

Abstract
Cellular circuits sense the environment, process signals, and compute decisions using networks of interacting proteins. To model such a system, the abundance of each activated protein species can be described as a stochastic function of the abundance of other proteins. High-dimensional single-cell technologies, like mass cytometry, offer an opportunity to characterize signaling circuit-wide. However, the challenge of developing and applying computational approaches to interpret such complex data remains. Here, we developed computational methods, based on established statistical concepts, to characterize signaling network relationships by quantifying the strengths of network edges and deriving signaling response functions. In comparing signaling between naïve and antigen-exposed CD4+ T-lymphocytes, we find that although these two cell subtypes had similarly-wired networks, naïve cells transmitted more information along a key signaling cascade than did antigen-exposed cells. We validated our characterization on mice lacking the extracellular-regulated MAP kinase (ERK2), which showed stronger influence of pERK on pS6 (phosphorylated-ribosomal protein S6), in naïve cells compared to antigen-exposed cells, as predicted. We demonstrate that by using cell-to-cell variation inherent in single cell data, we can algorithmically derive response functions underlying molecular circuits and drive the understanding of how cells process signals.
INTRODUCTION
Cells process external cues through the biological circuitry of signaling networks wherein each protein species processes information pertaining to other proteins whose activities themselves are determined by biochemical modifications (e.g., phosphorylation) or other allosteric interactions. Signaling networks can be remarkably attuned to distinguishing subtle features of stimuli to enable key decisions regarding cellular response or fate. For example, naïve CD4+ T cells take into account both dose and duration of T cell Receptor (TCR) engagement, the strength of peptide binding in the MHC cleft, and co-receptor cues in making a decision to differentiate into either regulatory or helper T cells (1-4). With this example as one amongst many, it follows then that to properly understand normal cellular responses and how these are dysregulated in disease, robust quantitative characterizations of signaling relationships will be required to enable more accurate models of signaling.
Despite progress in the quest to understand and represent the complexities of signaling biology, graph diagrams typically used as depictions of signaling relationships only offer qualitative abstractions. In such graphs the vertices correspond to proteins and a directional edge indicates the influence of one protein or molecular species on another and, as such, fail to capture many of the more complex ways through which signaling networks process information. Further, such representations are not designed to readily enable predictions of response to stimuli or therapeutic intervention. Although quantitative models have been proposed to describe signaling networks (3, 5, 6), these are specific to each system and require measurements of biochemical rates and many additional parameters. To scale to a large number of signaling networks and cell types, a robust data-driven approach that can quantify signaling interactions in molecular circuits is required. A data-driven approach would take advantage of statistically relevant differences in complex cell populations to better inform the function that is encoded by an inferred circuit diagram.
To this end, single-cell measurement technologies can offer quantitatively precise, even absolute (given appropriate probes and experimental design), measures of dozens of cellular components representing important biochemical functions. Variation in a complex cell population can be discerned in a functionally relevant context, and thereby enable unique insights into the underlying relationships between signaling molecules. Mass cytometry, for example, can assay the abundance of dozens of internal and surface protein epitopes simultaneously in millions of individual cells (7, 8), offering an opportunity to quantitatively characterize signaling at circuit-wide scales. Modeling a signaling network as a computational system, where each signaling protein computes a stochastic function of other proteins, and treating each single cell as an example of possible input-out enables the recovery of how a signaling network functions. With many thousands of individual cells, each providing a point of data about relationships between proteins, we can infer the network function.
However, a major challenge in deciphering single-cell signaling data is developing computational methods that can handle the complexity, noise (which can be either natural stochasticity or actual instrument noise), and bias in the measurements. First, because cell populations are rarely homogeneous, different cell subpopulations can manifest distinct behaviors – and therefore the relationships between signaling proteins may be obscured beneath a mixture of multiple network states. For example, naïve primary B cells can have weak and stochastic responses to stimuli such that only a small fraction of the population responds (via activation of signaling pathways), whereas memory B cells are considered primed and even evolved towards a more avid binding of antigen. Similarly, naïve T cells manifest different kinetics of response to T cell receptor engagement than do effector T cells. Second, technical noise in the measurements can further confound the quantification of molecular interactions. Third, marker abundance (which often correlates with cell size) can lead to biased correlations and thereby be misinterpreted as an influence between the assayed signaling proteins.
We addressed these challenges by developing an algorithm, based on the statistical concepts of conditional probability (9) and density estimation (10), termed conditional-Density Resampled Estimate of Mutual Information (DREMI) to quantify the strength of molecular interactions. Given a relationship between two proteins, where X influences Y, DREMI considers the abundance or activity of protein Y as a stochastic function of the abundance or activity of protein X. DREMI uses the variation in a population of individually measured single cells to quantify the amount of information transmitted from protein X to protein Y in the signaling network.
A key conceptual shift compared to previous approaches to single -cell analysis is that DREMI computes mutual information on the estimated conditional probability of Y∣X rather than the joint probability of X and Y (the latter being the preferred approached in most other mutual information-based metrics). Joint probability describes the density of cell states (like in a traditional scatter or density plot), whereas conditional probability describes how the state of Y varies with different states of X. To explore such relationships, we couple DREMI to an algorithm we term conditional-Density Rescaled Visualization (DREVI). We use DREVI to visually render the estimated conditional density function as a rescaled heat map, which allows us to visualize the function underlying the molecular interaction in the cell population.
T cells offer an opportune system within which to characterize signaling relationships because several well-characterized but subtly distinct T cell subsets exist (e.g., regulatory, effector and memory) – each of which have a distinct function and yet the signaling distinctions between them are not well appreciated. For example, in naïve CD4+ T cells, which have not been exposed to antigen, the activation of the TCR (dependent upon the engagement of co-receptors and availability of certain cytokines) leads to differentiation into functionally distinct effector cell types (11). In response to the same TCR activation, antigen-experienced effector or memory T cells tend to proliferate and mount responses more rapidly and in greater magnitude than naïve T cells (12-14). However, although the wiring diagrams that broadly describe the basic pathways are partly understood, the molecular mechanisms that lead to these differences are far from fully resolved.
