

Abstract
The Indian hedgehog (Ihh) pathway plays an essential role in facilitating chondrocyte hypertrophy and bone formation during skeletal development. Nkx3.2 is initially induced in chondrocyte precursor cells, maintained in early-stage chondrocytes, and down-regulated in terminal-stage chondrocytes. Consistent with these expression patterns, Nkx3.2 has been shown to enhance chondrocyte differentiation and cell survival, while inhibiting chondrocyte hypertrophy and apoptosis. Thus, in this work, we investigate whether Nkx3.2, an early stage chondrogenic factor, can be regulated by Ihh, a key regulator for chondrocyte hypertrophy. Here, we show that Ihh signaling can induce proteasomal degradation of Nkx3.2. In addition, we found that Ihh can suppress levels of Lrp (Wnt co-receptor) and Sfrp (Wnt antagonist) expression, which, in turn, may selectively enhance Lrp-independent non-canonical Wnt pathways in chondrocyte. In agreement with these findings, Ihh-induced Nkx3.2 degradation requires Wnt5a, which is capable of triggering Nkx3.2 degradation. Finally, we found that Nkx3.2 protein levels in chondrocytes are remarkably elevated in mice defective in Ihh signaling by deletion of either Ihh or Smoothened. Thus, these results suggest that Ihh/Wnt5a signaling may play a role in negative regulation of Nkx3.2 for appropriate progression of chondrocyte hypertrophy during chondrogenesis.
Introduction
Nkx3.2, also called Bapx1, is the vertebrate homologue of Drosophila bagpipe, and its initial expression can be observed in chondrocyte precursor cells during embryonic development and, later, its expression is maintained in chondrocytes of various skeletal elements [1-7]. Nkx3.2-deficient mice are perinatal lethal and exhibit significant skeletal phenotypes, including major hypoplasia in vertebrae [8-11]. Interestingly, during cartilage development, Nkx3.2 expression is maintained at high levels in early-stage chondrocytes, while it is down-regulated in terminal-stage chondrocytes [1, 2, 12, 13]. Consistent with these expression patterns, Nkx3.2 has been reported to support cell viability for early-stage chondrocytes [13, 14], whereas chondrocyte hypertrophy can be inhibited by Nkx3.2 [2, 15, 16]. Thus, while these previous studies clearly indicate that Nkx3.2 may function in a stage-specific manner during cartilage development, the molecular mechanism for regulation of Nkx3.2 during cartilage development is not well understood.
Indian Hedgehog (Ihh) is a member of the Hedgehog (Hh) family, which has been shown to regulate various developmental processes and cancer pathogenesis; in particular, Ihh signaling plays an essential role in both chondrocyte hypertrophy and bone formation during skeletal development [17-29]. These previous studies have demonstrated that binding of hedgehog ligand to the cell surface receptor patched (Ptc), which is normally associated with smoothened (Smo) to suppress signal transmission from Smo, releases Smo from Ptc; subsequently, the liberated Smo triggers downstream signaling cascades to activate glioma-associated oncogene homolog (Gli) family members, which, in turn, control various hedgehog-dependent cellular events. During cartilage development, expression of Ihh in growth plates is highly restricted in the border areas between proliferative chondrocytes and hypertrophic chondrocytes (i.e., pre-hypertrophic chondrocytes), and a remote crosstalk between Ihh in pre-hypertrophic chondrocytes and parathyroid hormone related protein (PTHrP) in the perichondrium has been shown to regulate chondrocyte proliferation, hypertrophic maturation, and bone formation [21-25, 30-35]. Besides, Ihh has also been suggested to function to promote chondrocyte hypertrophy in a PTHrP-independent manner [24, 36]. Thus, while previous studies have verified the importance of the Ihh signaling pathway in endochondral bone development, the precise role of Ihh has not been fully elucidated.
It has been demonstrated that various Wnts can control chondrocyte differentiation, proliferation, and hypertrophic maturation during cartilage development; in particular, Wnt5a has been shown to play a critical role in chondrocyte hypertrophy regulation [37-49]. In addition, a broad range of intracellular factors such as calcium ion, protein kinases, transcription factors, and ubiquitin E3 ligases have been indicated in various non-canonical Wnt signaling pathways [43, 44, 50-61]. In this study, we found that Ihh signaling can induce proteasomal degradation of Nkx3.2 via a Wnt5a-dependent pathway employing CK2 protein kinase and Siah1 ubiquitin E3 ligase. Furthermore, we support the physiological relevance of these findings by analyzing knockout mice with deletion of Ihh or Smoothened.
Experimental
Cell culture and transfection
ATDC5 cells were obtained from RIKEN BRC Cell Bank and maintained in Dulbecco's modified Eagle's medium (DMEM)-Ham's F-12 (1:1) supplemented with 5% fetal bovine serum (FBS). HEK 293T cells were purchased from American Type Culture Collection (ATCC) and grown in DMEM supplemented with 10% FBS. Cells were transfected under serum-free conditions using VivaMagic (Vivagen) according to the manufacturer's instructions. An empty vector, pCS2, was used to adjust total DNA amounts where necessary.
Chemical reagents and antibodies
Purmorphamine (PMP), cyclopamine (CCP), 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB), 4,5,6,7-tetrabromo-1H-benzotriazole (TBB), 4′,6-diamidino-2-phenylindole (DAPI), MG132, and Inonomycin were purchased from Calbiochem. Rabbit polyclonal and monoclonal (9E10) Myc antibodies were purchased from Upstate Biotechnology. HA monoclonal antibody was purchased from Roche, and anti-V5 was obtained from Invitrogen. Flag monoclonal M2 antibody was obtained from Sigma, and anti-Ihh was from Santa Cruz Biotechnology. Anti-Gli1, anti-Wnt5a, and anti-CK2α were purchased from Cell Signaling, and anti-PCNA was obtained from BD Biosciences. Anti-Nkx3.2 and anti-CK2β were obtained from Abcam and anti-GAPDH was purchased from AbFrontier. Secondary antibodies for western blotting and immunohistochemistry were purchased from Cell Signaling and Jackson ImmunoResearch, respectively.
Short hairpin RNA (shRNA) and reverse transcription-polymerase chain reaction (RT-PCR)
shRNAs for mouse Gli1, Gli2, Gli3, Wnt5a, CK2, and Siah1 were purchased from Open Biosystems and were transfected using Fugene 6 (Roche). Total RNA was isolated using the RNAspin Mini kit (GE Healthcare). The Superscript III cDNA synthesis kit (Invitrogen) was used for cDNA synthesis. PCR reactions were performed with the following oligonucleotides consisting of targeted mouse cDNA sequences:
| GAPDH: | 5′-TTTGTGATGGGTGTGAACCACG-3′ (sense) |
| 5′-TTGTGAGGGAGATGCTCAGTGTTG-3′ (antisense) | |
| Rps18: | 5′-TGCGAGTACTCAACACCAACATCG-3′ (sense) |
| 5′-GCCAGTGGTCCTGGTGTGCTGA-3′ (antisense) | |
| Smo: | 5′-TGCCACCAGTGCAGTTCCTCG-3′ (sense; endogenous-specific) |
| 5′-GGTGTGCTCTCAGGAAGAGCCAT-3′ (antisense; endogenous/exogenous) | |
| SmoA1: | 5′-CCCATGGAGAAGTCCGAGTC-3′ (sense; exogenous-specific) |
| 5′-GGTGTGCTCTCAGGAAGAGCCAT-3′ (antisense; endogenous/exogenous) | |
| Gli1: | 5′-TCACCCTGCCATGAAACTTTCACC-3′ (sense) |
| 5′-TCATGGGAAAGAGGAGGGCTCA-3′ (antisense) | |
| Nkx3.2: | 5′-AACCGTCGCTACAAGACCAAACG-3′ (sense) |
| 5′-GGGACGCAGGAATCCTTCTTTG-3′ (antisense) | |
| Ihh: | 5′-CATGACCCAGCGCTGCAAGG-3′ (sense) |
| 5′-CCTGGAAAGCTCTCAGCCGG-3′ (antisense) | |
| Col X: | 5′- CCCCATCCCATTTATGAGATTCTG-3′ (sense) |
| 5′- GAGCCATACCTGGTCATTTTCTGTG-3′ (antisense) | |
| Wnt1: | 5′-CCTCACGACCTCGTCTA-3′ (sense) |
| 5′-AGTGGAAGGTGCAGTTG-3′ (antisense) | |
| Wnt3a: | 5′-GGAGTTTGCCGATGCCA-3′ (sense) |
| 5′-CGCACCCATCTATGCCA-3′ (antisense) | |
| Wnt4: | 5′-TTTGACGGTGCCACGGA-3′ (sense) |
| 5′-TTGCGCTGTGTGGAAGC-3′ (antisense) | |
| Wnt5a: | 5′-TCCGGACTACTGTGTGC-3′ (sense) |
| 5′-AGCAGCACCAGTGAAAC-3′ (antisense) | |
| Wnt7a: | 5′-ATATCGAGAAGTCACCCA-3′ (sense) |
| 5′-AGCACCAGTGGAATTTG-3′ (antisense) | |
| Lrp5: | 5′-AACGTGGACGTGTTTTA-3′ (sense) |
| 5′-AGTCCGAATTCAAGTCC-3′ (antisense) | |
| Lrp6: | 5′-GTGACTATGCTCCTAGCC-3′ (sense) |
| 5′-TGGTGGGAGTAACTCCT-3′ (antisense) | |
| Sfrp1: | 5′-GGCCCATCAAGAAGAAG-3′ (sense) |
| 5′-GGGACACTCGTGGTTTT-3′ (antisense) | |
| Sfrp2: | 5′-GAGACACCAAGATCATCC-3′ (sense) |
| 5′-GTTTCACGGAGGTGATC-3′ (antisense) | |
| Sfrp3: | 5′-GTGACTATGCTCCTAGCC-3′ (sense) |
| 5′-TGGTGGGAGTAACTCCT-3′ (antisense) |
Immunoprecipitation (IP) and immunoblotting
Cells were washed in phosphate-buffered saline (PBS) and lysed in buffer containing 50 mM Tris (pH 7.8), 150 mM NaCl, 1 mM EDTA, 2 mM imidazole, 1 mM NaF, 1.15 mM Na2MoO4, 1 mM Na3VO4, 4 mM C4H4Na2O6, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT), 10% glycerol, 0.5% NP40, and protease inhibitors (Complete, EDTA-free Protease Inhibitor Cocktail Tablets; Roche). Cell extracts were centrifuged for 10 min at 13,000 g at 4°C, and then supernatants were subjected to IP and western blotting (WB) analysis. Visualization of the immunoblots was performed using the ECL Detection kit from GE Healthcare. For standard western blotting analysis, cells were lysed in buffer containing 50 mM Tris (pH 6.8), 1 mM DTT, 8.75% glycerol, and 2.5% sodium dodecyl sulfate (SDS). After boiling, protein concentrations were determined using the Bio-Rad protein assay kit, and equal amounts of protein were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and WB.
