

Abstract
Inheritance of the ε4 allele of apolipoprotein E (APOE) is the strongest genetic risk factor associated with the sporadic form of Alzheimer's disease (AD), whereas the rare APOE ε2 allele has the opposite effect. However, the mechanisms whereby APOE confers risk and protection remain uncertain. We used a gene transfer approach to bathe the cortex of amyloid plaque–bearing transgenic mice with virally expressed human APOE. We monitored amyloid-β (Aβ) with multiphoton imaging, in vivo microdialysis, and postmortem array tomography to study the kinetics of human APOE-mediated changes in Aβ-related neurotoxicity in a mouse model of AD. We observed that human APOE4 increased the concentrations of oligomeric Aβ within the interstitial fluid and exacerbated plaque deposition; the converse occurred after exposure to human APOE2. Peri-plaque synapse loss and dystrophic neurites were also worsened by APOE4 or attenuated by APOE2. Egress of Aβ from the central nervous system (CNS) into the plasma was diminished by APOE3 and APOE4 compared to APOE2, in accord with isoform-specific retention of Aβ in the CNS. Overall, our data show a differential effect of human APOE isoforms on amyloid deposition and clearance in transgenic mice and, more importantly, on Aβ-mediated synaptotoxicity. These results suggest that the APOE genetic risk is mediated by Aβ, and that therapeutic approaches aimed at decreasing APOE4, or increasing APOE2, may be beneficial in AD.
INTRODUCTION
Alzheimer's disease (AD) is the most frequent age-related neurode-generative disorder and a major public health concern. Among the susceptibility genes associated with the late-onset sporadic form of AD, the apolipoprotein E ε4 (APOE ε4) allele is by far the most significant genetic risk factor. The presence of one APOE ε4 copy substantially increases the risk of developing the disease by a factor of 3 compared with the most common APOE ε3 allele, whereas two copies lead to a 12-fold increase (1, 2). Intriguingly, APOE ε2 has an opposite impact and decreases the age-adjusted risk of AD by about a half (3). The average age of onset of dementia corresponds to these risk profiles, with APOE4/4 carriers having an onset in their mid-60s and APOE2/3 carriers in their early 90s, a shift of almost three decades, whereas APOE3/3 individuals have an age of onset in the mid-70s (1).
The accumulation of amyloid-β (Aβ)–containing senile plaques in the hippocampus and cortex of patients is believed to play a central role in AD (4) because all the known genes responsible for the rare autosomal dominant forms of the disease participate in the production of Aβ peptides (5). Whether APOE affects AD via an effect on Aβ is controversial. APOE genotype strongly affects the extent of amyloid deposition in patients (6), and genetically engineered animals expressing human APOE2, APOE3, and APOE4 have a similar rank order of amyloid burden as humans (7), consistent with the hypothesis that different APOE isoforms affect plaque initiation and growth. APOE variants have also been suggested to modify the amount of neurotoxic soluble oligomeric Aβ (8) and to differentially influence cerebrovascular integrity and Aβ efflux through the blood-brain barrier (BBB) (9, 10). Finally, APOE has been implicated directly in neurode-generation and in synaptic integrity (11, 12). How APOE2, which is protective against AD, influences these processes is unknown.
Because most of the previous studies of APOE effects used genetic ablation of murine Apoe or lifelong expression of human APOE in transgenic mice, the impact of introducing human APOE after plaque deposition has begun is unknown. Yet, it is critical to address this issue considering that new therapeutic trials aiming at boosting APOE expression are currently being evaluated (13) and that amyloid deposition can be observed decades before cognitive deficits emerge. To overcome this knowledge gap, we used a gene transfer approach in which an adeno-associated viral (AAV) vector expressing the various human APOE alleles [or the green fluorescent protein (GFP) gene as control] were injected into the lateral ventricles of a transgenic mouse model of AD to transduce the ependymal layer. Human APOE proteins diffused into the cerebrospinal fluid (CSF) and interstitial fluid (ISF) of the mouse central nervous system (CNS) (14). Using intravital multiphoton microscopy, in vivo microdialysis, and array tomography, we found that human APOE isoforms affected the concentrations of soluble oligomeric Aβ in the ISF, the pace of Aβ fibrillization and deposition, and the extent of peri-plaque neurotoxic effects. Indeed, AD mice exposed to human APOE4 showed an increase in soluble Aβ, a higher density of fibrillar plaques, an exacerbation of synaptic loss, and an increased number of dystrophic neurites around each deposit, whereas a relative protective effect was observed with APOE2.
RESULTS
Intraventricular injection of AAV4-APOE leads to sustained production of human APOE in the brain
APOE is a naturally secreted protein, produced mainly by astrocytes and microglial cells, which can diffuse throughout the cerebral parenchyma (15). We took advantage of this property by injecting AAV serotype 4 (AAV4) vectors carrying a gene encoding GFP or one of three human APOE alleles into the lateral cerebral ventricles of 7-month-old APP/PS1 transgenic mice. Considering the large cerebral areas affected by the characteristic amyloid plaque lesions of AD, this strategy might offer an advantage compared with multiple intraparenchymal injections.
Two months after injection, transduced cells were detected in the choroid plexus and ependyma lining the ventricle (Fig. 1A). Using species-specific antibodies, we also detected both human and murine APOE proteins by enzyme-linked immunosorbent assay (ELISA) (Fig. 1, B and C, and fig. S1B) and Western blot (fig. S1A). The concentration of human APOE reached 20 μg/mg of total protein on average (Fig. 1B), representing about 10% of the endogenous murine Apoe (Fig. 1C). This modest additional amount of human APOE did not significantly alter the levels of endogenous murine mRNA or protein (fig. S1, A to C). A small but statistically significant decrease in human APOE was observed between 2 and 5 months after AAV4 injection (fig. S1D). Nonetheless, the amounts of human protein remained readily detectable, suggesting that AAV4-mediated transduction provided a platform for sustained production of the secreted protein throughout the parenchyma. Indeed, human APOE protein was present around amyloid deposits throughout the cortical mantle where APOE protein is known to accumulate (Fig. 1D) (6, 16).
Fig. 1. AAV4-APOE gene transfer by intracerebroventricular injection of APP/PS1 mice.