Using a combination of DREMI and DREVI we modeled signaling in pathways downstream of the TCR. We find that the functional form of interactions between signaling proteins demonstrates sigmoidal behaviors that are dynamically and systematically altered upon activation and with increases in stimulus strength, leading to complex but definable outcomes. We applied this to a physiological case using mass cytometry to track the abundance of 11 phospho-proteins post stimulation, ranging from 30 seconds to several hours, of murine TCR-activation in over 2 million individual cells. A comparison of T cell subtypes reveals subtle reconfigurations in the strength and functional shape of signal transfer between previously known pairs of interacting signaling molecules, all of which impacts the response to TCR activation. Our model enabled predicting the outcome of a perturbation that quantitatively differed between T cell subtypes that we subsequently verified experimentally. The computational framework developed here can be generally applied to model any signaling system for which appropriate antibodies, binding agents, or surrogate readouts are available. Notably, the approach discerns signaling behaviors that exist in complex cell populations, and in departure from our previous work (15), uses the inherent stochasticity to quantify and characterize the underlying signaling behaviors, demonstrating that computation can help reveal mechanistic insight from multi-dimensional single cell datasets.
RESULTS
The dynamics of T cell signaling was characterized and compared across murine T cell subsets. Time-series data was collected for phospho-signaling proteins in T-lymphocyte populations of B6 mice (16) after stimulation of the TCR (by crosslinking TCR and the CD28 co-stimulator with biotinylated antibodies). We assayed the abundance of 9 surface markers and 11 phospho-epitopes, among which were key nodes in TCR signaling and related pathways (Supplementary Fig. 1 and Table 1). Samples were collected at 13 time points after TCR activation ranging from 30 seconds to 80 minutes with two different types of stimuli (CD3/CD28 and CD3/CD4/CD28). We measured ~10,000 cells in each sample, cumulatively resulting in deep profiling of signaling data for more than 2 million individual cells.
The 9 surface markers in our panel distinguish between T cell subsets, including naïve CD44low and antigen-experienced CD44high T cells (17, 18) (Supplementary Fig. 2). Overall, with 20 markers per individual cell, these studies simultaneously resolve 6 T cell subsets in the lymph nodes along with their signaling responses – thus reducing technical limitations inherent in the assay of post-sorted T cell subsets.
Conditional Density-Rescaled Visualization of Single-Cell Data
To analyze these data, we consider each individual cell in our mass cytometry data as an instance of co-occurring protein states and use this information to quantify the signaling relationship between the assayed protein parameters. We demonstrate our approach using a signaling relationship between two measured protein epitopes in our data. A primary locus for intracellular signaling initiation of the TCR activation cascade is a protein scaffold known as the cluster of differentiation 3 zeta-chain (CD3ζ). Upon engagement with antigen, CD3ζ is phosphorylated (to pCD3ζ), which creates a scaffold target for the zeta-chain-associated protein kinase 70 (ZAP-70), which in turn phosphorylates SLP76 (SH2 domain containing leukocyte protein of 76kDa) and LAT (the adaptor protein SH2 domain containing leukocyte protein and Linker of activated T cells, respectively (19)).
When the relation between pCD3ζ and pSLP76 pre-stimulation and 30 seconds after stimulation (Fig. 1A) is visualized as a scatterplot (a traditional means used to analyze flow cytometry data), it is difficult to identify any clear characteristics of the data besides the range of expression values. Visualizing the data using a density plot (Fig 1B) (10), one observes that the abundance of pCD3ζ and pSLP76 is concentrated at a particular value, but there is no clear statistical dependency between the two molecules, nor does this visualization method illuminate the nature of the well-researched influence that pCD3ζ has on pSLP76 (20).
Figure 1. Outline of the DREVI method.
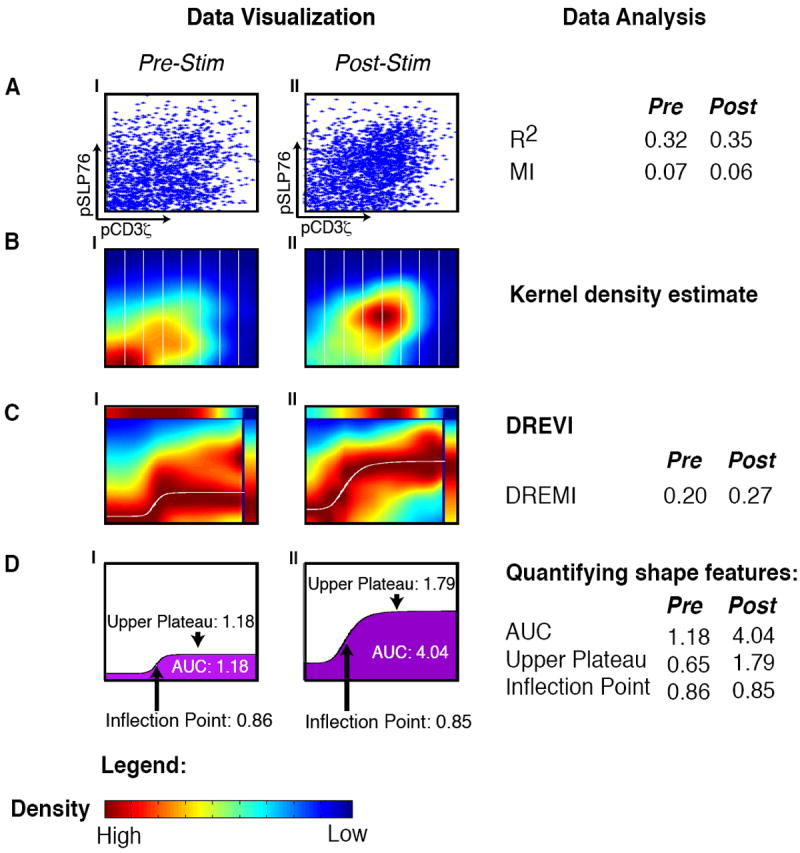
All panels represent the relationship between pCD3ζ and pSLP76 in CD4+ naïve T-lymphocytes from B6 mice with CD3 and CD28 before stimulation (AI-DI) and 30 seconds after stimulation (AII-DII). (A) Each dot in the scatterplot represents the value of a single cell, where the x-axis represents the amount of pCD3ζ levels and the y-axis represents the amount of pSLP76 levels. (B) The same data is shown using kernel density estimation with red corresponding to dense regions and blue corresponding to sparse regions. The x-axis is partitioned into slices (in practice we use 256 such slices). (C) A DREVI plot of the same data, renormalizing the density estimate to obtain a conditional-density estimate of the abundance of pSLP76 levels given the abundance of pCD3ζ levels. Within each column, dark red (maximal color) represents the highest density in that slice. A response function (white curve) is fit to the region of highest conditional density. The top bar plots the marginal distribution of pSLP76 levels and the right bar plots the marginal distribution of pS6 levels. (D) Following curve fitting, the inflection point, upper plateau and area under the curve can be computed and compared between conditions.