Immunohistochemistry (IHC)
For Figure 7A, E16.5 mouse limbs were fixed in 4% paraformaldehyde (PFA), and for Figures 7E and 7F, E18.5 mouse limbs were fixed in 10 % buffered formalin overnight at room temperature. The fixed embryos were then processed and embedded in paraffin before sectioning at 4-μm (Figure 7A) and 6-μm (Figure 7E and 7F) and mounted on silane-coated slides (Muto-Glass). For E18.5 embryo limbs, decalcification was performed with 14 % EDTA/PBS (pH7.4) for 48 hours after formalin fixation. Deparaffinization was performed using xylene, a xylene/ethanol mixture, and ethanol. For antigen retrieval, sections were incubated with 10 mM citrate (pH 6.0) and 0.05% Tween-20 for 20 min at 80°C, followed by a double wash in 1× PBS with 0.05% Tween-20. Sections were then pre-incubated with blocking buffer containing 5% goat (or bovine) serum, 0.05% sodium azide, and 0.1% Triton X-100 in 1× PBS for 45 min at 25°C, and incubated overnight at 4°C with primary antibodies in antibody dilution buffer containing 1% goat (or bovine) serum, 0.05% sodium azide, and 0.02% Tween-20. Prior to microscopy, mounting was performed with anti-fading solution containing 25 mM Tris (pH 8.7), 10% polyvinyl alcohol, 5% glycerol, and 2.5% DABCO.
Figure 7. Nkx3.2 degradation triggered by Ihh during chondrogenesis.
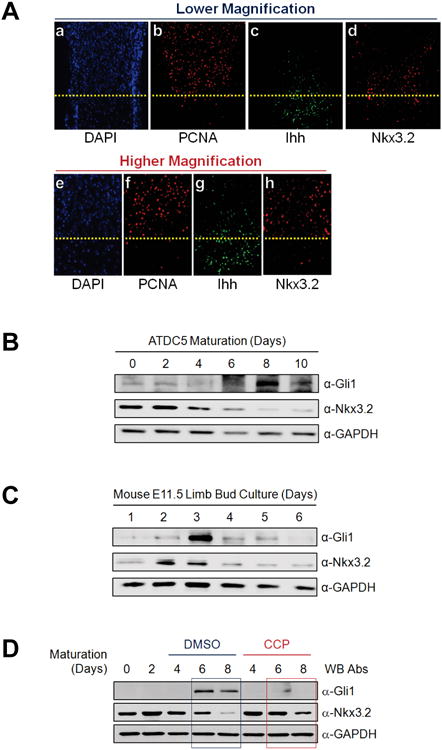
(A) E16.5 mouse humeri sections were stained with DAPI or processed for F-IHC to detect Nkx3.2, Ihh and PCNA. The border between proliferative and pre-hypertrophic chondrocytes is indicated with a yellow dotted line.
(B) Confluent ATDC5 cultures were subjected to in vitro chondrocyte maturation with Insulin-Transferrin-Selenium (ITS) supplements and 10 ng/ml of BMP-2. Total cell lysates were harvested after 0, 2, 4, 6, 8, or 10 days, and protein levels of Gli1, Nkx3.2, and GAPDH were evaluated by western blotting.
(C) E11.5 mouse limb bud culture was performed for 1, 2, 3, 4, 5 and 6 days. At each time point, total cell lysates were prepared and analyzed by western blotting for Gli1, Nkx3.2, and GAPDH.
(D) ATDC5 in vitro maturation assay as described in (B) was performed in the presence of 0.5% DMSO (lanes 3-5) or 5 μM cyclopamine (CCP) (lanes 6-8). Total cell lysates were harvested after 0, 2, 4, 6, or 8 days, and protein levels of Gli1, Nkx3.2, and GAPDH were determined by western blotting.
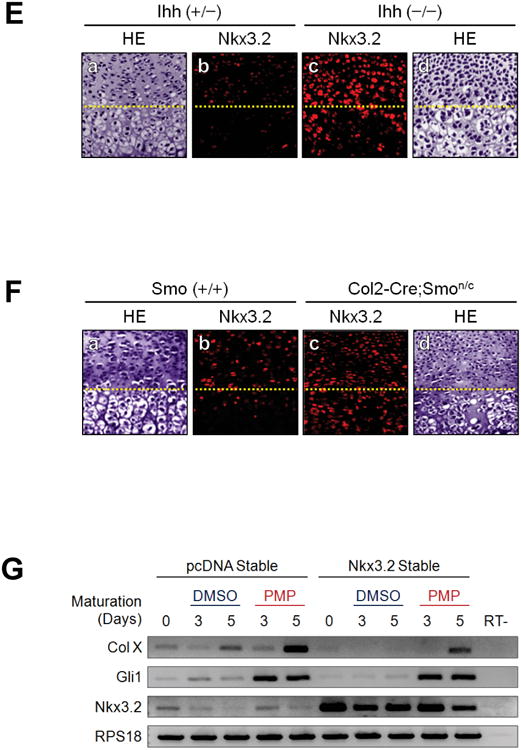
(E) Hematoxylin and eosin (HE) staining or F-IHC for Nkx3.2 were performed with the sections of E18.5 femur obtained from Ihh (+/−) or Ihh (−/−) mouse embryos. The border between the proliferative zone and maturing zone is indicted with a yellow dotted line.
(F) E18.5 femur sections prepared from Smo (+/+) or Col2-Cre;Smonc mouse embryos were processed for HE staining or F-IHC for Nkx3.2. The border between the proliferative zone and hypertrophic zone is indicted with a yellow dotted line.
(G) ATDC5 cells stably expressing pcDNA empty vector or Nkx3.2-HA were subjected to in vitro chondrocyte maturation assays with ITS supplements and 10 ng/ml of BMP-2. For indicated samples, 10 μM PMP was additionally included for the final 3 days and RT-PCR analyses were performed for chondrogenesis markers as shown.
Mouse embryo limb bud culture
E11.5 mouse embryos were isolated and the limb buds were dissected and subjected to micromass cultures. Mesenchymal cells were isolated by digestion with 0.1% dispase at 37°C for 90 min, and isolated cells were dissociated by pipetting and filtering through a 40-μm nylon cell strainer. Filtered cells were then centrifuged at 120 g for 10 min, and cell pellets were resuspended in DMEM/F12 (1:1) supplemented with 10% FBS. The resuspension volume was 5 μl for each limb bud, and 10-μl aliquots were used for each micromass culture. The cultures were maintained in DMEM/F12 (1:1) supplemented with 10% FBS in a humidified, 5% CO2, 37°C incubator for various times up to 6 days.
Results
Nkx3.2 protein degradation can be induced by Indian Hedgehog signaling
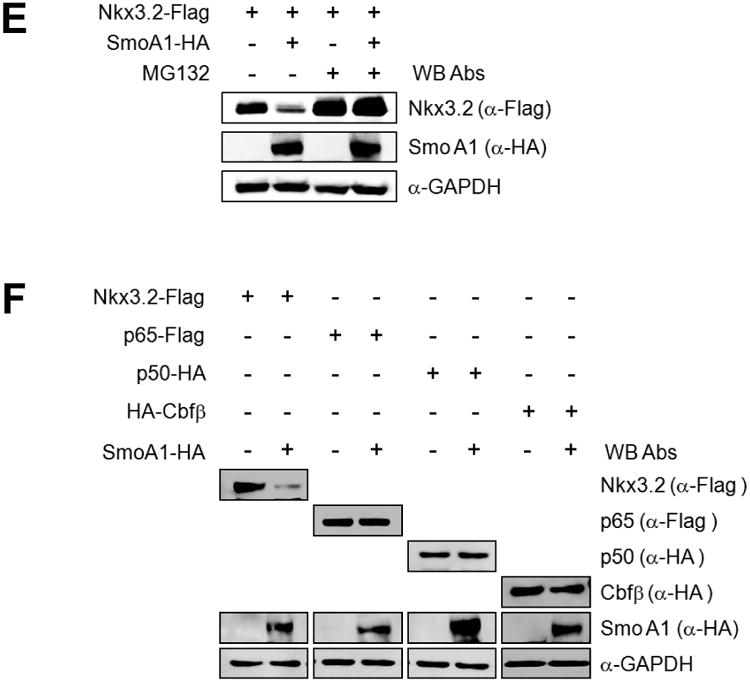
To explore the possibility that Ihh, which enhances chondrocyte hypertrophy, may negatively regulate Nkx3.2, an inhibitor of chondrocyte maturation, we first tested whether a Hedgehog agonist, purmorphamine (PMP) [62-64] can modulate Nkx3.2. Interestingly, PMP treatment dramatically reduced the levels of endogenous Nkx3.2 protein in ATDC5 murine chondrogenic cells [65]; the functional efficacies of PMP were shown by confirming induction of Gli1, a typical Hedgehog-dependent target gene (Figure 1A). In addition, we observed that transient transfection of a constitutively active form of Smoothened (SmoA1) can significantly decrease the levels of co-transfected Nkx3.2 proteins dose-dependently (Figure 1B). Then, to clarify whether the Hedgehog signaling modulates the expression of Nkx3.2 mRNA expression or Nkx3.2 protein stability, we examined the effects of PMP or SmoA1 overexpression on Nkx3.2 mRNA levels, and found that the levels of Nkx3.2 mRNA were not significantly altered by either PMP treatment or lentiviral infection of SmoA1 (Figures 1C and 1D). Consistent with these results, SmoA1-induced Nkx3.2 protein degradation was effectively blocked by MG132, a proteasome inhibitor (Figure 1E), suggesting that the Hedgehog signaling regulates Nkx3.2 at the level of protein degradation. Finally, we have verified that Ihh signaling does not cause random destabilization of cellular proteins by showing that SmoA1 overexpression does not alter the levels of co-transfected NF-κB p65, NF-κB p50, or Cbfβ proteins (Figure 1F). These results indicate that Ihh signaling can induce proteasome-dependent degradation of Nkx3.2.