(A) Immunohistological labeling of GFP. (Top) Low-magnification image of the whole mouse brain ventricle area after AAV4-GFP infusion showing GFP deposition (white arrows). (Bottom) Higher-magnification images of the ependymal layer of the mouse brain ventricle (indicated by white arrows) after AAV4-APOE or AAV4-GFP injections. (B) A species-specific ELISA assay was used to quantify the concentrations of human APOE protein in mouse brain homogenates after 2 and 5 months. (C) Percentage of human APOE compared to endogenous mouse Apoe after 2 or 5 months. The ratio between human and murine APOE was calculated for each animal. (D) Immunostaining of human APOE protein (using the specific anti-human APOE antibody 3H1) showing its association with some amyloid deposits in the cortical parenchyma of APP/PS1 mice injected with AAV4 carrying GFP, APOE4, APOE3, or APOE2. (E) Detection of human APOE by Western blot in ISF samples from Apoe knockout mice injected with AAV4-APOE4 using the sensitive (but non–species-specific) goat anti-APOE antibody (AB947, Millipore). Albumin was used as a control. Scale bars, 100 μm. n = 4 to 6 animals per group. *P < 0.05 using nonparametric Kruskal-Wallis test followed by Dunn's multiple comparison test.
Next, we assessed the presence of humanAPOE in the ISF, an extracellular compartment that contains highly biologically active soluble Aβ (17, 18). Becauseof the relatively small amount of APOE detected in the entire brain lysate, we injected Apoe knockout mice with AAV4-APOE2/3/4 and tracked the presence of human APOE protein using highly sensitive but non–species-specific antibodies. Microdialysis confirmed the presence of APOE in the ISF of injected Apoe knockout animals (Fig. 1E).
Overall, these data confirm that a single intracerebroventricular injection of AAV4-APOE2/3/4 leads to sustained production of a protein of interest throughout the entire brain parenchyma and ISF, and that the ependyma/choroid plexus can be used to deliver proteins to the brain (19).
Infusion of each APOE isoform differentially affects amyloid peptides and plaque deposition
Seven-month-old APP/PS1 mice with substantial amyloid deposition were injected with vectors expressing GFP or each human APOE iso-form for 5 months before euthanasia. An analysis of the amyloid plaque load revealed a significant increase in the density of amyloid deposits in the cortex of animals injected with AAV4-APOE4 compared with those injected with AAV4-APOE2 (AAV4-APOE2: 31 ± 4 and AAV4-APOE4: 77 ± 5 plaques/mm2 of cortex; P < 0.01). Plaque densities in GFP- and APOE3-exposed mice reached an intermediate level between APOE4 and APOE2 (AAV4-GFP: 54 ± 15; AAV4-APOE3: 47 ± 3 plaques/mm2 of cortex; Fig. 2, A and B) and were also equivalent to that of uninjected animals.
Fig. 2. Aβ peptides and amyloid deposits are modulated by different APOE alleles.

(A) Representative images of amyloid deposition in APP/PS1 mice 5 months after intraventricular injection of AAV4-GFP or AAV4-APOE2/3/4 after immunostaining for amyloid plaques (rabbit anti-human Aβ antibody, Immuno-Biological Laboratories, 18584). Scale bar, 200 μm. (B) Analysis of the density of amyloid deposits in the cortex (left panel) and hippocampus (right panel) of APP/PS1 transgenic mice injected with AAV4-GFP or AAV4-APOE2/3/4. (C) Determination by ELISA assay of the concentrations of Aβ40 and Aβ42 in the formic acid fraction of mouse brain. (D) Quantification by ELISA assay of the concentrations of Aβ40 and Aβ42 peptides in the TBS-soluble fraction of mouse brain. (E) Plasma concentrations of Aβ40 peptides in APP/PS1 mice infused with AAV4-GFP or AAV4-APOE2/3/4. (B to E) n = 4 AAV4-GFP, n = 5 AAV4-APOE2, n = 6 AAV4-APOE3, and n = 5 AAV4-APOE4 mice. **P < 0.01, *P < 0.05 using non-parametric Kruskal-Wallis test followed by Dunn's multiple comparison test.
The concentrations of Aβ40 and Aβ42 peptides measured from the formic acid extracts of mouse brain mimicked the changes observed histologically so that an increase in amyloid peptides was found in mice expressing APOE4 (Aβ40: 1733 ± 338 pmol/mg of protein and Aβ42: 8720 ± 1155 pmol/mg of protein), and an opposite effect was detected with APOE2 (Aβ40: 468 ± 105 pmol/mg of protein and Aβ42: 5156 ± 1318 pmol/mg of protein; P < 0.05; Fig. 2C). The content of Aβ peptides in the tris-buffered saline (TBS)–soluble fraction was similarly affected, and higher concentrations of both soluble Aβ40 and Aβ42 were measured after APOE4 exposure (Aβ40: 94 ± 18 pmol/mg of protein and Aβ42: 12.9 ± 1.5 pmol/mg of protein) compared with APOE2 (Aβ40: 20 ± 8 pmol/mg of protein and Aβ42: 2.3 ± 0.8 pmol/mg of protein; P < 0.05; Fig. 2D).
Expression of each APOE isoform led to smaller effects at 2 months than at 5 months, implying that APOE's impact on amyloid formation or clearance took several months to emerge when assessed in postmortem tissue. Nevertheless, a significant increase in amyloid plaque density within the cortex of AAV4-APOE4–injected mice was observed compared with the other groups (AAV4-GFP: 25 ± 4; AAV4-APOE2: 22 ± 6; AAV4-APOE3: 26 ± 5; and AAV4-APOE4: 56 ± 4 plaques/mm2 of cortex; P < 0.05; fig. S2, A and B), paralleled by the amount of Aβ40 contained in the formic acid fraction (AAV4-GFP: 725 ± 92; AAV4-APOE2: 492 ± 62; AAV4-APOE3: 681 ± 140; and AAV4-APOE4: 1108 ± 238 pmol/mg of protein; P < 0.05; fig. S2D). The concentrations of TBS-soluble Aβ40/42 species did not change, and no change in the ratio of dense core plaques to total Aβ immunoreactivity was detected (fig. S2C). Further control experiments did not reveal any impact of AAV4-APOE injection on amyloid precursor protein (APP) metabolism, glial activation, or amount of insulin-degrading enzyme (fig. S3, A to C). Overall, comparison of the data at 2- and 5-month exposure suggested that alterations in the APOE isoform shift Aβ-soluble and fibrillar deposits over months, rather than acting acutely.
APOE affects the BBB and alters the efflux of Aβ peptides (9, 20, 21). To examine how APOE exposure modulates the equilibrium of Aβ peptides across the BBB, we measured the plasma concentrations of Aβ40 after 5 months. The plasma content of Aβ in mice injected with AAV4-APOE3 or AAV4-APOE4 was lower compared with AAV4-APOE2 and AAV4-GFP (AAV4-GFP: 1167 ± 213; AAV4-APOE2: 1247 ± 160; AAV4-APOE3: 736 ± 78; and AAV4-APOE4: 799 ± 67 pmol/ml of plasma; P < 0.05; Fig. 2E), suggesting that APOE3 and APOE4 helped to retain Aβ in the CNS, consistent with the relative increased Aβ brain concentrations and with previous data reporting a longer CNS half-life of Aβ because of APOE (10).