Why do traditional methods of visualizing data, such as scatterplots or density estimations, fail to reveal clear relationships between molecules? In Figure 1, the majority of cells can be found in a narrow range of values for each marker. Therefore, the joint density of pCD3ζ and pSLP76 is dominated by a narrow set of values and does not reveal the overall functional response, i.e., how protein Y changes as a function of the activity of protein X. To understand Y as a function of X, we have to explore X’s full dynamic range and how values of Y are dependent on values of X.
To visualize and characterize signal transfer between proteins X and Y, we developed DREVI (conditional Density-Rescaled Visualization) (Fig. 1, BOX 1, Methods), which represents in a visual form the stochastic function of how X influences Y. By using the natural variation in the amount of X and Y from cell to cell, we empirically learn the probability density P(X, Y) from the data. To characterize the influence that X extends on Y, we shift from the joint density and empirically estimate the conditional density P(Y∣X) (See Methods), by means of a kernel density estimation method based on heat diffusion (10, 21). The conditional density enables us to ascertain how Y’s values are changing with respect to the values of X, regardless of where the majority of cells are concentrated in the joint distribution. We note that while statisticians have previously developed sophisticated methods to directly compute the conditional density (22), these perform poorly on our data (Supplementary Fig 3).
BOX 1. conditional Density-Rescaled Visualization.
DREVI reveals the influence of protein X on protein Y. Most methods for visualizing pairwise data (such as scatterplots and density contours) depict the joint probability of X and Y. By contrast, we reveal signaling behavior along its full dynamic range by visualizing conditional probability. The main steps of DREVI are as follows:
Compute the joint kernel density estimate f̂(x, y) (10).
Compute the marginal density of X, f̂(x) and the conditional density estimate of Y given X as on a fine grid of points G = {(xi, yj), 1<i<n, 1<j<m}(2, 12, 15) that span the range of X and Y.
Rescale each value of the conditional density estimate by its column-maximum to obtain f̂*(yj∣xi)=f̂(yj∣xi)/maxk(f̂(yk∣xi))
Visualize f̂ * (G) as a heatmap, adding side-bars depicting the marginal densities of X and Y.
Fig. 1C shows DREVI plots that depict pSLP76 abundance (Y) when conditioned on pCD3ζ (X) abundance. This view of the data reveals a distinct difference in the relation between pCD3ζ and pSLP76 before and after TCR activation that was not apparent in the scatterplots or joint distribution. Given an equal amount of pCD3ζ, we observe that pSLP76 has a stronger response to increasing pCD3ζ levels after TCR activation (note the increased values of the median response represented by the white line in Figure 1C). This suggests that following TCR activation, additional factors (perhaps ZAP70 or LAT) modulate the relationship between pCD3ζ and pSLP76. Similarly, DREVI can help clarify the relationship between additional protein pairs (Supplementary Fig. 4) and visualize how the relationships change through time (Supplementary Fig. 5).
Conditional Density-Resampled Estimate of Mutual Information
To systematically compare between conditions, time points, and cell types, a measure that quantifies the relationship between two proteins is needed. Often, mutual information-based metrics are used to evaluate relationships between gene or protein pairs. However, mutual information (MI) is difficult to compute on continuous data. To resolve this, a first step is to discretize the data. Currently, adaptive partitioning (23) is one of the most widely used approaches for such discretization (24, 25). However, adaptive partitioning assumes that denser regions of the data are more important than sparser regions, and therefore the dense regions are partitioned more finely than sparse regions and dominate the resultant mutual-information metric. By contrast, we developed a measure that allows for sparser populations to be accounted for – thus preventing bias against small cell populations that could have distinct and interesting biology that might inform understanding of the signaling relations in question.
To quantify the strength of the influence protein X has on protein Y, we developed conditional Density-Resampled Estimate of Mutual Information (DREMI) (see BOX 2, Fig. 2, Methods). Like MI (26), DREMI is a shape-agnostic measure that scores how predictive X is of Y, but – unlike MI – it is not symmetric (and therefore might inform directionality) and also captures the strength of this relationship over all populated regions of the dynamic range, regardless of the (often peaked) distribution of X in the data. This is achieved by computing mutual information on the conditional density estimate of the data, rather than the raw data itself. DREMI begins with the conditional density estimate, computed for DREVI (See Box 1). The data is then resampled evenly through the range of the conditional probability density, and MI is computed on the resampled data, using equi-partitioned binning spanning the entire range.
BOX 2. Conditional Density-Resampled Estimate of Mutual Information.
DREMI provides a score for the strength of the influence protein X has on protein Y. In many physiological conditions, only a small fraction of the cells have activated protein X in response to stimuli and these active populations have little influence on the mutual information metric. DREMI explicitly factors these populations by computing a score based on the conditional distribution of Y∣X rather than joint distribution. DREMI estimates the computation Ic, given by:
DREMI is computed as follows:
Begin with the rescaled conditional-density estimate f̂*(G) as in DREVI (BOX 1), computed on the grid of points G = {(xi, yj), 1<i<n, 1<j<m}.
Round values f̂*(xi, yj)< ε to 0, for user-defined threshold ε, to eliminate technical noise from the measurements.
Resample from G according to the conditional density estimate f̂(G).
Equi-partition the full range of the data, and calculate mutual information using this partition as the discretization.
Figure 2. Outline of the DREMI method.