Figure 1. Indian Hedgehog can induce Nkx3.2 protein degradation.
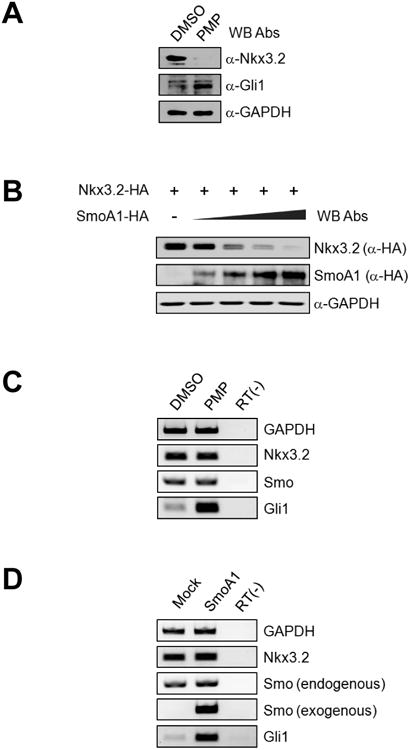
(A) ATDC5 cells were treated with 0.5% DMSO or 10 μM purmorphamine (PMP) for 24 h, and analyzed by western blotting to evaluate levels of endogenous Nkx3.2 protein.
(B) Nkx3.2-HA expression construct (1 μg) was transfected into ATDC5 cells with increasing amounts of SmoA1-HA expression plasmid (0, 0.2, 0.5, 1, and 1.5 μg), and after 48 h, total cell lysates were analyzed by western blotting.
(C) ATDC5 cells were treated with DMSO or 10 μM PMP for 24 h, and mRNA expression levels of GAPDH, Nkx3.2, Smoothened, and Gli1 were determined by RT-PCR.
(D) ATDC5 cells were infected with control or SmoA1 lentivirus. At 48 h post-infection, cells were harvested and then mRNA expression levels of GAPDH, Nkx3.2, endogenous Smoothened, exogenous SmoA1, and Gli1 were determined by RT-PCR.
(E) Nkx3.2-Flag expression plasmid was transfected into ATDC5 cells in the absence or presence of SmoA1-HA expression construct for 48 h. Transfected cells were exposed to 20 μM MG132 for the final 12 h of incubation. Total cell lysates were analyzed by western blotting.
(F) ATDC5 cells were transfected with Nkx3.2-Flag, p65-Flag, p50-HA, or HA-Cbfβ expression constructs in the absence or presence of SmoA1-HA expression plasmid for 48 h, and then total cell lysates were analyzed by western blotting.
Ihh can enhance non-canonical Wnt signaling in chondrocytes
As we found that Ihh can trigger Nkx3.2 protein degradation, we next probed whether a Wnt pathway may participate in this process as well. We suspected a role of Wnt signaling in Ihh-induced Nkx3.2 degradation because various Wnt pathways, along with Ihh, have also been well documented to critically regulate chondrocyte hypertrophy [37, 40, 41, 45-49]. As an initial step, we sought to determine whether Sfrp3 (Frzb), a well-known Wnt antagonist [66-69], can affect Nkx3.2 degradation by Ihh. Interestingly, we found that Nkx3.2 degradation triggered by SmoA1 overexpression can be effectively abolished by Sfrp3 co-transfection (Figure 2A); the anti-Wnt functionality of Sfrp3 was verified by confirming its ability to suppress Wnt3a-induced TOP-FLASH reporter activation (data not shown). These results suggest that Ihh-induced Nkx3.2 degradation requires intact Wnt signaling.
Figure 2. Ihh can modulate Wnt signaling pathways in chondrocytes.
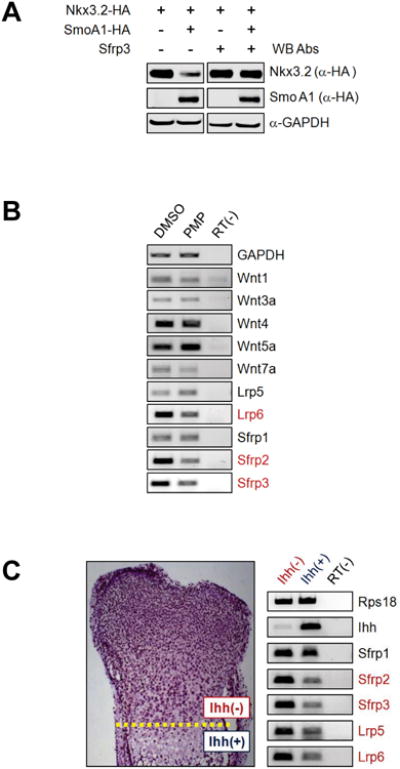
(A) ATDC5 cells were transfected with Nkx3.2-HA expression construct in the absence or presence of SmoA1-HA and/or Sfrp3 expression plasmid for 48 h, and total cell lysates were analyzed by western blotting.
(B) ATDC5 cells were treated with DMSO or 10 μM PMP for 24 h, and analyzed by RT-PCR for indicated genes.
(C) Mouse E16.5 growth plate cartilages were dissected to separate the tissues above and (D) below the dotted line, and subjected to RT-PCR analyses for indicated genes.
To further elucidate the relationship between Ihh and Wnt, we next investigated whether Ihh signaling can modulate expression levels of various Wnt ligands (Wnt1, 3a, 4, 5a, 7a), Wnt co-receptors (Lrp5, 6) and Wnt antagonists (Sfrp1, 2, 3) in chondrocytes. Then, we found that PMP treatment can cause marginal up-regulation of Wnt5a and slight down-regulation of Wnt4 in ATDC5 cells (Figure 2B). In addition, we interestingly noticed that PMP treatment can remarkably suppress levels of Lrp6, Sfrp2 and Sfrp3 expression (Figure 2B). On the other hand, expression levels of Wnt1, Wnt3a, Wnt7a, Lrp5, and Sfrp1 were quite low and not significantly altered by PMP treatment in ATDC5 cells (Figure 2B). Besides, lentivirus-mediated overexpression of SmoA1 was also capable of similarly modulating expression patterns of these genes (data not shown). Taken together, these results indicate that Ihh can cause significant down-regulation of Lrp6 (Wnt co-receptor) and Sfrp2/Sfrp3 (Wnt antagonist) in chondrocytes.
To assess the physiological relevance of our results (i.e., down-regulation of Lrp and Sfrp by Ihh), we analyzed expression of Sfrp1/2/3 and Lrp5/6 in Ihh expression negative and positive tissues isolated from E16.5 mouse growth plate; the tissues above and below the dotted line were dissected separately as Ihh expression negative (i.e., proliferating zone) and positive (i.e., maturing zone) chondrocytes, respectively (see left panel in Figure 2C) from E16.5 mouse hind limbs. Consistent with the results obtained from ATDC5 in vitro cultures, expression levels of Sfrp2, Sfrp3, Lrp5, and Lrp6 were remarkably reduced in Ihh expression enriched chondrocytes (see right panels in Figure 2C). Therefore, these results show that elevated levels of Ihh expression and decreased levels of Lrp (Wnt co-receptor) and Sfrp (anti-Wnt) expression have a tight correlation in vivo as well as in vitro.
As a matter of fact, these findings suggest the following intriguing scenario. First, down-regulation of Sfrp (anti-Wnt) expression by Ihh would, in principle, increase the efficacies of both canonical and non-canonical Wnt signaling because reduced levels of anti-Wnts can indirectly enhance the interactions between Wnt ligands and Wnt receptors [70, 71]. Second, suppression of Lrp (Wnt co-receptor) expression by Ihh would selectively attenuate canonical Wnt pathways since Lrp co-receptors are required for Wnt receptors to optimally interact with canonical Wnt ligands and transmit canonical Wnt signals [70, 72, 73]. On the other hand, non-canonical Wnt ligands can bind to the receptors in the absence of Lrp co-receptors and can transmit non-canonical (i.e. β-catenin independent) downstream signals via a Lrp5/6-independent manner [70, 72, 73]. Therefore, simultaneous suppression of Wnt co-receptors and Wnt antagonists (i.e., Lrps and Sfrps) would be beneficial only to non-canonical Wnt signaling pathways because reduced levels of Lrp co-receptors would result in substantial attenuation of canonical Wnt signaling pathways in spite of decreased levels of Sfrp anti-Wnts.