APOE4 carriers are more susceptible to neurovascular dysfunction (22), and BBB breakdown was shown to be favored in APOE4 trans genic mice (9), even in the absence of amyloid deposition. To assess whether AAV4-APOE injections compromised the integrity of the BBB, we performed postmortem staining with Prussian blue. Despite the presence of a few hemosiderin-positive focal areas, no effect was observed among any of the experimental groups.
Expression of APOE isoforms modulates the kinetics of amyloidosis
To assess how APOE variants affect the dynamic progression of amyloidosis (23), we used in vivo two-photon imaging and followed the kinetics of amyloid plaque formation and clearance. Mice received an intraventricular injection of AAV4 carrying GFP or each APOE variant at 7 months of age, and a cranial window was implanted 1 week thereafter (T0). After 1 month (T1) or 2 months (T2), amyloid deposits were imaged, following the same cortical areas in the brain over time.
Most of the amyloid deposits remained stable, although new plaques could occasionally be detected in the small viewing volume (Fig. 3A). However, on rare occasions, a methoxy-positive plaque that was imaged at the beginning could not be detected after 1 or 2 months, suggesting that some plaques could be cleared (Fig. 3A). The rate of amyloid progressionwas faster in APP/PS1 miceexposed toAPOE4 but was decreased and even reversed in animals exposed to APOE2. After 2 months, APOE2 reduced the density of amyloid deposits relative to GFP by 0.66 (P = 0.002), relative to APOE3 by 0.67 (P < 0.0001), and relative to APOE4 by 0.74 (P < 0.0001) (Fig. 3, B and C).
Fig. 3. Human APOE variants differentially modulate amyloidosis progression.

(A) Representative in vivo two-photon images of amyloid deposition in APP/PS1 mice 1 week (T0), 1 month (T1), and 2 months (T2) after injection of AAV4 carrying GFP, APOE2, APOE3, or APOE4. An intravenous injection of Texas red dextran (70,000 daltons) was performed before imaging so that the same fields of view could be followed over time. Few new amyloid plaques (in green, detected after intraperitoneal injection of the amyloid fluorescent probe methoxy-XO4) could be detected (blue arrows), whereas occasional deposits initially visible were not detected after 1 or 2 months (yellow arrows). Scale bar, 100 μm. (B) Evaluation of the volumetric cortical density of amyloid deposits over a 2-month period after intraventricular injection of AAV4-GFP, AAV4-APOE2, AAV4-APOE3, or AAV4-APOE4. Six to eight fields of view were longitudinally imaged for each animal. The density of plaques was calculated per volume of cortex and was normalized to the initial value for each animal at baseline (T0). (C) Linear regression fit of amyloidosis progression over 2 months after gene transfer shows that only AAV4-APOE4 induced a significant positive slope during this period. n = 4 AAV4-GFP, n = 4 AAV4-APOE2, n = 6 AAV4-APOE3, and n = 5 AAV4-APOE4 mice. *P < 0.05 using mixed-effects model, with a random effect for mouse and fixed effects for vector, time, and baseline volumetric density.
We next assessed single amyloid plaque growth by measuring the ratio of the cross-sectional area of individual deposits between T1/T0 and T2/T1 (Fig. 4A). Differences were detected among groups at T1 (ratio T1/T0; Fig. 4B) such that amyloid deposits grew significantly more in APOE4-exposed animals compared to GFP-, APOE2-, or APOE3-exposed animals (T1/T0 ratios: 1.175 ± 0.048 for AAV4-GFP, 0.97 ± 0.043 for AAV4-APOE2, 1.063 ± 0.041 for AAV4-APOE3, and 1.29 ± 0.065 for AAV4-APOE4; P < 0.05). This effect was, however, not observed after the second month (ratio T1/T2; Fig. 4C).
Fig. 4. Change in size of amyloid deposits 1 or 2 months after infusion with APOE2, APOE3, or APOE4.

(A) Representative images (maximal z projection of z stacks) of amyloid plaques (in green, detected after intraperitoneal injection of the amyloid fluorescent probe methoxy-XO4) that were longitudinally imaged for 2 months after AAV4 intracerebroventricular injection of APP/PS1 mice. Specific areas within each field of view were selected, and the same deposits could be followed over time. The cross-sectional area of each plaque was measured with ImageJ, and the ratio of the area between two time points was calculated (T1 versus T0 and T2 versus T1). Most amyloid deposits remained stable over time (white arrows), but some plaques grew (yellow arrows) or shrank (blue arrows). Scale bar, 100 μm. (B and C) Scatter dot plots representing the ratio of plaque sizes (B) between T1 and T0 and (C) between T2 and T1. n > 50 plaques measured per group of three to four animals. *P < 0.05 using two mixed-effects models fitted for log of the ratio of two consecutive time points, with random effects for mouse and fixed effects for log baseline size (T0 in the first analysis, T1 in the second analysis).
APOE exposure modulates synaptic loss and neuritic dystrophies around amyloid deposits
Synapse loss is known to correlate with cognitive impairment (24, 25). We recently showed that the presence of APOE4 is associated with higher concentrations of synaptic oligomeric Aβ and a decreased synapse density around amyloid plaques in the brains of human AD patients compared to APOE3 (8, 26). In addition, recent in vitro evidence demonstrated that APOE4 failed to protect against Aβ-induced synapse loss (27). We therefore hypothesized that APOE may differentially affect not only the kinetics of Aβ deposition and clearance but also the integrity of synapses surrounding amyloid deposits.
The densities of pre- and postsynaptic elements (synapsin-1 and PSD95) were determined using array tomography, a high-resolution technique based on immunofluorescence staining of ultrathin tissue sections (28, 29) (Fig. 5A). Because amyloid oligomeric species were shown to be highly concentrated in the vicinity of amyloid deposits, synapsin-1 and PSD95 puncta were quantified either far (>50 μm) or close (<50 μm) to plaques (29). Loss of presynaptic elements near plaques was exacerbated when either APOE3 or APOE4 was expressed, which was not the case after injection of AAV4-APOE2 or AAV4-GFP (Fig. 5B). The density of postsynaptic puncta remained unchanged among mice injected with AAV4-GFP, AAV4-APOE2, and AAV4-APOE3, whereas AAV4-APOE4–exposed animals showed a bigger loss of PSD95 around amyloid deposits, thus reinforcing the deleterious effect of APOE4 on Aβ neurotoxicity (Fig. 5, A to C). When the density of synaptic elements was evaluated in areas located far from amyloid deposits (>50 μm), no difference could be detected among the groups, suggesting that the relative synaptic loss observed with APOE3 and APOE4 was directly related to the presence of Aβ peptides surrounding each plaque.
Fig. 5. Neuropathological changes associated with amyloid deposits are differentially affected by the APOE variants.