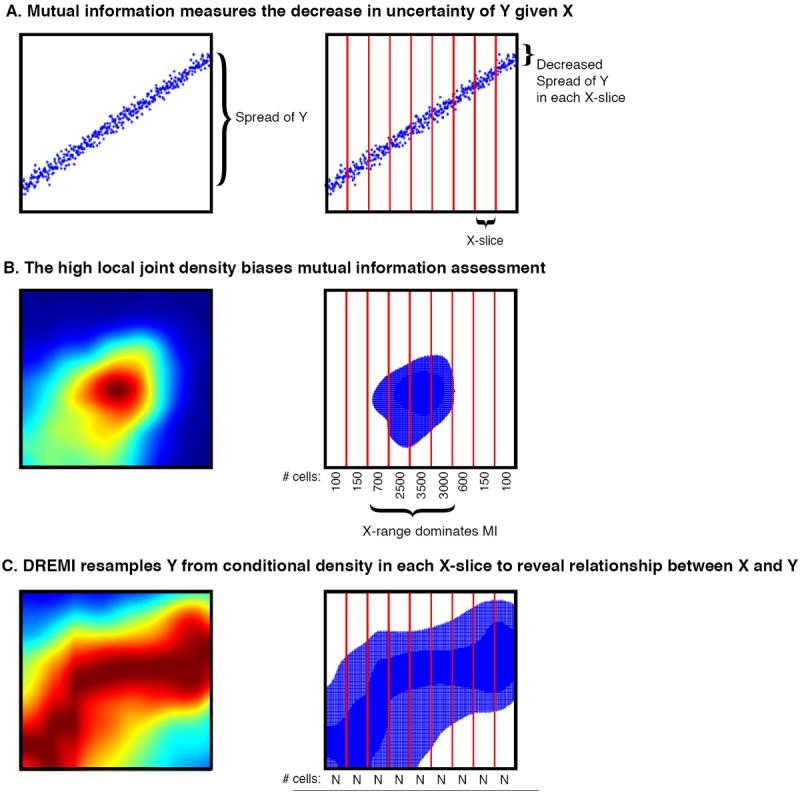
(A) Simulated data depicting a relationship with high mutual information (MI). Across the entire range of X, Y has a wide range of values (left). After conditioning on X (red lines), the range of Y within each x-slice is significantly smaller (right). Thus knowledge of X’s value provides information on Y’s likely value. (B) The data represents the relationship between pCD3ζ and pSLP76 30 seconds after stimulation of the TCR as in Fig 1BII. Most of the cells are concentrated around a single peak as shown in the 2D density estimate (left). If we resample from this density estimate, samples will fall within a narrow range where there is little change in Y’s range. This narrow range dominates the MI metric. (C) Instead, DREMI samples from the renormalized conditional density estimate, which covers entire X-range. So there is the same number of cells in each X-slice, as long as there are sufficient cells in the original data. MI information is calculated over this resampled data.
To understand the differences between MI and DREMI, we use the data from Figure 1. Ordinary MI works well for data that is well distributed across the range of X and Y (see toy example in Fig 2A) – by equi-partitioning slices of X, the range of Y drastically drops within each slice and therefore knowing the value of X provides substantial information on the likely value of Y. However, in distributions typical of single cell data (Fig 2B), MI is dominated by a peak in density (a narrow range of X) in which only minor changes in Y are observed, thus the relationship between X and Y is obscured. By contrast, DREMI (Fig. 2C) resamples from the conditional density estimate and equally weights all regions along the entire range of X, as long as there are enough cells to form a robust conditional density estimate. Thus, DREMI takes into account sparse populations along the X-axis and factors for the full range.
DREMI takes advantage of mass cytometry’s ability to collect large sample sizes (data for millions of cells), facilitating the estimation of the conditional distribution in relatively sparse regions. For example, with 50,000 cells, an X-range with only 1% of the data (typically treated as an outlier) still contains 500 cells, a sufficient number to robustly estimate P(Y∣X=x). To further ensure a robust estimation of P(Y∣X=x), DREMI incorporates an automated noise-filtering step where points are eliminated if they have a low rescaled conditional density.
We evaluated the robustness of DREMI to noise (Supplementary Fig. 6) and subsampling (Supplementary Fig. 7). Supplementary Fig. 6 shows that DREMI values generally decrease linearly as the standard deviation of the noise increases and that the noise-elimination step can be used to adjust signal detection in noisy experimental conditions. A key strength of DREMI is that it is shape agnostic and is similarly resilient to noise, whether the underlying function is linear or sigmoid, making it well suited to capture the range of behaviors observed in biology. Further, DREMI is a robust measure that remains consistent under multiple subsamples of the data (Supplementary Fig. 7).
Characterization of Signaling Relations by Curve-Fitting
In the case of a strong relationship (high DREMI score), we can derive a response function from the conditional density using curve fitting. Conditional density is particularly suited for the derivation of an edge response function because it allows for identification of the consensus Y-response (densest region in DREVI; the deep red in the heat maps) for each value of X. We compare curve-fitting of the conditional density to directly fitting the raw data to demonstrate the superiority of the former – both in terms of fit (RMSE root mean squared error) and in terms of the interpretability of the parameters (Supplementary Fig. 8). For a well-fit curve, we expect most of the data points to fall in close proximity to this curve, as measured by RMSE. A fit to the conditional mean results in a sigmoidal curve that closely follows the data points (indicated by low RMSE), whereas the raw data best fits a line – but most data points are distant from this line (indicated by high RMSE). An optimal sigmoidal fit on the raw data results in degenerate curves, where most data points reside at a significant distance.
Fitting a curve in this manner allows for a parametric description of the relationship between the two proteins X and Y. Such a functional description of the relationship between X and Y enables us to potentially predict the value of Y, if the value of X is altered by an intervention or drug. To derive the edge-response function, we fit points sampled from the conditional density to one of three models using regularized regression: linear, sigmoidal, or double sigmoidal (27). If none of the models result in a good fit, the model is then fit to a free-form curve.
