss: a review of their principles, safety and efficacy. Obesity Research and Clinical Practice. 2008;2(1):1–13. doi: 10.1016/j.orcp.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 165.Lagiou P, Sandin S, Lof M, Trichopoulos D, Adami HO, Weiderpass E. Low carbohydrate-high protein diet and incidence of cardiovascular diseases in Swedish women: prospective cohort study. The British Medical Journal. 2012;344 doi: 10.1136/bmj.e4026.e4026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Moran LJ, Pasquali R, Teede HJ, Hoeger KM, Norman RJ. Treatment of obesity in polycystic ovary syndrome: a position statement of the androgen excess and polycystic ovary syndrome society. Fertility and Sterility. 2009;92(6):1966–1982. doi: 10.1016/j.fertnstert.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 167.Diamanti-Kandarakis E, Bourguignon J, Giudice LC, et al. Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocrine Reviews. 2009;30(4):293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]