(A) Representative images of array tomography sections of mouse cortex immunostained for PSD95 (postsynaptic element; green) and amyloid deposits (stained with antibody NAB61 that preferentially labels Aβ oligomeric species; red) in APP/PS1 mice 2 months after intraventricular injection of AAV4-GFP or AAV4-APOE2/3/4. Scale bar, 50 μm. (B and C) Percentage of presynaptic (synapsin-1) (B) and postsynaptic (PSD95) (C) loss in the vicinity of amyloid plaques. (D) The number of dystrophic neurites per amyloid plaque was evaluated in the brain of injected APP/PS1 mice after immunostaining for the axonal marker SMI312 and Thio-S for amyloid plaques. Scale bar, 100 μm. (E) Bar graph representing the mean number of dystrophic neurites per surface of amyloid plaques. n = 4 AAV4-GFP, n = 4 AAV4-APOE2, n = 6 AAV4-APOE3, and n = 5 AAV4-APOE4 mice. *P < 0.05, one-way analysis of variance (ANOVA) test.
We also evaluated the number of dystrophic neurites associated with amyloid deposits because neuritic plaques reflect a more general alteration of the neuropil with an increased neurite curvature and the appearance of swollen dystrophies (30). These pathological changes are likely attributable to soluble oligomeric Aβ species (31, 32) that are enriched in a region within 50 μm of the plaque surface (29). Expression of APOE4 exacerbates the formation of SMI312-positive dystrophic neurites associated with amyloid deposits compared with GFP, APOE2, and APOE3 (Fig. 5, D and E). This result confirms the observation that the APOE4 isoform affects amyloid neurotoxicity.
Human APOE proteins modify the amount of oligomeric Aβ species in the ISF in Tg2576 mice
We next addressed the question of whether the presence of different human APOE isoforms within the ISF may alter the amount of soluble amyloid species in that same extracellular compartment. We injected Tg2576 mice to validate our previous findings in a different AD mouse model. Tg2576 mice present a much milder phenotype than APP/PS1 mice of the same age. We treated cohorts of 16- to 18-month-old animals in which amyloid deposition had already begun. Three months after gene transfer, a microdialysis probe was inserted into the hippocampus, and samples were collected to characterize early changes associated with each human APOE variant.
We observed that the concentration of Aβ oligomers measured using the specific 82E1/82E1 ELISA assay was significantly higher (by 42 ± 7%; P < 0.05) after injection of AAV4-APOE4 compared with AAV4-APOE2 (Fig. 6), demonstrating that the presence of APOE may modulate the nature of amyloid aggregates in the extracellular compartment. When total Aβ40 and Aβ42 were assessed in the ISF, the same trends were observed, but were not significant (fig. S4A).
Fig. 6. Changes in oligomeric Aβ species in ISF after injection of Tg2576 mice.
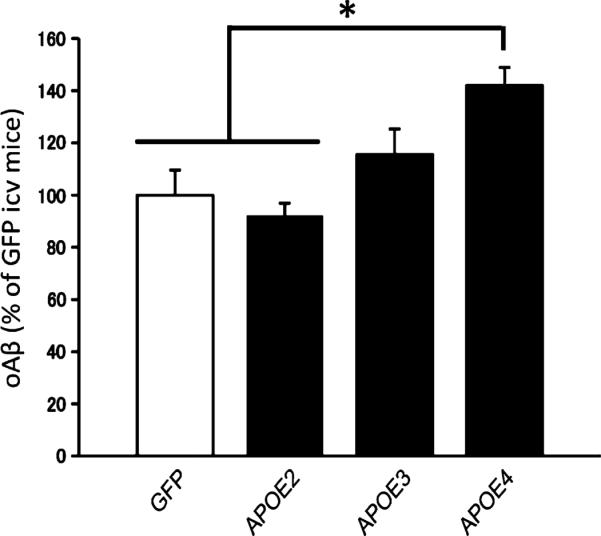
Quantification of the ISF content of oligomeric Aβ (oAβ) as a percentage of GFP-injected mice using the 82E1/82E1 ELISA assay after injection of AAV4-GFP, AAV4-APOE2, AAV4-APOE3, and AAV4-APOE4. n = 3 AAV4-GFP, n = 3 AAV4-APOE2, n = 5 AAV4-APOE3, and n = 5 AAV4-APOE4 mice. *P < 0.05 using nonparametric Kruskal-Wallis test followed by Dunn's multiple comparison test. icv, intracerebroventricular.
As expected, postmortem biochemical analyses of brains from Tg2576 mice showed that the concentration of Aβ42 in the formic acid fraction was increased in APOE4-exposed animals (fig. S4B), confirming, in a second transgenic model, our previous observations in APP/PS1 mice.
DISCUSSION
The marked genetic association between inheritance of APOE alleles and AD (1, 2) has led to multiple suggestions as to how this risk is mediated. APOE binds to Aβ and has been implicated in Aβ clearance (10, 21), but studies in Apoe knockout mice surprisingly show that Aβ deposits are substantially lower in the absence of Apoe. Replacement of endogenous murine Apoe with human APOE2, APOE3, or APOE4 modifies amyloid deposits in the same order as in AD patients, presumably through effects on plaque initiation or fibril formation (33, 34). Alternative hypotheses focus on differential effects on neuritic outgrowth, or the proposal that the effect of APOE genotype on AD phenotype is a consequence of another gene in genetic disequilibrium with APOE on chromo-some 19 (35).
Using a recently developed gene transfer technology (19) and two mouse models of AD, our data addressed these issues by using a combination of in vivo multiphoton imaging, standard quantitative immunohistopathology, array tomography, and high– molecular weight microdialysis approaches that allow examination of oligomeric Aβ within the ISF. Our study demonstrated that a modest (~10%) increase in APOE4 levels altered Aβ aggregation and clearance kinetics, leading to increased retention of Aβ and exacerbating synaptic loss and neuritic dystrophies around plaques. Conversely, APOE2 decreases Aβ deposition and has a marked neuroprotective impact. The observed effects did not correlate with changes in the content of APP proteolytic products, suggesting that Aβ production per se was unaffected. Thus, these results argue that modest amounts of APOE4 and APOE2 in the ISF affect amyloid clearance, deposition, and neurotoxicity in opposite directions. Because lower amounts of Aβ were measured in the plasma of mice exposed to APOE3 or APOE4 solely in the CNS, we hypothesize that our results may be due to differential Aβ clearance within the brain. This effect may be potentially due to the lipidation status of each APOE variant produced by AAV-transduced ependymal cells. Indeed, previous studies have shown that less APOE4 is lipoprotein-associated compared with APOE2 and APOE3 so that increasing the levels of delipidated versus lipidated lipoproteins may be one mechanism for the differential effects of APOE isoforms on Aβ accumulation and retention in the brain (36, 37).