In Figure 1, we fit DREVI plots of the pCD3ζ-pSLP76 edge before and after TCR activation to a sigmoid curve (Fig. 1D1-II). Among other characteristics, the parameters of the fitted sigmoid specify the lower and upper asymptote that correspond to a digital “low” an “high” mode of activation for the protein Y, and the inflection point (i.e., the activation threshold of X at which the protein Y transitions from low to high state). Comparing the edge before TCR activation to after, there is a similar inflection point activation threshold in the pCD3ζ axis but a considerably higher response on the SLP76 axis, and a corresponding increase in total activation of SLP76 (Fig. 1D). The post-activation state results in higher area-under-the-curve (AUC), which is proportional to the average amount of pSLP76 generated per quantity of pCD3ζ. A larger AUC implies more signal transfer activity such that after stimulation less pCD3ζ is required to induce a response in pSLP76. Thus, when the TCR is activated, not only do pCD3ζ amounts increase, but this change is also more impactful (than a similar increase would have been before stimulation). This increase in edge strength indicates that the pairwise relation between the two proteins is boosted by changes in the recruitment and localization of additional and potentially unknown proteins (the effects of which are successfully captured by DREVI and DREMI).
Not only do the DREMI and AUC scores increase after T cell stimulation, but they also enable distinguishing between different strengths and forms of stimulus (Fig. 3). Murine T cells were stimulated with either CD3/CD28 or CD3/CD28/CD4, and mass cytometry analysis was undertaken as per above. CD4-costimulus, in addition to CD3/CD28, is known to boost signaling responses in CD4+ T cells by engaging additional pathways that reinforce the signal transmission (28). When comparing these two forms of stimuli, we derive a higher DREMI score under the stronger stimulus in all three of the edges shown in Fig. 3. The CD3/CD28/CD4 stimulus clearly leads to a higher activation (magnitude) and/or a lower activation threshold in the edge-response functions. This translates to a higher AUC in both the sigmoidal (pCD3ζ-pSLP76, Fig. 3A, Panel IV) and linear (pERK-pS6, Fig. 3C, Panel IV) cases. Interestingly, the pSLP76-pERK edge (Fig. 3B) can be interpreted as a stochastic “digital” response – with a larger percentage of cells responding upon CD3/CD28/CD4 stimulus (Fig. 3B, panel IV) (5). Together, DREMI, DREVI, and the edge-response function form a powerful toolbox for the analysis of signaling interactions.
Figure 3. Comparison between stimulation conditions.
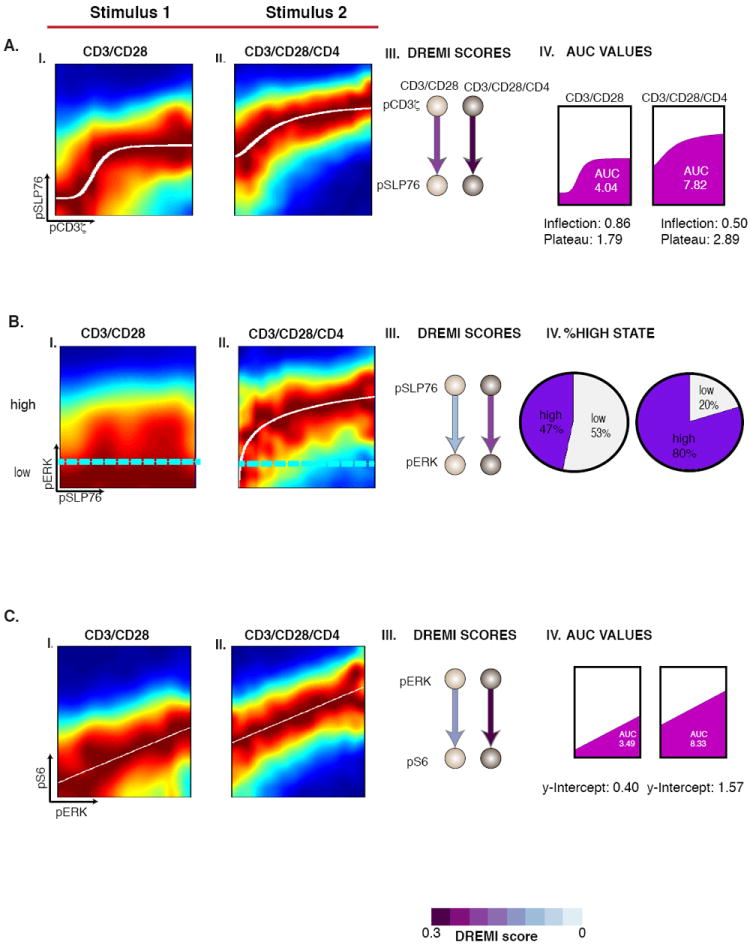
The cells are CD4+ naïve T-lymphocytes of B6 mice. (A) I-II) The pCD3ζ-pSLP76 edge at 30s after stimulation under both stimulation conditions. III) As stimulation strength increases, DREMI increases. IV) The inflection threshold moves to the left and the upper plateau of the response curve also increases resulting in a higher AUC. B) The pSLP76-pERK edge 2 minutes after stimulation. The cyan lines indicate a partitioning between “low” and “high” states of pERK. The pie charts in IV show increase in the percentage of cells that reach the high state at the higher dose. C) The pERK-pS6 edge has a linear edge-response function. Here, increasing stimulation strength results in an increase in DREMI shown in III and AUC, shown in IV.
Canonically Ordered Signaling Transfer in the TCR Pathway
We used DREMI to analyze the dynamics of the pCD3ζ-pSLP76-pERK-pS6 signaling cascade downstream of the TCR (Fig. 4). The edge strengths and shapes were highly dynamic within just a few minutes following TCR activation. The DREMI scores pinpoint the peak timing of signal transduction, meaning the timing when protein activation of Y is most sensitive to changes in the activity of protein X. Notably, the peak timings of all edges in this signaling cascade followed the expected canonical order (Fig. 4). For example, a pCD3ζ and pSLP76 relationship, or edge, was detectable in a small population of cells when pCD3ζ is at its highest value even before stimulation (Fig. 4, top left panel). However, after TCR activation, pCD3ζ reached maximum signal transfer within 30 seconds and decreased soon after. The pSLP76-pERK edge scored highest at 1 minute after activation, and the pERK-pS6 edge peaked at 2 minutes after TCR activation. Thus, the peak response follows the canonical ordering of the pathway in a manner analogous to the “just-in-time” transcriptional networks of metabolic pathways in Zaslaver et al. (29).