Wnt5a is required for Ihh-induced Nkx3.2 degradation
So far, our results indicate that Wnt signaling is required for Ihh-induced Nkx3.2 degradation (Figure 2A) and Ihh can enhance and attenuate non-canonical and canonical Wnt signaling, respectively (Figures 2B and 2C). Therefore, it is a decent possibility that Ihh may employ a non-canonical Wnt pathway to trigger Nkx3.2 protein degradation. In this context, we hypothesized that Wnt5a, a non-canonical Wnt, which has been shown to be a key regulator for chondrocyte hypertrophy [37, 40, 41, 45-49], may modulate Nkx3.2 protein stability. In agreement with this hypothesis, we found that the protein levels of Nkx3.2 can be significantly decreased by Wnt5a co-transfection, while Cbfβ protein levels are unaffected (Figure 3A). Thus, these results prompted us to examine whether Wnt5a is required for Ihh-induced Nkx3.2 degradation. Interestingly, Wnt5a knockdown was effective in suppressing both overexpressed Nkx3.2 degradation by SmoA1 co-transfection (Figure 3B) and endogenous Nkx3.2 protein destabilization by PMP treatment (Figure 3C); the efficacies of Wnt5a shRNAs were verified as shown in Figure 3D.
Figure 3. Wnt5a is required for Ihh-induced Nkx3.2 degradation.
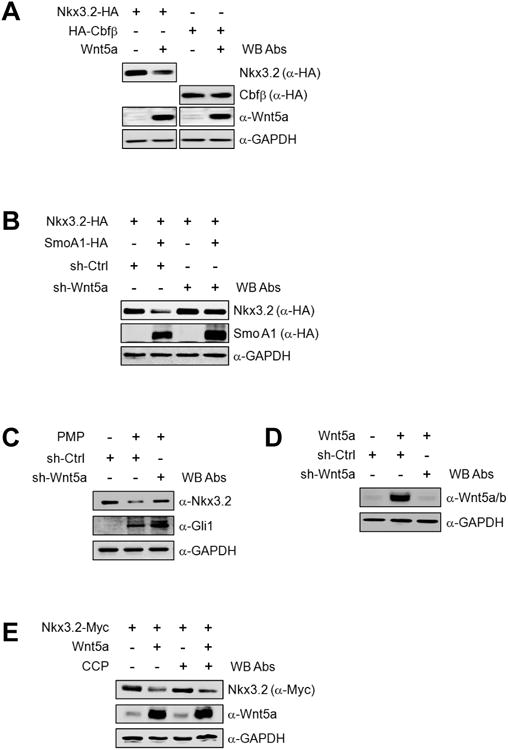
(A) Nkx3.2-HA or HA-Cbfβ expression construct was transfected into ATDC5 cells in the absence or presence of Wnt5a expression plasmid for 48 h. Total cell lysates were analyzed by western blotting.
(B) Expression vehicles for control or Wnt5a shRNA were transfected into ATDC5 cells with Nkx3.2-HA expression construct in the absence or presence of SmoA1-HA expression plasmid for 48 h. Total cell lysates were analyzed by western blotting.
(C) ATDC5 cells were infected with control or Wnt5a shRNA-harboring lentivirus. At 24 h post-infection, cells were treated with DMSO or 10 μM PMP. After an additional 24 h, total cell lysates were analyzed by western blotting.
(D) Wnt5a expression plasmid was transfected into ATDC5 cells in the absence or presence of control or Wnt5a shRNA constructs, and total cell lysates were analyzed by western blotting after 48 h.
(E) ATDC5 cells were transfected with Nkx3.2-Myc or Wnt5a expression plasmids. At 24 h post-transfection, cells were treated with 0.5% DMSO or 5 μM CMP for an additional 24 h, and total cell lysates were analyzed by western blotting.
As these results indicate that Wnt5a is required for Ihh to destabilize Nkx3.2 proteins, we next evaluated whether Ihh is necessary for Wnt5a to trigger Nkx3.2 degradation. In contrast to the results in Figures 3B and 3C, Ihh signaling inhibition by using cyclopamine (CCP), a well established Hedgehog antagonist, did not abrogate Nkx3.2 protein degradation caused by Wnt5a overexpression (Figure 3E). Thus, these results indicate that endogenous Wnt5a is required for exogenously forced Ihh signals (i.e., SmoA1 overexpression or PMP treatment) to cause Nkx3.2 destabilization, whereas endogenous Ihh is not necessary for overexpressed Wnt5a to trigger Nkx3.2 degradation.
Taken together, our results in Figure 2 and Figure 3 support the following hypothesis; Ihh enhances non-canonical Wnt (e.g., Wnt5a) signaling efficacies via parallel inhibition of Lrp co-receptors and Sfrp antagonists (Figures 2B and 2C), and, as a result, augmented Wnt5a signaling can trigger Nkx3.2 protein degradation (Figure 3A). Furthermore, for this hypothesis to be correct, it is indeed expected that Wnt5a is required for Ihh signaling to cause Nkx3.2 degradation (Figures 3B and 3C), while Nkx3.2 degradation can occur in the absence of Ihh signaling if Wnt5a is provided in excess (Figure 3E).
CK2 is necessary for Ihh/Wnt5a to induce proteasomal degradation of Nkx3.2
Because our results indicated that Wnt5a can render Nkx3.2 protein degradation, we next sought to identify intracellular components employed in this process. To do this, we first examined various intracellular components including protein kinase C (PKC), TAK1 kinase, casein kinase 1 (CK1), casein kinase 2 (CK2), and calcium ion, all of which have been indicated in various Wnt5a-mediated signaling pathways [50-52, 54, 56-58]. Then, we found that PKCα, Ca2+, TAK1, and CK1α/δ/ε may not be closely associated with Nkx3.2 protein stability control (Figures 4A-4D). However, interestingly, ectopic expression of CK2α/β dramatically reduced the levels of co-expressed Nkx3.2 proteins, but not Runx2 (Figure 4E). In addition, we observed the same effects on Nkx3.2 protein stability by co-expressing CK2α′/β (data not shown). Conversely, kinase-dead forms of CK2α and/or CK2α′ could not induce Nkx3.2 protein degradation (data not shown), suggesting that the catalytic activity of CK2 is necessary for CK2 to cause Nkx3.2 protein destabilization.
Figure 4. CK2 overexpression can destabilize Nkx3.2 protein.
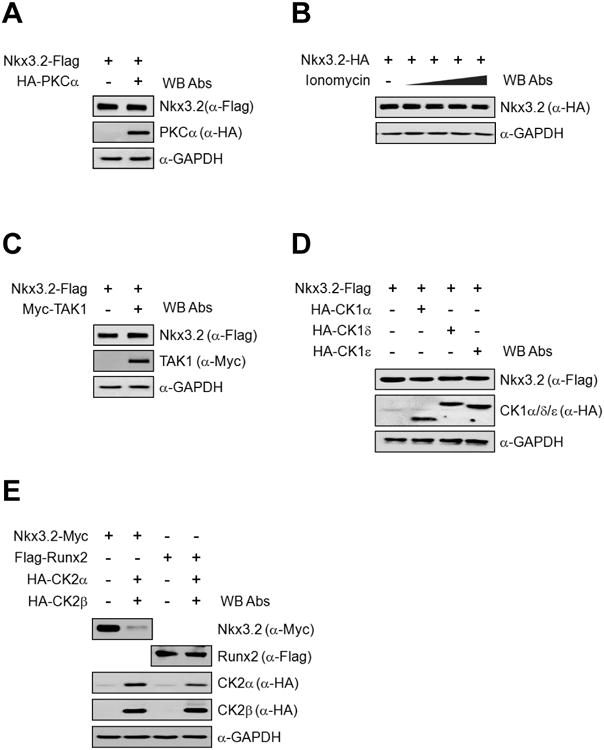
(A) Nkx3.2-Flag encoding plasmid was co-transfected with HA-PKCα into ATDC5 cells. After 48 h of transfection, cells were harvested and total cell lysates were analyzed by western blotting.
(B) ATDC5 cells were transfected with Nkx3.2-HA expression vector for 48 h, and then cells were treated with increasing doses (0, 0.5, 1, 2, and 3 μM) of Inonomycin for the final 6 h. Total cell lysates were then analyzed by western blotting.
(C) Nkx3.2-Flag encoding expression vector was co-transfected with Myc-TAK1 expression plasmid into ATDC5 cells, and, after 48 h of transfection, cells were harvested and total cell lysates were analyzed by western blotting.
(D) ATDC5 cells were transfected with Nkx3.2-Flag expression plasmid in the presence of HA-CK1α, δ, or ε expression vectors. At 48 h post-transfection, cells were processed for western blot analyses.
(E) Nkx3.2-Myc or Flag-Runx2 expression constructs were transfected into ATDC5 cells in the absence or presence of HA-CK2α and HA-CK2β expression plasmids. After 48 h of transfection, cells were harvested and total cell lysates were analyzed by western blotting.
We then tried to determine whether CK2 is required for Ihh/Wnt5a-induced Nkx3.2 degradation. Interestingly, we found that kinase-dead forms of CK2α and/or CK2α′ can efficiently abolish Nkx3.2 protein degradation triggered by SmoA1 (Figure 5A) or by Wnt5a (Figure 5B). Consistent with these findings, pharmacological inhibition of CK2 using DRB or TBB resulted in successful blockage of Nkx3.2 protein degradation caused by SmoA1 (Figure 5C) or by Wnt5a (Figure 5D). In addition, we observed that endogenous Nkx3.2 protein destabilization by PMP treatment can be effectively attenuated by pharmacological inhibition of CK2 using DRB or TBB treatment (Figure 5E) or by RNA knockdown of CK2α/β (Figure 5F). Finally, we found that CK2α/β overexpression can remarkably elevate the levels of Nkx3.2 ubiquitination (Figure 5G). Together, these results indicate that CK2 is engaged in Ihh-induced Wnt5a-dependent proteasomal degradation of Nkx3.2.
Figure 5. CK2 is required for Ihh/Wnt5a-induced Nkx3.2 protein degradation.