One limitation of our study is the possibility that expression of human APOE2 lowers or displaces murine Apoe in a critical compartment, thereby producing an effect analogous to that seen in Apoe-null animals in which less Aβ accumulates (38, 39). Nonetheless, because our direct measures of murine Apoe protein and mRNA do not show a global effect of human APOE expression on the endogenous protein, and because APOE2 and APOE4 have opposing impacts on amyloid deposition, we favor a direct effect of human APOE isoforms on amyloid deposition.
Because modest changes in the levels of ISF APOE have such marked consequences, these results may also lead to insight into the effects of a wide variety of environmental and genetic factors that might alter the risk or progression of AD by influencing APOE expression. Increases in APOE greater than the magnitude we demonstrate here can occur after traumatic brain injury, epilepsy, ischemia (40, 41), and a high cholesterol diet (42), all of which have been associated with elevated cerebral Aβ. Moreover, promoter polymorphisms that have been found to be in genetic disequilibrium with the APOE4 allele (43) affect APOE expression.
Other manipulations that affect APOE-lipoprotein homeostasis in the CNS clearly change Aβ deposition. For example, in experiments with focal gene transfer with APOE lentiviruses (primarily expressed in hippocampal neurons), APOE4 overexpression exerts a stronger effect on amyloid relative to APOE3 (44). Previous studies also showed that retinoid X receptor agonists that enhance endogenous APOE synthesis led to clearance of Aβ from the brain (13, 20). In addition, brain transduction with CYP46A1, which metabolizes cholesterol in the CNS and lowers its levels, reduces Aβ deposition (45), as does increasing low-density lipo-protein receptor in the brain, which is known to decrease APOE levels (46). Alternatively, Aβ-mimicking peptides that block the interaction between APOE3/4 and Aβ (47) or that convert the structure of APOE4 into an APOE2- or APOE3-like conformation (48) also showed effects on the accumulation of amyloid. Finally, genetic manipulations suggest that decreasing endogenous Apoe expression by 50% strongly affects Aβ phenotype over the animal's lifetime (38). Our current results, with human APOE expressed on the background of murine Apoe, may, in part, result from competition between human APOE and murine Apoe for Aβ interactions. Regardless, these data suggest that human isoform–dependent changes can have marked effects even after pathological changes are well established.
Our data directly address four important areas of controversy in the APOE AD literature. We demonstrate a clear effect of APOE iso-forms on neurotoxicity assessed by synapse loss and neuritic dystrophies, both likely related to impairment of neuronal system function (24, 25). Because these effects were evident in the immediate vicinity of plaques but not in areas distant from plaques, the protection of synapses by APOE2 is likely to be mediated by effects on peri-plaque Aβ rather than due to a direct impact of APOE on synaptic stability. Longitudinal multiphoton in vivo imaging of amyloidosis shows that APOE4 enhances plaque deposition and growth, whereas APOE2 is associated with resolution of plaques, arguing that APOE isoforms have a powerful impact on the pace and progression of disease beyond an initial effect on fibrillar plaque formation. These results reinforce the idea that APOE4 may accelerate the disease process in terms of both amyloid deposition and neurotoxicity (and hence lead to an earlier age of onset), whereas APOE2 does the opposite, which raises the possibility that introduction of APOE2 (or an APOE2 mimetic) into the CNS might have therapeutic value. APOE has variously been suggested as a mechanism to clear Aβ from the brain or as a retention molecule that decreases clearance half-life; our current results show that introduction of modest amounts of APOE3/4 into the ISF is sufficient to enhance retention of Aβ in the CNS. The mechanism of APOE2's remarkable protective action in AD has long been unclear, in part because APOE2 binds to APOE receptors relatively poorly.
Our current data suggest that APOE2 has a gain of function and can reverse established Aβ deposits, as well as support synaptic and neuritic plasticity, in addition to a likely neutral or beneficial effect (compared to other alleles) on CNS retention of Aβ. This suggests that the decade-long difference in age of onset between patients who inherit APOE4 versus APOE2 alleles may reflect both a different initiation point and continuous differences in the kinetics of Aβ deposition and clearance, as well as allele-specific differences in the extent of neurotoxicity associated with the deposits. This dual function of APOE2 may lead to therapeutic approaches aimed at mimicking its plaque clearing and synaptic restoration capacity.
These results are consistent with a model in which APOE acts as a scaffold for Aβ oligomerization. These results also are consistent with our recent observation that oligomeric Aβ is elevated in the CNS of human patients with AD with APOE4 > APOE3 (even when plaque burden is normalized across cases) (26). If APOE, especially APOE4, mediates formation of neurotoxic oligomeric Aβ, the prediction would be that enhanced APOE4 expression would lead to increased synaptic and neuritic alterations, as appears to be the case from our current data. On the basis of these results, caution should be exercised with regard to agents that would increase APOE concentrations in the brains of APOE4 carriers.
Finally, our data highlight the utility of AAV-mediated transduction of the brain's ependymal layer for delivery of genes encoding secreted proteins that then diffuse through the entire cortical mantle (19). We suggest that gene transfer or other approaches that decrease APOE4, or increase APOE2, may be useful for affecting AD disease progression.
MATERIALS AND METHODS
Study design
The present study aimed to investigate how introduction of each human APOE isoform would influence amyloid progression and Aβ-associated neurotoxicity, after plaque deposition has started. To address this question, we did a single infusion with AAV vectors coding for each APOE variant within the intracerebroventricular space of two different AD transgenic mouse models. Mice were exposed for a short (2 to 3 months) or long (5 months) period. Using in vivo imaging, in vivo microdialysis, and array tomography, we evaluated the impact of human ApoE isoforms not only on amyloid deposition and oligomerization within the ISF but also on Aβ-related neurotoxicity (spine loss and dystrophies). Mice were randomly assigned to treatment groups. The nature of the injected vector was kept blinded until statistical analyses. All animals that successfully recovered from the surgical procedures were included in the postmortem analyses. Sample sizes were predetermined on the basis of previous experience. Replication with a second transgenic line was carried out to further test the hypotheses.
Animals
APPswe/PS1dE9 (APP/PS1) (49) (The Jackson Laboratory) and Tg2576 (50) mice express a human mutant APP gene containing the Swedish mutation K594N/M595L under the control of the prion promoter. The APP/PS1 model also overexpresses the presenilin 1 gene deleted for exon 9, which leads to a severe phenotype with amyloid deposition at 6 months of age. The Tg2576 line is a milder model that develops amyloid plaques around1yearofage.Weexposed7-month-oldAPP/PS1foreither2months (n = 4 AAV4-GFP, n = 4 AAV4-APOE2, n = 6 AAV4-APOE3, and n = 5 AAV4-APOE4 animals) or 5 months (n = 4 AAV4-GFP, n = 5 AAV4-APOE2, n = 6 AAV4-APOE3, and n = 5 AAV4-APOE4 animals) with each AAV vector. In addition, a cohort of 16-month-old Tg2576 mice (n = 3 AAV4-GFP, n = 3 AAV4-APOE2, n = 5 AAV4-APOE3, and n = 5 AAV4-APOE4 animals) was included to measure the levels of Aβ within the ISF. APOE-deficient mice (The Jackson Laboratory) were also used. Experiments were performed in accordance with the National Institutes of Health (NIH) and institutional guidelines.