Figure 4. DREVI and DREMI reveal the dynamics of TCR signaling.
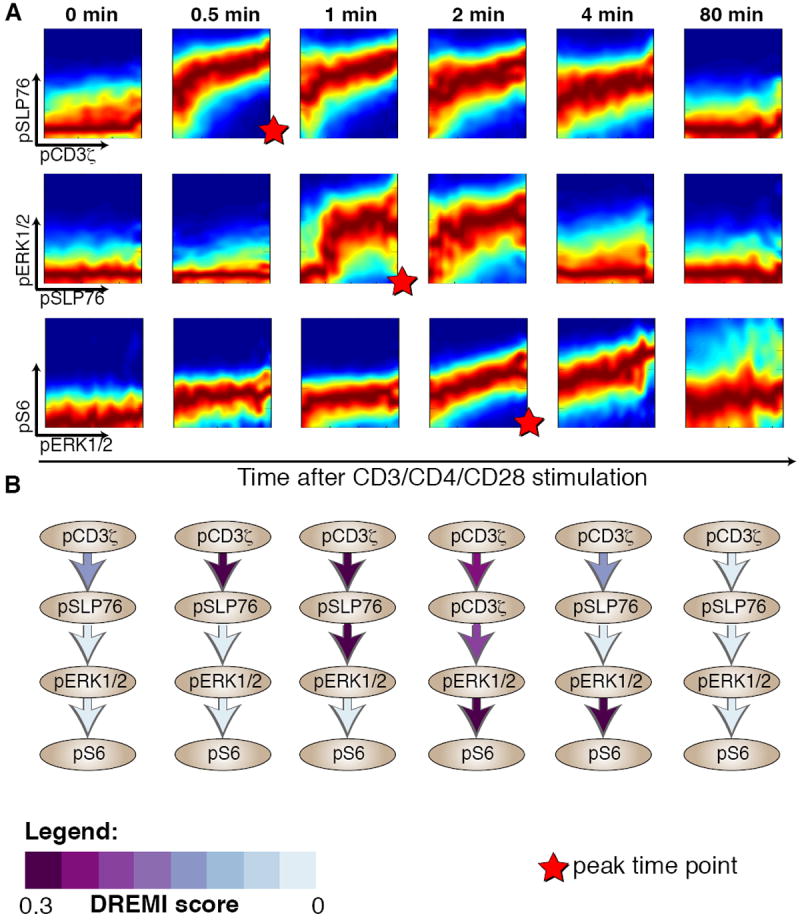
The pCD3ζ -pSLP76-pERK-pS6 cascade at various time points following after TCR activation. All panels represent data from CD4+ naïve T-lymphocytes from B6 mice with CD3, CD28 and CD4 crosslinking. (A) Each row represents an edge in the cascade and each column a time point after TCR activation. DREVI plots show the dynamics of TCR signaling; edges change their strength and shape as signaling proceeds. The scale used is the same as in figure 1, with dark red representing the behavior of the majority of cells, whereas the other colors depict the heterogeneity and spread of the response. (B) Network diagrams with DREMI values indicated on each edge, columns matching the same time points as in A. For each edge, the color indicates the computed DREMI values, more purple indicating a higher DREMI score. DREMI captures peak signal transfer timings in each of the 3 edges.
The observed dynamics highlight the importance of time series data and the impact of signaling dynamics on the relationships that can be derived from each individual time point. For instance, the pSLP76-pERK edge (Fig. 4A) is only active in the 1 to 2 minute time frame with a strong sigmoidal shape. The pSLP76-pERK edge is flat at other times. Such signaling dynamics are suggestive of the observed negative feedback loop resulting in de-phosphorylation of ERK perhaps by PTPN22 or SHP-1 (30, 31), as the influence of pSLP76 on pERK drastically decreased at 4 minutes, although pSLP76 remained activated at the 4 minute time point (Fig. 4A, top row, panel 5). In summary, the DREMI scores quantify the intuitive visual interpretation of the DREVI plots (Fig. 4B).
This canonical pathway ordering of peak timing is demonstrated again under the CD3/CD28 stimulation (Supplementary Fig. 9). However, we see that under the CD3/CD28/CD4 stimulation, signals are transferred downstream faster, and the signal transfer is sustained for longer periods. For example, the pCD3ζ-pSLP76 edge is only activated up until two minutes under CD3/CD28 stimulation, whereas it remains active until 4 minutes under a CD3/CD28/CD4 stimulatory environment. Additionally, the pSLP76-pERK edge activates only after 2 minutes under the CD3/CD28 stimulation but is activated within one minute under CD3/CD28/CD4 stimulation. This is in accordance with upstream influences that change the nature and speed of the signaling behavior downstream as reflected in the edges more distal to the TCR. This increased activation is possibly due to additional signaling pathways that converge through other signaling network components downstream of the TCR.
DREMI quantified the strength of each known edge in a given network and the dynamics of how the molecular interactions underlying this edge changed over time. Together they enabled an elucidation of a classically understood pathway in a single experiment, while providing subtle insights into the timing of signaling relationships within the network.
Performance evaluation of DREMI
To compare the performance of DREMI to other approaches that quantify relations typically used for this data type, we utilized the canonical timing of signaling edges described in the previous section as a basis for comparison. We compared these metrics on 6 edges to evaluate the ability of each method to rank edge strengths at various time points after TCR stimulation. DREMI outperformed Pearson correlation and recently-developed dependency measures such as MIC (32) and adaptively partitioned MI (23) (Fig. 5I, Supplementary Fig. 10). DREMI identified the peak timing of signal transfer in all six test cases. Adaptive partitioning missed the peak timing in four of the six cases, including two cases where adaptive partitioning picked the pre-stimulation time as the peak, and typically does not perform well when the joint density is concentrated in a narrow range. In such cases, adaptive partitioning places many partitions in the dense region (so that the partitions are often finer than the sensitivity of the experimental technology) and only a few in the sparse regions. MIC tends to pick peaks similarly to adaptive partitioning, identifying the correct peak in only one case. MIC has a further limitation in that there is very little range in the scores, possibly because it aims for equitability (33), which is a condition that does not hold in this data. Pearson correlation missed the peak timing in two cases and typically did not perform well for noisy and non-linear relationships. We also compared the performance of DREMI in an additional biological system using published mass-cytometry data focused on human B-cells following B-cell receptor stimulation, as described in (8). Similar to T cell signaling, DREMI was able to quantify and rank relationships better than metrics typically used for such data (Supplementary Fig. 11).