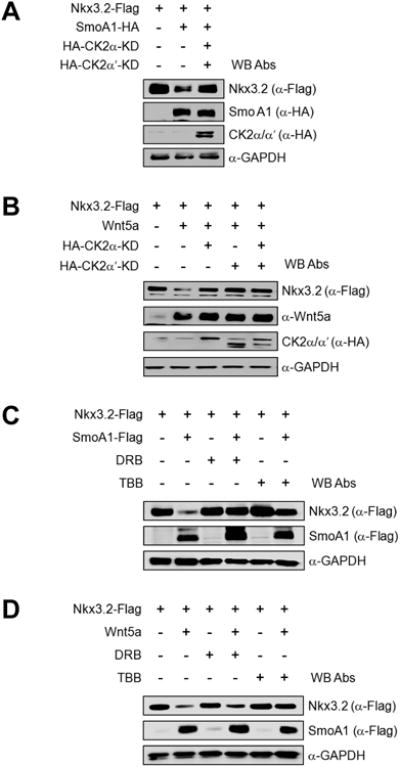
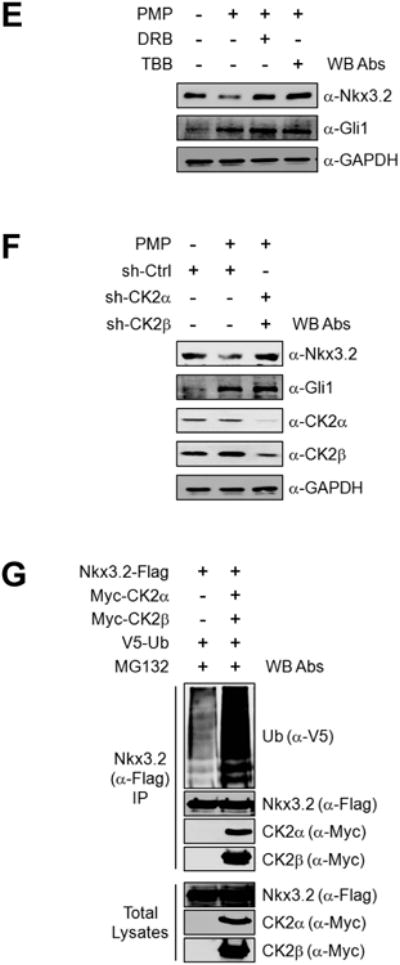
(A) Nkx3.2-Flag expression construct was co-transfected into ATDC5 cells with empty vector or SmoA1-HA expression vector in the absence or presence of HA-CK2α-KD and HA-CK2α′-KD expression plasmids. After 48 h of transfection, cells were harvested and total cell lysates were analyzed by western blotting.
(B) ATDC5 cells were transfected with Nkx3.2-Flag expression construct together with empty vector or Wnt5a expression vector in the absence or presence of HA-CK2α-KD and/or 3HA-CK2α′-KD expression plasmids for 48 h, and then total cell lysates were analyzed by western blotting.
(C) Nkx3.2-Flag expression vector was transfected into ATDC5 cells in the absence or presence of SmoA1-Flag expression plasmid. At 36 h post-transfection, cells were treated with DMSO, 100 μM DRB, or 100 μM TBB for an additional 12 h. Cells were then harvested and total cell lysates were analyzed by western blotting.
(D) ATDC5 cells were transfected with Nkx3.2-Flag expression plasmid in the absence or presence of Wnt5a expression vehicle for 48 h. DMSO, 100 μM DRB, or 100 μM TBB was included for the final 12 h prior to harvesting, and then total cell lysates were analyzed by western blotting.
(E) ATDC5 cultures were pre-treated with DMSO or 10 μM PMP for 24 h prior to addition of 100 μM DRB or 100 μM TBB. After an additional 12 h, cells were harvested and total cell lysates were analyzed by western blotting.
(F) Control or CK2α/β shRNA-harboring lentivirus was infected into ATDC5 cells. After 24 h, cells were treated with DMSO or 10 μM PMP for an additional 24 h, and then total cell lysates were analyzed by western blotting.
(G) Expression constructs for Nkx3.2-Flag and V5-Ubiquitin were transfected into 293T cells in the absence or presence of Myc-CK2α and Myc-CK2β expression plasmid for 24 h. Transfected cells were exposed to 20 μM MG132 for the final 6 h of transfection. Anti-Flag immunoprecipitates and total cell lysates were analyzed by western blotting.
Siah1 plays a role in Nkx3.2 ubiquitination triggered by Ihh signaling
Because our results suggest that proteasomal degradation of Nkx3.2 can be induced by Ihh signaling, we next sought to identify the E3 ligase engaged in this process. To this end, we first examined involvement of Siah1, the mammalian homolog of Drosophila Sina [74, 75], because Ihh-induced Nkx3.2 degradation requires Wnt5a signals, and Siah1 has been shown to play a role in various Wnt5a-induced proteasomal degradation processes in chondrocytes [43, 61, 76]. Interestingly, we found that overexpression of Siah1, but not PTrCP1, substantially reduced the protein levels of co-expressed Nkx3.2 (Figure 6A). As we observed that Siah1 is capable of inducing Nkx3.2 protein degradation, we next investigated whether Siah1 is required for Ihh-induced Nkx3.2 degradation. We found that a dominant-negative form of Siah1 (i.e., a ring domain deletion mutant) can abolish SmoA1-induced Nkx3.2 degradation (Figure 6B), and Siah1 knockdown disabled SmoA1 to destabilize Nkx3.2 proteins (Figure 6C); the efficacies of Siah1 shRNAs were estimated as shown in Figure 6D. Together, these results indicate that Siah1 may function in the Ihh-induced Nkx3.2 degradation pathway.
Figure 6. Siah1 is an E3 ligase for Nkx3.2 degradation.
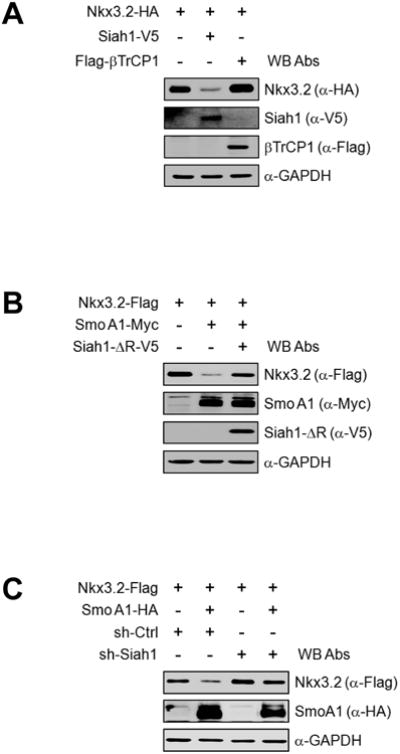
(A) Nkx3.2-HA encoding plasmid was transfected into ATDC5 cells with Siah1-V5 or Flag-βTrCP1 expression constructs. At 48 h post-transfection, cells were harvested and total cell lysates were analyzed by western blotting.
(B) Nkx3.2-Flag expression construct was transfected into ATDC5 cells in the absence or presence of SmoA1-Myc or Siah1-ΔR-V5 expression plasmid. After 48 h, cells were harvested and total cell lysates were analyzed by western blotting.
(C) Nkx3.2-Flag encoding plasmid was transfected into ATDC5 cells with empty vector or SmoA1-HA expression plasmid in the absence or presence of control or Siah1 shRNA expression vehicles for 48 h. Total cell lysates were analyzed by western blotting.
(D) ATDC5 cells were transfected with Siah1-V5 expression plasmid in the absence or presence of Siah1 shRNA construct. After 48 h of transfection, cells were harvested and total cell lysates were analyzed by western blotting.
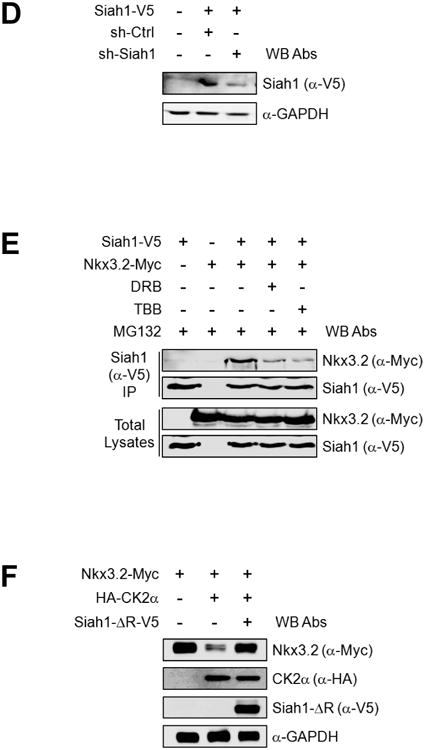
(E) Expression constructs for Nkx3.2-Myc and/or Siah1-V5 were transfected into ATDC5 cells for 48 h. For the final 12 h, 20 μM MG132, 100 μM DRB, or 100 μM TBB was included where indicated. Anti-V5 immunoprecipitates and total cell lysates were analyzed by western blotting.
(F) Nkx3.2-Myc expression vehicles were transfected into ATDC5 cells with or without HA- CK2α expression plasmid in the presence or absence of Siah1-ΔR-V5 expression construct for 48 h, and total cell lysates were analyzed by western blotting.
We next explored the possibility that CK2 may regulate the interaction between Nkx3.2 and Siah1 E3 ligase. Intriguingly, co-IP assays revealed that Nkx3.2 could form a complex with Siah1 in the presence of the proteasome inhibitor MG132, and this interaction could be remarkably attenuated by pharmacological inhibitors of CK2 such as DRB and TBB (Figure 6E). In addition, we have confirmed that CK2-induced Nkx3.2 protein degradation can be effectively protected by dominant-negative Siah1 (Figure 6F). Therefore, these results suggest that CK2 can enhance the interaction between Nkx3.2 and Siah1 to cause Nkx3.2 ubiquitination, which, in turn, promotes proteasomal degradation of Nkx3.2.