Viral vector construction and production
APOE2, APOE3, and APOE4 complementary DNAs (cDNAs), provided by M. J. LaDu at the University of Illinois (Chicago), were amplified by polymerase chain reaction (PCR), digested by Bam HI, and inserted into an AAV2-pCMV-hrGFP backbone. AAV serotype 4 vectors were produced by the Gene Transfer Vector Core at the University of Iowa, Iowa City. The AAV viral titers were determined by quantitative PCR.
Stereotactic intracerebroventricular injections
AAV intraventricular injections were performed as described previously (14, 30). Animals were anesthetized (ketamine/xylazine: 100 and 50 mg/kg, respectively) and positioned on a stereotactic frame (David Kopf Instruments). Injections were performed in each lateral ventricle with 5 ml of viral preparation (titer of 2 × 1012 viral genomes/ml) using a 33-gauge needle attached to a 10-μl Hamilton syringe (Hamilton Medical) at 0.25 μl/min. Stereotactic coordinates were calculated from bregma (anteroposterior +0.3 mm, mediolateral ±1 mm, and dorsoventral −2 mm).
Cranial window implantation and multiphoton imaging
Mice were anesthetized with isoflurane (1.5%), and a cranial window was implanted by replacing a piece of skull by a 5-mm glass coverslip (30). For imaging, a wax ring was built to create a well of water for the objective (20×, numerical aperture of 0.95, Olympus). Twenty-four hours before surgery, amyloid deposits were visualized after intraperitoneal injection of methoxy-XO4 (5 mg/kg), a fluorescent compound that crosses the BBB and binds to plaques (51). Texas red dextran [70,000 daltons; 12.5 mg/ml in phosphate-buffered saline (PBS); Molecular Probes] was injected into the lateral tail vein to provide a fluorescent angiogram and follow the exact same fields of view over time. Mice were imaged 1 week after AAV injection and then 1 and 2 months after.
A mode-locked Ti:sapphire laser (Mai Tai, Spectra-Physics) mounted on a multiphoton imaging system (Bio-Rad 1024ES, Bio-Rad) generated 860-nm two-photon fluorescence excitation light. Emitted light was collected through an external detector containing three photomultiplier tubes (PMTs) (Hamamatsu Photonics) in the range of 380 to 480, 500 to 540, and 560 to 650 nm. Two-color images were acquired for plaques and angiography simultaneously. Low-magnification in vivo images (615 × 615 μm; z step, 2 μm; depth, ~200 μm) were taken, and six to eight fields of view were imaged.
Image processing and analysis
Plaque density was quantified with ImageJ by reporting the number of amyloid deposits per volume of cortex imaged. We considered the cortical volume starting from the first slice of the z stack at the surface to the last slice where an amyloid deposit could be detected. The size of amyloid deposits was evaluated over time by measuring their cross- sectional area from the maximal intensity after two-dimensional projection. For each plaque, the ratio of the area between the initial time point and the first month (T1/T0), or between the second and first months (T2/T1), was calculated. The settings of the multiphoton microscope (laser power and PMTs) were maintained throughout the imaging sessions.
In vivo microdialysis sampling
In vivo microdialysis sampling of ISF was performed on Tg2576 mice 3 months after AAV injection (17). The microdialysis probe had a 4-mm shaft with a 3.0-mm, 1000-kD molecular weight cutoff polyethylene membrane (PEP-4-03, Eicom). Before use, the probe was washed with artificial CSF (aCSF) (122 mM NaCl, 1.3 mM CaCl2, 1.2 mM MgCl2, 3.0 mM KH2PO4, 25.0 mM NaHCO3). The preconditioned probe's outlet and inlet were connected to a peristaltic pump (ERP-10, Eicom) and a microsyringe pump (ESP-32, Eicom), respectively, using fluorinated ethylene propylene tubing (ϕ 250-μm inside diameter).
Probe implantation was performed as previously described (17, 18). Anesthetized animals (1.5% isoflurane) were stereotactically implanted with a guide cannula (PEG-4, Eicom) in the hippocampus (bregma −3.1 mm, −2.5 mm lateral to midline, −1.2 mm ventral to dura). The guide was then fixed to the skull using dental cement. Four days after surgery, the mice were placed in a microdialysis cage, and a probe was inserted through the guide. The probe was perfused with aCSF for 240 min at a flow rate of 10 μl/min before sample collection. Samples were collected at a flow rate of 0.25 μl/min (for Aβ) or 0.1 μl/min (for ApoE). Mice were awake and free-moving in the microdialysis cage (AtmosLM microdialysis system, Eicom).
Immunohistological analysis
Mice were euthanized by CO2 inhalation. One cerebral hemisphere was fixed in 4% paraformaldehyde in PBS and embedded in paraffin wax. A 1-mm3 piece of cortex was processed for array tomography, whereas the rest was snap-frozen. Paraffin-embedded sections (10 μm) were deparaffinized in xylene, rehydrated in ethanol, treated in citrate buffer [10 mM sodium citrate, 0.05% Tween 20 (pH 6.0)], permeabilized in PBS with 0.5% Triton, and blocked in PBS with 3% bovine serum albumin (BSA) for 2 hours. Incubation with primary antibodies was done overnight at 4°C: Bam10 (1:1000, Sigma) and R1282 (1:500, provided by D. Selkoe) for amyloid plaques; mouse monoclonal antibody 3H1 (1:250, Ottawa Heart Institute) for human APOE; chicken anti-GFP (1:500, Aves) and SMI-312 (1:500, Covance) for neuritic dystrophies; and IBA1 (1:500, Wako) and glial fibrillary acidic protein (GFAP) (1:1000, Sigma) for microglial cells and astrocytes, respectively. Incubation with the secondary antibody was done for 2 hours. Amyloid dense core plaques were labeled by 0.05% Thio-S (Sigma-Aldrich) in 50% ethanol before mounting.
Sample preparation, immunostaining, and imaging for array tomography
Array tomography analyses were performed as previously described (29). A piece of cortical tissue (1 mm3) adjacent to the ventricular region was dissected and fixed for 3 hours in 4% paraformaldehyde and 2.5% sucrose in 0.01 M PBS. After dehydration in ethanol, samples were incubated in LR White resin (Electron Microscopy Sciences) overnight at 4°C before polymerization at 53°C. Ribbons of sections (70 nm) were then cut on an ultracut microtome (Leica) with a Jumbo Histo Diamond Knife (DiATOME).