Figure 5. Comparison of DREMI with three other metrics.
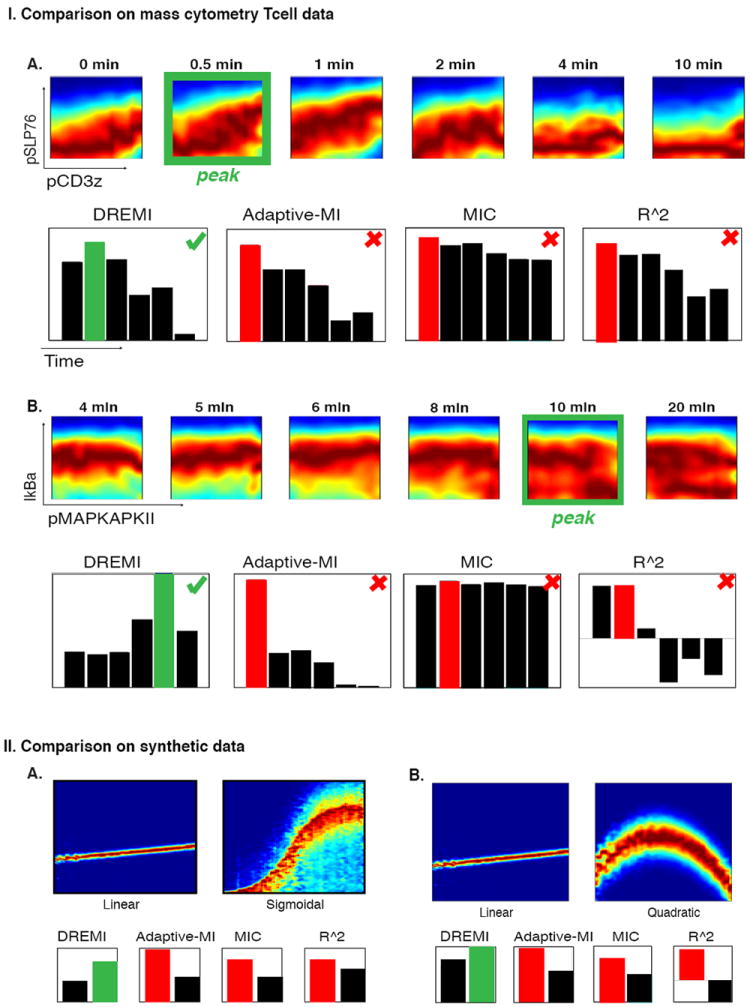
The metrics compared are mutual information using adaptive partitioning (23), MIC (32), and Pearson correlation. (I) Comparisons are performed on edges in naïve CD4+ T cells under CD3 and CD28 stimulation. In each case the green outline indicates the point with the strongest relationship upon visual inspection of the DREVI plot. (A) The pCD3ζ-pSLP76 edge: Only DREMI identifies the post-stimulation 0.5m time point as having a stronger signaling relationship than the pre-stimulation data. (B) The pMAPKAPKII-Ikba relationship as an example of a negative relationship. DREMI successfully identifies this negative relationship, while other methods are unable to detect it. Four additional edges are compared in Supplementary Fig 10. (II) Comparison of dependency metrics on synthetic data: both parts (A) and (B) show comparisons between a noiseless but weak dependence of Y on X, compared to a noisy but strong dependence of Y on X, (A) sigmoidal and (B) quadratic relationships. The other three metrics score the linear, weaker dependence higher than the noisier but stronger dependence. Since single-cell signaling data is often noisy and non-linear, DREMI is better suited.
Finally, we compared DREMI with other approaches on synthetic data that was designed to exemplify features of realistic data (Fig. 5II). We compared two weak, low-noise linear relationships against two strong non-linear noisy relationships. Specifically, we included a sigmoidal relationship and a non-monotonic quadratic function typical of systems that simultaneously induce both protagonist and antagonistic pathways (34). The weak relationships were characterized by only slight changes in the distribution of Y as X increases (small slope) while the stronger non-linear relationships have more pronounced change in the distribution of Y with changes in X. In both examples, the other methods scored the weaker linear relationship higher (Fig. 5II).
A key advantage of single-cell technologies such as mass cytometry and flow cytometry are the provision of millions of data-points in each experiment. Therefore, the scalability of the analysis is imperative to effectively analyze such data. DREMI is faster and more scalable than either MIC or adaptive partitioning (Supplementary Fig. 12). On a dataset with 500,000 cells, DREMI analysis can be computed within less than 10 seconds. MI with adaptive partitioning takes over ninety minutes to perform the same computation and MIC is limited to datasets of less than 10,000 cells. Thus, DREMI is a versatile metric that can handle a variety of characteristics, including large sample size, non-linearity and different degrees of stochasticity.
Unlike the other metrics, DREMI is a directional metric, i.e., DREMI(X, Y) is not necessarily equal to DREMI(Y, X) (Supplementary Fig. 13). Here, we based our analysis on the known network diagram and scored DREMI in the causal direction of influence. Evaluating DREMI in the reverse direction shows a slight degradation of performance compared to DREMI in the correct direction (Supplementary Figs. 10, 11, 14). However, reverse DREMI outperforms the three symmetric measures used in our comparison and has the advantage that it can handle non-linearity and noise (Supplementary Fig. 14). The correct causal direction scores approximately equal or higher in 3 of the 4 edges explored at peak signaling times (Supplementary Fig. 13). The exception was pERK-pS6, where pS6 is known to have many additional factors influencing its activity, a recurring situation in cases in which DREMI in the reverse-causal direction scores higher. We show that increasing the number of parents decreases the DREMI in the causal direction more than in the reverse direction both in linear and non-linear functions (Supplementary Fig. 15).