Ihh signaling regulates Nkx3.2 protein levels during chondrogenesis
To validate the physiological relevance of Ihh-induced Nkx3.2 protein degradation phenomena, we first performed fluorescence immunohistochemistry (F-IHC) of mouse E16.5 growth plates for Ihh and Nkx3.2 proteins. Consistent with our in vitro findings, Nkx3.2 protein levels were notably reduced in Ihh expression-positive tissues (compare panels c and d, or g and h, in Figure 7A.); the staining patterns of DAPI and PCNA indicate that the areas above and below the dotted line represent proliferating and maturing zones of the growth plates, respectively (See panels a and b, or e and f, in Figure 7A). To provide additional evidence for this link, we next monitored the relationship between levels of Nkx3.2 protein and Ihh signaling during in vitro chondrocyte maturation; here, we considered the induction of Gli1 as a marker for Ihh signaling (see top panels in Figures 7B and 7C). In agreement with the in vivo expression patterns observed from mouse embryonic tissues (Figure 7A), levels of Nkx3.2 protein were rapidly decreased upon the appearance of Ihh signaling (i.e., Gli1 expression) during ATDC5 maturation (see second panel in Figure 7B) or mouse E11.5 limb bud culture (see second panel in Figure 7C). In addition, this typical reduction of Nkx3.2 protein levels during chondrocyte maturation can be significantly delayed by the presence of CCP, a Hedgehog antagonist (compare lanes 4-5 with 7-8 in second panel of Figure 7D); as expected, CCP treatment blocked naturally occurring Gli induction (i.e., Ihh signal activation; top panel in Figure 7D). Thus, these results suggest that Nkx3.2 down-regulation during chondrocyte maturation can be closely associated with Ihh signaling.
To further substantiate this connection between Nkx3.2 protein levels and Ihh signaling in vivo, we next analyzed mouse E18.5 cartilage growth plate from Ihh knockout mice [24] or Col2-Cre;Smoothenedn/c mice [23]. Briefly, Smoothenedn/c mice contain one copy of null allele and one copy of conditional allele, which can be deleted by Cre recombinase. As previously reported, both of these mutant mice displayed severe skeletal phenotypes, including significant inhibition of hypertrophic maturation of chondrocyte and osteoblast differentiation in endochondral bones. Most relevant to this study, we have carefully analyzed the expression patterns of Nkx3.2 proteins in these mutant mice. Consistent with our results so far, the characteristic decrease in Nkx3.2 protein levels in hypertrophic chondrocytes of normal growth plates (see panel b in Figure 7E for Ihh (+/−) mice and panel b in Figure 7F for Smo (+/+) mice) was not observed in either the Ihh (−/−) growth plate (see panel c in Figure 7E) or the Col2-Cre;Smon/c growth plate (see panel c in Figure 7F). Instead, in these mutant mice, Nkx3.2 protein levels were substantially elevated throughout the growth plate, including both proliferative and hypertrophic chondrocytes.
Finally, to support the biological importance of Nkx3.2-Ihh crosstalk, we examined the effects of Nkx3.2 overexpression and/or PMP administration on chondrocyte maturation. First, stable overexpression of Nkx3.2 substantially diminished type X collagen induction during ATDC5 maturation (compare lanes 3 and 8 in top panel of Figure 7G), suggesting that forced expression of Nkx3.2 can inhibit chondrocyte maturation. Second, as would be expected, the elevation of Ihh signaling by PMP treatment significantly potentiated the induction both type X collagen, a typical marker for chondrocyte hypertrophy, and Gli1, a typical marker for Ihh signaling (compare lanes 3 and 5 in top and second panels of Figure 7G). Third, blockage of type X collagen induction by Nkx3.2 overexpression was partially rescued by PMP treatment (compare lanes 8 and 10 in top panel of Figure 7G), suggesting that inhibition of chondrocyte maturation caused by the elevated levels of Nkx3.2 can be attenuated by Ihh-induced down-regulation of Nkx3.2. Forth, unlike type X collagen, Gli1 induction by PMP was not significantly altered by stable overexpression of Nkx3.2 (compare lanes 4-5 with 9-10 in second panel of Figure 7G), suggesting that Nkx3.2 may not inhibit Ihh signaling (e.g. Gli12 induction by Ihh) per se. Taken together, Nkx3.2 degradation triggered by Ihh may be a necessary event for optimal progression of hypertrophic maturation in chondrocytes.
Discussion
Chondrocyte maturation control via Ihh-induced Nkx3.2 degradation
While many previous studies have indicated that Ihh plays an essential role in endochondral bone development, complete understanding of precise actions of Ihh pathways will require further studies. In this study, we have revealed that Ihh signaling can negatively regulate Nkx3.2 by triggering proteasomal degradation of Nkx3.2. In fact, these results provide an additional molecular explanation regarding how Ihh functions to promote chondrocyte hypertrophy during cartilage development. As stated earlier, defects in Ihh signaling have been shown to result in disorganized and incomplete chondrocyte hypertrophy [20, 22-25], and our results may suggest that inappropriate accumulation of Nkx3.2 could be one of the consequences caused by loss of Ihh signaling. Thus, it is a decent possibility that Nkx3.2-Ihh crosstalk may have contributed to cause the phenotypes observed from Ihh signaling-defective mice. Consistent with this hypothesis, our current and previous studies have indicated that forced expression of Nkx3.2 can delay chondrocyte hypertrophy in vitro [2, 15]. Thus, these findings together logically support the biological significance of Nkx3.2-Ihh crosstalk in normal progression of chondrocyte maturation during cartilage development.
On the other hand, previous reports have demonstrated that Nkx3.2 (Bapx1) deletion in mice results in severe defects in axial skeletons, while limb development is relatively normal [8-11]. Nonetheless, our results suggest a possibility that a functional crosstalk between Ihh and Nkx3.2 may play a role in appendicular cartilage development as well. Thus, it is conceivable that a redundant factor may have compensated the absence of Nkx3.2 for appendicular cartilage development in previously reported Nkx3.2 knockout mice. Our future studies may focus on understanding the effects of chondrocyte-specific gain-of-function or loss-of-function mutations of Nkx3.2 on Ihh signaling and cartilage development in vivo.
A crosstalk between Ihh and Wnt pathways during cartilage hypertrophy
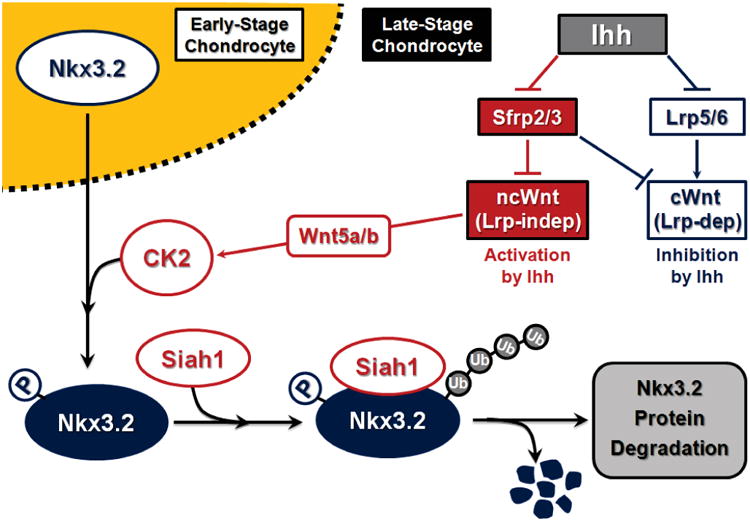
Functional interactions between Ihh and Wnt pathways have been previously indicated in diverse events, including cartilage growth plate control and joint segmentation [17, 20, 27, 36, 42, 44, 46, 55, 77-80]. In this study, we found that Ihh can cause parallel inhibition of Lrp (Wnt co-receptors) and Sfrp (Wnt antagonists) in chondrocytes, which, in turn, may result in selective enhancement of Wnt5a signaling in pre-hypertrophic zone of cartilage growth plate. Consistent with these observations, we demonstrate that Ihh-induced Nkx3.2 degradation requires intact Wnt5a expression (Figure 4). On the other hand, it has been indicated that the levels of Wnt5a expression in Ihh KO or Col2-Cre;Smonc mice were not notably altered [23, 24]. Nevertheless, we found that Nkx3.2 protein levels in the growth plates are evidently up-regulated in these Ihh signaling deficient mice (Figure 8). Thus, these results indicate that Wnt5a expression alone is not sufficient to down-regulate Nkx3.2 in Ihh KO or Col2-Cre;Smonc mice because Ihh-mediated enhancement of Wnt5a signaling would be absent in these Ihh signal-defective mice. Taken together, it is plausible that a collaborative action involving Ihh-Wnt5a crosstalk may play a role in optimal progression of chondrocyte hypertrophy during cartilage development (Figure 8). Additionally, cellular components such as CK2 and Siah1, which we have revealed to be necessary factors for Ihh-induced Wnt5a-dependent Nkx3.2 protein degradation, may target other proteins as well during cartilage development to mediate the signals involving Ihh-Wnt5a crosstalk (Figure 8).
Figure 8. A schematic diagram of Ihh-induced Wnt5a-dependent Nkx3.2 degradation.
Involvement of PTHrP signaling in Ihh-induced Nkx3.2 degradation
The Ihh and PTHrP pathways have been demonstrated to closely communicate to control both chondrocyte proliferation and hypertrophic maturation via a remote crosstalk in the growth plate [18, 20-22, 24, 29, 77, 81-83]. However, as would be expected, to demonstrate that Ihh could induce Nkx3.2 protein degradation, additional modulation of PTHrP signaling was not required in our routine experimental settings. Nevertheless, alterations of Ihh signaling might have modified PTHrP signaling by affecting expression of PTHrP and/or PTH/PTHrP receptor (PPR) or the activity of various cellular components. To clarify this issue, we have investigated whether exogenous Ihh signaling (i.e., PMP treatment or SmoA1 overexpression) can modulate expression of PTHrP and/or PPR under the experimental conditions employed in this study. Then, we found that neither PTHrP nor PPR expression is appreciably altered by providing exogenous Ihh signaling in ATDC5 cells as revealed by RT-PCR and microarray analyses (data not shown). Consistent with these findings, we have observed that pharmacological inhibition of protein kinase A (PKA), a well-established PTHrP downstream component, did not block Ihh-induced Nkx3.2 protein degradation (data not shown). Thus, these results together suggest that PTHrP signaling is unlikely to be necessary for Ihh to trigger Nkx3.2 protein degradation.