After rehydration in 50 mM glycine in TBS for 5 min, sections were blocked in 0.05% Tween and 0.1% BSA in tris for 5 min, and primary antibodies were applied at a 1:50 dilution in blocking buffer for 2 hours [PSD95 (Abcam, Ab12093), synapsin-1 (Millipore, AB1543), and NAB61 (from V. Lee), which preferentially stains oligomeric Aβ species (52)], followed by secondary antibodies (Invitrogen). Images were obtained on 7 to 30 serial sections and were acquired with a Zeiss Axioplan LSM510 confocal/multiphoton microscope (63×/1.4 numerical aperture Plan Apochromat oil objective).
Images were analyzed as previously described with ImageJ (NIH) and MATLAB (MathWorks) (29). Each set of images was converted to stacks and aligned with ImageJ MultiStackReg and StackReg plug-ins (courtesy of B. Busse and P. Thevenaz, Stanford University). Known volumes were selected, and an automated, threshold-based detection program was used to count both PSD95 and synapsin puncta that appeared in more than one consecutive section (WaterShed program, provided by B. Busse, S. Smith, and K. Micheva, Stanford University). WaterShed exported a thresholded image stack (separate for each channel) showing puncta that were present in more than one slice of the array. Several sites in the cortex were sampled per mouse, and their distance from the edge of a plaque was measured.
Aβ quantification
The concentrations of Aβ40 and Aβ42 were determined by BNT-77/BA-27 (for Aβ40) and BNT-77/BC-05 (for Aβ42) sandwich ELISA (Wako) according to the manufacturer's instructions. Aβ oligomers were quantified with the 82E1/82E1 sandwich ELISA (Immuno-Biological Laboratories), in which the same N-terminal (residues 1 to 16) antibodies were used for both capture and detection (53).
Immunoblot analysis
Brain lysates and microdialysates (20 mg of protein) were electrophoresed on 4 to 12% Novex Bis-Tris gels (Invitrogen) in Mops running buffer for SDS–polyacrylamide gel electrophoresis (Invitrogen). Gels were transferred onto polyvinylidene difluoride membranes and blocked for 60 min in 5% milk/TBS–Tween 20. The membranes were probed with goat anti-APOE antibody (1:1000, Millipore) to detect APOE in the ISF of Apoe-null animals, whereas albumin was used as a control. Blots for human and mouse APOE were respectively probed with EP1373Y antibody (1:1000, Novus Biologicals) and rabbit polyclonal APOE antibody (1:1000, Abcam). APP proteolytic products (sAPP: 22C11, 1:4000, Millipore and C83/C99: APP Cter, 1:4000, Sigma) and the insulin-degrading enzyme (1:1000, Santa Cruz Biotechnology) were also analyzed. Incubation with horseradish peroxidase (HRP)–conjugated goat immunoglobulin G antibodies (Vector) was done for 2 hours. Immuno-reactive proteins were developed with ECL kit (Western Lightning, PerkinElmer) and detected on Hyperfilm ECL (GE Healthcare).
Quantitative reverse transcription PCR
Total RNA from brain was extracted with TRIzol Reagent (Life Technologies, 15596-026), and cDNA was then synthesized with the SuperScript III One-Step RT-PCR System (Life Technologies, 12574-018). PCR primers were designed to amplify human APOE mRNA, endogenous apoE, and Gapdh mRNAs [Apoe: 5′-AGCTCCCAAGTCACACAAGA (forward) and5′-GTTGCGTAGATCCTCCATGT(reverse);APOE:5′-CCAGCGACAATCACTGAAC (forward) and 5′-GCGCGTAATACGACTCACTA (reverse); Gapdh: 5′-ATGACATCAAGAAGGTGGTG (forward) and 5′-CATACCAGGAAATGAGCTTG (reverse)].
APOE ELISA
Samples were extracted in 2% Triton in TBS, allowing the mobilization and quantification of the membrane-bound ApoE. ELISA plates were coated overnight with goat anti-APOE antibody (1.5 μg/ml) (murine APOE) or WUE4 antibody (1.5 μg/ml) (human APOE) and blocked with 1% milk in PBS for 1.5 hours. Human recombinant ApoE (human assay, BioVision) or in-house mouse standards from brain extract (murine assay) were used, and samples were incubated overnight in ELISA buffer (0.5% BSA and 0.025% Tween 20 in PBS). Detection antibodies specific for human (goat-apoe, Millipore; 1:10,000) or mouse (Abcam; 1:2000) were used, followed by HRP-conjugated secondary antibodies. Revelation of the signal was done using the 3,3′,5,5′-tetramethylbenzidine (TMB) substrate before stopping the solution using H3PO4, which was measured at 450 nm.
Statistical analyses
Statistical analyses were performed with the Prism software. Because of the small size of the samples, normality could not be assumed for most of the analyses. For all the postmortem analysis, nonparametric Kruskal-Wallis test followed by Dunn's multiple comparison test was performed. In vivo imaging data of amyloid progression were analyzed using a mixed-effects model, with a random effect for mouse and fixed effects for vector, time, and baseline volumetric density. An interaction between time and vector was considered in this analysis but was not significant. For the analysis of the plaque size over time, two mixed-effects models were fitted for log of the ratio of two consecutive time points, with random effects for mouse and fixed effects for log baseline size (T0 in the first analysis, T1 in the second analysis). Samples were blinded for each analysis.
Supplementary Material
Acknowledgments
We thank M. J. LaDu (University of Illinois, Chicago) for providing the APOE2, APOE3, and APOE4 cDNAs. Funding: This work was supported by NIH grants R00AG33670, NS34568, HD33531, and DK54759; the NIH Challenge Grant (1RC1AG026265-01); the Roy J. Carver Trust (B.L.D.); the Lefler Fellowship (Harvard Medical School); the French Bettencourt-Schueller Foundation; and the Dreyfoos Program.
Footnotes
Author contributions: E.H. designed and performed the experiments and wrote the manuscript. J.D. performed the postmortem immunohistological study of amyloid deposition. A.D.R. and T.H. performed the ELISA for human and mouse APOE. S.T. performed the ISF sampling and analyses. R.M.K. performed the array tomography assay. M.S. produced the AAV vector stocks. M.A.-O. helped to acquire in vivo imaging data. T.S.-J., R.B., B.L.D., and B.T.H. contributed to design of the experiments and wrote the manuscript.
Competing interests: M.A.-O. was a consultant in 2012 for Neurophage Pharmaceuticals. The other authors declare that they have no competing interests. The University of Iowa has filed a patent with MGH as a secondary on the idea of APOE gene transfer as a treatment of AD.