From a different perspective, it has been demonstrated that appropriate expression of PTHrP in vivo is dependent on intact Ihh signaling [21, 22, 24, 25, 29]. In addition, prior work has indicated that Nkx3.2 mRNA expression in the proliferating columnar chondrocytes is specifically and remarkably diminished in PTHrP signaling deficient mice [2]. Thus, based on these findings, it could be predicted that Nkx3.2 mRNA levels may be very low in Ihh (−/−) or Col2-Cre;Smon/c mice. Nonetheless, we have found that Nkx3.2 protein levels in these mutant mice are substantially elevated (Figure 8). Therefore, these previous and current findings together made us speculate the following. First, PTHrP signaling may play a role in potentiating and maintaining Nkx3.2 expression rather than initial induction per se; thus, much reduced but appreciable levels of Nkx3.2 mRNA may still exist in Ihh signaling-deficient chondrocytes. Second, while Ihh deletion would result in a reduction of Nkx3.2 mRNA expression via PTHrP inhibition, Ihh-induced Nkx3.2 protein degradation would also be blocked in Ihh signaling-defective chondrocytes; therefore, under these circumstances, low levels of Nkx3.2 mRNA expression might be sufficient to give rise to detectable accumulation of Nkx3.2 proteins. Third, it is also feasible that Nkx3.2 expression may require PTHrP signaling only when Ihh signaling is normally operated.
Acknowledgments
Funding: This study was supported by the grants (2011-0020409; FPR08B1-260; 2011-0001029; 2011-0027837) funded by the Korean Ministry of Education, Science and Technology.
Footnotes
Author Contribution Seung-Won Choi contributed to performing the experiments in Figures 1 to 6 and to writing the paper. Da-Un Jeong, Boyoung Lee, and Jeong-Ah Kim conducted the experiments for Figure 7. Kyu Sang Joeng and Fanxin Long provided paraffin sections for Figures 7E and 7F. Dae-Won Kim supervised the experiments and wrote the paper.
References
- 1.Murtaugh LC, Zeng L, Chyung JH, Lassar AB. The chick transcriptional repressor Nkx3.2 acts downstream of Shh to promote BMP-dependent axial chondrogenesis. Developmental Cell. 2001;1:411–422. doi: 10.1016/s1534-5807(01)00039-9. [DOI] [PubMed] [Google Scholar]
- 2.Provot S, Kempf H, Murtaugh LC, Chung UI, Kim DW, Chyung J, Kronenberg HM, Lassar AB. Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development. 2006;133:651–662. doi: 10.1242/dev.02258. [DOI] [PubMed] [Google Scholar]
- 3.Tribioli C, Lufkin T. Molecular cloning, chromosomal mapping and developmental expression of BAPX1, a novel human homeobox-containing gene homologous to Drosophila bagpipe. Gene. 1997;203:225–233. doi: 10.1016/s0378-1119(97)00520-9. [DOI] [PubMed] [Google Scholar]
- 4.Zeng L, Kempf H, Murtaugh LC, Sato ME, Lassar AB. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev. 2002;16:1990–2005. doi: 10.1101/gad.1008002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim DW, Kempf H, Chen RE, Lassar AB. Characterization of Nkx3.2 DNA binding specificity and its requirement for somitic chondrogenesis. J Biol Chem. 2003;278:27532–27539. doi: 10.1074/jbc.M301461200. [DOI] [PubMed] [Google Scholar]
- 6.Kim DW, Lassar AB. Smad-dependent recruitment of a histone deacetylase/Sin3A complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of Nkx3.2. Mol Cell Biol. 2003;23:8704–8717. doi: 10.1128/MCB.23.23.8704-8717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshiura KI, Murray JC. Sequence and chromosomal assignment of human BAPX1, a bagpipe-related gene, to 4p16.1: a candidate gene for skeletal dysplasia. Genomics. 1997;45:425–428. doi: 10.1006/geno.1997.4926. [DOI] [PubMed] [Google Scholar]
- 8.Lettice LA, Purdie LA, Carlson GJ, Kilanowski F, Dorin J, Hill RE. The mouse bagpipe gene controls development of axial skeleton, skull, and spleen. Proc Natl Acad Sci USA. 1999;96:9695–9700. doi: 10.1073/pnas.96.17.9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akazawa H, Komuro I, Sugitani Y, Yazaki Y, Nagai R, Noda T. Targeted disruption of the homeobox transcription factor bapx1 results in lethal skeletal dysplasia with asplenia and gastroduodenal malformation. Genes Cells. 2000;5:499–513. doi: 10.1046/j.1365-2443.2000.00339.x. [DOI] [PubMed] [Google Scholar]
- 10.Tribioli C, Lufkin T. The murine Bapx1 homeobox gene plays a critical role in embryonic development of the axial skeleton and spleen. Development. 1999;126:5699–5711. doi: 10.1242/dev.126.24.5699. [DOI] [PubMed] [Google Scholar]
- 11.Herbrand H, Pabst O, Hill R, Arnold HH. Transcription factors Nkx3.1 and Nkx3.2 (Bapx1) play an overlapping role in sclerotome development of the mouse. Mech of Dev. 2002;117:217–224. doi: 10.1016/s0925-4773(02)00207-1. [DOI] [PubMed] [Google Scholar]
- 12.Church V, Yamaguchi K, Tsang P, Akita K, Logan C, Francis-West P. Expression and function of Bapx1 during chick limb development. Anat Embryol (Berl) 2005;209:461–469. doi: 10.1007/s00429-005-0464-z. [DOI] [PubMed] [Google Scholar]
- 13.Park M, Yong Y, Choi SW, Kim JH, Lee JE, Kim DW. Constitutive RelA activation mediated by Nkx3.2 controls chondrocyte viability. Nat Cell Biol. 2007;9:287–298. doi: 10.1038/ncb1538. [DOI] [PubMed] [Google Scholar]
- 14.Yong Y, Choi SW, Choi HJ, Nam HW, Kim JA, Jeong DU, Kim DY, Kim YS, Kim DW. Exogenous Signal-Independent Nuclear IKKbeta Activation Triggered by Nkx3.2 Enables Constitutive Nuclear Degradation of IkappaB-alpha in Chondrocytes. Mol Cell Biol. doi: 10.1128/MCB.00253-10. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lengner CJ, Hassan MQ, Serra RW, Lepper C, van Wijnen AJ, Stein JL, Lian JB, Stein GS. Nkx3.2-mediated repression of Runx2 promotes chondrogenic differentiation. J Biol Chem. 2005;280:15872–15879. doi: 10.1074/jbc.M411144200. [DOI] [PubMed] [Google Scholar]
- 16.Yamashita S, Andoh M, Ueno-Kudoh H, Sato T, Miyaki S, Asahara H. Sox9 directly promotes Bapx1 gene expression to repress Runx2 in chondrocytes. Exp Cell Res. 2009;315:2231–2240. doi: 10.1016/j.yexcr.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman DM, Karhadkar SS, Maitra A, Montes De, Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 18.de Crombrugghe B, Lefebvre V, Nakashima K. Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol. 2001;13:721–727. doi: 10.1016/s0955-0674(00)00276-3. [DOI] [PubMed] [Google Scholar]
- 19.Dyer MA, Farrington SM, Mohn D, Munday JR, Baron MH. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development. 2001;128:1717–1730. doi: 10.1242/dev.128.10.1717. [DOI] [PubMed] [Google Scholar]
- 20.Iwamoto M, Enomoto-Iwamoto M, Kurisu K. Actions of hedgehog proteins on skeletal cells. Crit Rev Oral Biol Med. 1999;10:477–486. doi: 10.1177/10454411990100040401. [DOI] [PubMed] [Google Scholar]
- 21.Karp SJ, Schipani E, St-Jacques B, Hunzelman J, Kronenberg H, McMahon AP. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development. 2000;127:543–548. doi: 10.1242/dev.127.3.543. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi T, Chung UI, Schipani E, Starbuck M, Karsenty G, Katagiri T, Goad DL, Lanske B, Kronenberg HM. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 2002;129:2977–2986. doi: 10.1242/dev.129.12.2977. [DOI] [PubMed] [Google Scholar]
- 23.Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- 24.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 26.van den Brink GR. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol Rev. 2007;87:1343–1375. doi: 10.1152/physrev.00054.2006. [DOI] [PubMed] [Google Scholar]
- 27.Katoh Y, Katoh M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review) Int J Mol Med. 2008;22:271–275. [PubMed] [Google Scholar]
- 28.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 29.Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LH, Ho C, Mulligan RC, Abou-Samra AB, Juppner H, Segre GV, Kronenberg HM. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 30.Methot N, Basler K. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell. 1999;96:819–831. doi: 10.1016/s0092-8674(00)80592-9. [DOI] [PubMed] [Google Scholar]
- 31.Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, Vortkamp A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128:4523–4534. doi: 10.1242/dev.128.22.4523. [DOI] [PubMed] [Google Scholar]
- 32.Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3:439–449. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- 33.Kalderon D. Transducing the hedgehog signal. Cell. 2000;103:371–374. doi: 10.1016/s0092-8674(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 34.Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339–4351. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- 35.Joeng KS, Long F. The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development. 2009;136:4177–4185. doi: 10.1242/dev.041624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mak KK, Kronenberg HM, Chuang PT, Mackem S, Yang Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development. 2008;135:1947–1956. doi: 10.1242/dev.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergwitz C, Wendlandt T, Kispert A, Brabant G. Wnts differentially regulate colony growth and differentiation of chondrogenic rat calvaria cells. Biochim Biophys Acta. 2001;1538:129–140. doi: 10.1016/s0167-4889(00)00123-3. [DOI] [PubMed] [Google Scholar]