Citation: E. Hudry, J. Dashkoff, A. D. Roe, S. Takeda, R. M. Koffie, T. Hashimoto, M. Scheel, T. Spires-Jones, M. Arbel-Ornath, R. Betensky, B. L. Davidson, B. T. Hyman, Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 5, 212ra161 (2013).
REFERENCES
- 1.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 2.Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer's disease. Annu. Rev. Neurosci. 1996;19:53–77. doi: 10.1146/annurev.ne.19.030196.000413. [DOI] [PubMed] [Google Scholar]
- 3.West HL, Rebeck GW, Hyman BT. Frequency of the apolipoprotein E epsilon 2 allele is diminished in sporadic Alzheimer disease. Neurosci. Lett. 1994;175:46–48. doi: 10.1016/0304-3940(94)91074-x. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Alzheimer's disease: Genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 6.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer's disease: Allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 7.Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters Aβ metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol. Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 8.Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM, Holtzman DM, Hyman BT, Spires-Jones TL. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-β. Brain. 2012;135:2155–2168. doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe HS. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 2009;29:15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holtzman DM, Fagan AM. Potential role of apoE in structural plasticity in the nervous system; implications for disorders of the central nervous system. Trends Cardiovasc. Med. 1998;8:250–255. doi: 10.1016/s1050-1738(98)00017-6. [DOI] [PubMed] [Google Scholar]
- 13.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G, Martins IH, Chiorini JA, Davidson BL. Adeno-associated virus type 4 (AAV4) targets ependyma and astrocytes in the subventricular zone and RMS. Gene Ther. 2005;12:1503–1508. doi: 10.1038/sj.gt.3302554. [DOI] [PubMed] [Google Scholar]
- 15.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 16.Wisniewski T, Frangione B. Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 17.Takeda S, Sato N, Ikimura K, Nishino H, Rakugi H, Morishita R. Novel microdialysis method to assess neuropeptides and large molecules in free-moving mouse. Neuroscience. 2011;186:110–119. doi: 10.1016/j.neuroscience.2011.04.035. [DOI] [PubMed] [Google Scholar]
- 18.Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J. Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G, Martins I, Wemmie JA, Chiorini JA, Davidson BL. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J. Neurosci. 2005;25:9321–9327. doi: 10.1523/JNEUROSCI.2936-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachmeier C, Beaulieu-Abdelahad D, Crawford F, Mullan M, Paris D. Stimulation of the retinoid × receptor facilitates beta-amyloid clearance across the blood–brain barrier. J. Mol. Neurosci. 2013;49:270–276. doi: 10.1007/s12031-012-9866-6. [DOI] [PubMed] [Google Scholar]
- 21.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E e4 and cerebral hemorrhage associated with amyloid angiopathy. Ann. Neurol. 1995;38:254–259. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 23.Cruz L, Urbanc B, Buldyrev SV, Christie R, Gómez-Isla T, Havlin S, McNamara M, Stanley HE, Hyman BT. Aggregation and disaggregation of senile plaques in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7612–7616. doi: 10.1073/pnas.94.14.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 25.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, Mitani A, Joyner D, Thyssen DH, Bacskai BJ, Frosch MP, Spires-Jones TL, Finn MB, Holtzman DM, Hyman BT. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012;32:15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sen A, Alkon DL, Nelson TJ. Apolipoprotein E3 (ApoE3) but not ApoE4 protects against synaptic loss through increased expression of protein kinase Cε. J. Biol. Chem. 2012;287:15947–15958. doi: 10.1074/jbc.M111.312710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Micheva KD, Smith SJ. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid β induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry MC, Cheung BS, Rebeck GW, Paul SM, Bales KR, Hyman BT. Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid β-peptide deposition in homozygous APPV717F transgenic mice. Acta Neuropathol. 2000;100:451–458. doi: 10.1007/s004010000263. [DOI] [PubMed] [Google Scholar]
- 35.Roses AD. An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Arch. Neurol. 2010;67:536–541. doi: 10.1001/archneurol.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, Weeber EJ, Bu G, Yu C, Ladu MJ. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2012;287:41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J. Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeMattos RB. Apolipoprotein E dose-dependent modulation of β-amyloid deposition in a transgenic mouse model of Alzheimer's disease. J. Mol. Neurosci. 2004;23:255–262. doi: 10.1385/JMN:23:3:255. [DOI] [PubMed] [Google Scholar]
- 40.Iwata A, Browne KD, Chen XH, Yuguchi T, Smith DH. Traumatic brain injury induces biphasic upregulation of ApoE and ApoJ protein in rats. J. Neurosci. Res. 2005;82:103–114. doi: 10.1002/jnr.20607. [DOI] [PubMed] [Google Scholar]
- 41.Page KJ, Hollister RD, Hyman BT. Dissociation of apolipoprotein and apolipoprotein receptor response to lesion in the rat brain: An in situ hybridization study. Neuroscience. 1998;85:1161–1171. doi: 10.1016/s0306-4522(97)00661-1. [DOI] [PubMed] [Google Scholar]
- 42.Srivastava N, Averna M, Srivastava RA. Dietary cholesterol and estrogen administration elevate brain apolipoprotein E in mice by different mechanisms. Indian J. Biochem. Biophys. 2008;45:410–415. [PubMed] [Google Scholar]
- 43.Bekris LM, Galloway NM, Montine TJ, Schellenberg GD, Yu CE. APOE mRNA and protein expression in postmortem brain are modulated by an extended haplotype structure. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010;153B:409–417. doi: 10.1002/ajmg.b.30993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hudry E, Van Dam D, Kulik W, De Deyn PP, Stet FS, Ahouansou O, Benraiss A, Delacourte A, Bougnères P, Aubourg P, Cartier N. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol. Ther. 2010;18:44–53. doi: 10.1038/mt.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, Guridi M, Asuni AA, Sullivan PM, Holtzman DM, Sadowski MJ. Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumulation of Aβ and inhibits synaptic degeneration. Am. J. Pathol. 2013;182:1750–1768. doi: 10.1016/j.ajpath.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brodbeck J, McGuire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong DE, Pleiss M, McComas C, Hess F, Witter D, Peterson S, Childers M, Goulet M, Liverton N, Hargreaves R, Freedman S, Weisgraber KH, Mahley RW, Huang Y. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 2011;286:17217–17226. doi: 10.1074/jbc.M110.217380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 50.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 51.Bacskai BJ, Klunk WE, Mathis CA, Hyman BT. Imaging amyloid-β deposits in vivo. J. Cereb. Blood Flow Metab. 2002;22:1035–1041. doi: 10.1097/00004647-200209000-00001. [DOI] [PubMed] [Google Scholar]
- 52.Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-β peptide (Aβ) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Aβ precursor protein (APP) transgenic mice. J. Biol. Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 53.Xia W, Yang T, Shankar G, Smith IM, Shen Y, Walsh DM, Selkoe DJ. A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 2009;66:190–199. doi: 10.1001/archneurol.2008.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.