- 38.Bradley EW, Drissi MH. WNT5A regulates chondrocyte differentiation through differential use of the CaN/NFAT and IKK/NF-kappaB pathways. Mol Endocrinol. 2010;24:1581–1593. doi: 10.1210/me.2010-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Church VL, Francis-West P. Wnt signalling during limb development. Int J Dev Biol. 2002;46:927–936. [PubMed] [Google Scholar]
- 40.Du SJ, Purcell SM, Christian JL, McGrew LL, Moon RT. Identification of distinct classes and functional domains of Wnts through expression of wild-type and chimeric proteins in Xenopus embryos. Mol Cell Biol. 1995;15:2625–2634. doi: 10.1128/mcb.15.5.2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartmann C, Tabin CJ. Dual roles of Wnt signaling during chondrogenesis in the chicken limb. Development. 2000;127:3141–3159. doi: 10.1242/dev.127.14.3141. [DOI] [PubMed] [Google Scholar]
- 42.Spater D, Hill TP, O'Sullivan RJ, Gruber M, Conner DA, Hartmann C. Wnt9a signaling is required for joint integrity and regulation of Ihh during chondrogenesis. Development. 2006;133:3039–3049. doi: 10.1242/dev.02471. [DOI] [PubMed] [Google Scholar]
- 43.Topol L, Jiang X, Choi H, Garrett-Beal L, Carolan PJ, Yang Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J Cell Biol. 2003;162:899–908. doi: 10.1083/jcb.200303158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y, Topol L, Lee H, Wu J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development. 2003;130:1003–1015. doi: 10.1242/dev.00324. [DOI] [PubMed] [Google Scholar]
- 45.Chimal-Monroy J, Montero JA, Ganan Y, Macias D, Garcia-Porrero JA, Hurle JM. Comparative analysis of the expression and regulation of Wnt5a, Fz4, and Frzb1 during digit formation and in micromass cultures. Dev Dyn. 2002;224:314–320. doi: 10.1002/dvdy.10110. [DOI] [PubMed] [Google Scholar]
- 46.Church V, Nohno T, Linker C, Marcelle C, Francis-West P. Wnt regulation of chondrocyte differentiation. J Cell Sci. 2002;115:4809–4818. doi: 10.1242/jcs.00152. [DOI] [PubMed] [Google Scholar]
- 47.Daumer KM, Tufan AC, Tuan RS. Long-term in vitro analysis of limb cartilage development: involvement of Wnt signaling. J Cell Biochem. 2004;93:526–541. doi: 10.1002/jcb.20190. [DOI] [PubMed] [Google Scholar]
- 48.Rudnicki JA, Brown AM. Inhibition of chondrogenesis by Wnt gene expression in vivo and in vitro. Dev Biol. 1997;185:104–118. doi: 10.1006/dbio.1997.8536. [DOI] [PubMed] [Google Scholar]
- 49.Tuli R, Tuli S, Nandi S, Huang X, Manner PA, Hozack WJ, Danielson KG, Hall DJ, Tuan RS. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J Biol Chem. 2003;278:41227–41236. doi: 10.1074/jbc.M305312200. [DOI] [PubMed] [Google Scholar]
- 50.Boutros M, Paricio N, Strutt DI, Mlodzik M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell. 1998;94:109–118. doi: 10.1016/s0092-8674(00)81226-x. [DOI] [PubMed] [Google Scholar]
- 51.Bryja V, Schambony A, Cajanek L, Dominguez I, Arenas E, Schulte G. Beta-arrestin and casein kinase 1/2 define distinct branches of non-canonical WNT signalling pathways. EMBO Rep. 2008;9:1244–1250. doi: 10.1038/embor.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dejmek J, Safholm A, Kamp Nielsen C, Andersson T, Leandersson K. Wnt-5a/Ca2+-induced NFAT activity is counteracted by Wnt-5a/Yes-Cdc42-casein kinase 1alpha signaling in human mammary epithelial cells. Mol Cell Biol. 2006;26:6024–6036. doi: 10.1128/MCB.02354-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato T, Jr, Delhase M, Hoffmann A, Karin M. CK2 Is a C-Terminal IkappaB Kinase Responsible for NF-kappaB Activation during the UV Response. Mol Cell. 2003;12:829–839. doi: 10.1016/s1097-2765(03)00358-7. [DOI] [PubMed] [Google Scholar]
- 54.Katoh M. WNT/PCP signaling pathway and human cancer (review) Oncol Rep. 2005;14:1583–1588. [PubMed] [Google Scholar]
- 55.Katoh M, Katoh M. Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int J Mol Med. 2009;23:763–769. doi: 10.3892/ijmm_00000190. [DOI] [PubMed] [Google Scholar]
- 56.Lu X, Borchers AG, Jolicoeur C, Rayburn H, Baker JC, Tessier-Lavigne M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature. 2004;430:93–98. doi: 10.1038/nature02677. [DOI] [PubMed] [Google Scholar]
- 57.Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, Koshida I, Suzuki K, Yamada G, Schwabe GC, Mundlos S, Shibuya H, Takada S, Minami Y. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells. 2003;8:645–654. doi: 10.1046/j.1365-2443.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- 58.Price MA. CKI, there's more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006;20:399–410. doi: 10.1101/gad.1394306. [DOI] [PubMed] [Google Scholar]
- 59.Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci. 2009;66:1850–1857. doi: 10.1007/s00018-009-9153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ishitani T, Kishida S, Hyodo-Miura J, Ueno N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J, Matsumoto K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol Cell Biol. 2003;23:131–139. doi: 10.1128/MCB.23.1.131-139.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL, Matsunami N. Siah-1 mediates a novel beta-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell. 2001;7:927–936. doi: 10.1016/s1097-2765(01)00241-6. [DOI] [PubMed] [Google Scholar]
- 62.Sinha S, Chen JK. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat Chem Biol. 2006;2:29–30. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- 63.Wu X, Ding S, Ding Q, Gray NS, Schultz PG. A small molecule with osteogenesis-inducing activity in multipotent mesenchymal progenitor cells. J Am Chem Soc. 2002;124:14520–14521. doi: 10.1021/ja0283908. [DOI] [PubMed] [Google Scholar]
- 64.Wu X, Walker J, Zhang J, Ding S, Schultz PG. Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chem Biol. 2004;11:1229–1238. doi: 10.1016/j.chembiol.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 65.Atsumi T, Miwa Y, Kimata K, Ikawa Y. A chondrogenic cell line derived from a differentiating culture of AT805 teratocarcinoma cells. Cell Differ Dev. 1990;30:109–116. doi: 10.1016/0922-3371(90)90079-c. [DOI] [PubMed] [Google Scholar]
- 66.Hsieh JC, Kodjabachian L, Rebbert ML, Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB, Nathans J. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398:431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- 67.Leyns L, Bouwmeester T, Kim SH, Piccolo S, De Robertis EM. Frzb-1 is a secreted antagonist of Wnt signaling expressed in the Spemann organizer. Cell. 1997;88:747–756. doi: 10.1016/s0092-8674(00)81921-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mayr T, Deutsch U, Kuhl M, Drexler HC, Lottspeich F, Deutzmann R, Wedlich D, Risau W. Fritz: a secreted frizzled-related protein that inhibits Wnt activity. Mech Dev. 1997;63:109–125. doi: 10.1016/s0925-4773(97)00035-x. [DOI] [PubMed] [Google Scholar]
- 69.Uren A, Reichsman F, Anest V, Taylor WG, Muraiso K, Bottaro DP, Cumberledge S, Rubin JS. Secreted frizzled-related protein-1 binds directly to Wingless and is a biphasic modulator of Wnt signaling. J Biol Chem. 2000;275:4374–4382. doi: 10.1074/jbc.275.6.4374. [DOI] [PubMed] [Google Scholar]
- 70.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 71.Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- 72.Liu G, Bafico A, Aaronson SA. The mechanism of endogenous receptor activation functionally distinguishes prototype canonical and noncanonical Wnts. Mol Cell Biol. 2005;25:3475–3482. doi: 10.1128/MCB.25.9.3475-3482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006;4:e115. doi: 10.1371/journal.pbio.0040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carthew RW, Rubin GM. seven in absentia, a gene required for specification of R7 cell fate in the Drosophila eye. Cell. 1990;63:561–577. doi: 10.1016/0092-8674(90)90452-k. [DOI] [PubMed] [Google Scholar]
- 75.Hu G, Chung YL, Glover T, Valentine V, Look AT, Fearon ER. Characterization of human homologs of the Drosophila seven in absentia (sina) gene. Genomics. 1997;46:103–111. doi: 10.1006/geno.1997.4997. [DOI] [PubMed] [Google Scholar]
- 76.Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915–926. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- 77.Vortkamp A. Interaction of growth factors regulating chondrocyte differentiation in the developing embryo. Osteoarthritis Cartilage. 2001;(9 Suppl A):S109–117. [PubMed] [Google Scholar]
- 78.Katoh Y, Katoh M. Hedgehog signaling pathway and gastric cancer. Cancer Biol Ther. 2005;4:1050–1054. doi: 10.4161/cbt.4.10.2184. [DOI] [PubMed] [Google Scholar]
- 79.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743–754. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bui TD, Zhang L, Rees MC, Bicknell R, Harris AL. Expression and hormone regulation of Wnt2, 3, 4, 5a, 7a, 7b and 10b in normal human endometrium and endometrial carcinoma. Br J Cancer. 1997;75:1131–1136. doi: 10.1038/bjc.1997.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chung UI, Lanske B, Lee K, Li E, Kronenberg H. The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc Natl Acad Sci USA. 1998;95:13030–13035. doi: 10.1073/pnas.95.22.13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs CS, Lee K, Pirro A, Kronenberg HM, Juppner H. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci USA. 1997;94:13689–13694. doi: 10.1073/pnas.94.25.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, Mulligan RC. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]