Comparison of Signaling Interactions between T cell Subtypes
The high-dimensionality of mass cytometry enabled us to simultaneously treat and measure multiple T cell subsets, including both naïve and antigen-exposed CD4+ T cells. It is well documented that naïve T cells behave differently than cells that have previously seen and “remember” antigen. In naïve CD4+ T cells, the activation of the TCR and appropriate co-receptors under the right cytokine environment leads to differentiation towards different effector cell fates (11). In response to the same TCR activation, antigen-experienced effector or memory T cells proliferate and respond to the infection (12-14). Moreover, the activation response time is known to be faster and of greater magnitude in effector T cells than in naïve T cells. However, the signaling mechanisms that create these differences are not yet clear. The data set collected here enabled direct comparison of signaling responses in T cells before and after exposure to antigen, because the cell subsets are in the same analysis tube and are resolved post-measurement by gating techniques (such as the differential expression of the surface marker CD44).
We used DREMI and DREVI to analyze differences along the signaling cascade that starts at pCD3ζ and continues down through pSLP76, pERK, and pS6. Naive and antigen-exposed T cells had three main differences in their responses to TCR cross-linking. First, we found that along each edge in this pathway, naïve cells had higher peak-DREMI, indicating that signals were transferred more faithfully from the upstream molecule to the downstream molecule (Fig. 6 A-C). Second, we found that naïve cells had more sustained periods of high-DREMI (Fig. 6 B), which indicated that signal transduction events transpired longer. Third, despite the fact that there was a more faithful signal transfer in naïve T cells, the levels of most of the measured phospho-proteins along this cascade, except pCD3ζ, were observed to be higher (constitutively starting at time 0) in antigen-exposed cells (Fig. 6D).
Figure 6. Comparison of naïve with antigen-exposed T cells.
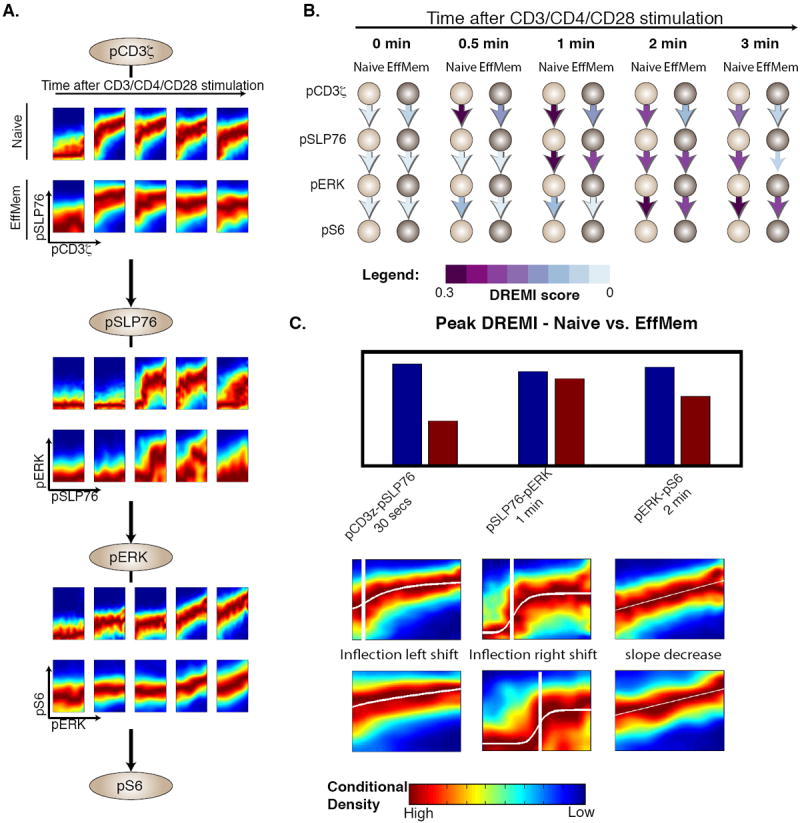
All panels in this figure represent data from CD4+ naïve (CD44low) and effector or memory (antigen-exposed, CD44high) T-lymphocytes from B6 mice with CD3, CD28, and CD4 crosslinking. We compare the pCD3ζ-pSLP76-pERK-pS6 cascade between the naïve and effector or memory CD4+ T cells using (A) DREVI and (B) DREMI. The time points in (A) match the indicated time points in (B). Both depictions show that signal transmission is sharper and more sustained in naïve cells. (C) Bar graph depicts the higher peak-DREMI detected in naïve cells, DREVI plots with edge response functions show alterations in edge behavior during peak signaling. The pCD3ζ-pSLP76 edge response function shows an earlier inflection point in effector or memory cells and is relatively flat after induction. This suggests only a small amount of pCD3ζ is needed to initiate a complete response. In contrast, pSLP76 needs more pCD3ζ for a similar response in naive cells. The pSLP76-pERK edge indicates that the pERK response is weaker, and requires more pSLP76 in these cells. Finally the pERK-pS6 edge shows a steeper slope in naïve cells indicating that pS6 responds more strongly to pERK levels in naïve cells.
The peak DREMI score (Fig. 6C) in either the naïve or the effector cascades occurs at 30 seconds for the pCD3ζ -pSLP76 edge, at 1 minute for the pSLP76-pERK edge, and at 2 minutes for the pERK-pS6 edge. All three of these were significantly higher in naïve cells than in effector-memory cells. Further, the pCD3ζ -pSLP76 relationship sustained signal transmission (high DREMI) for 2 minutes in naïve cells, compared to only 30 seconds in antigen-exposed cells. Similarly, the pSLP76-pERK edge had high DREMI between 1 and 3 minutes in naïve cells, whereas DREMI was only high between 1-2 minutes for antigen-exposed cells. Thus, at each stage in this cascade, Y transmitted more information about X, over a longer time period, in naïve cells than in antigen-exposed cells, suggesting tighter control in naïve cells. This difference between naïve and antigen-exposed cells was consistently observed across multiple replicates (Supplementary Fig. 16).
Although edge strength was higher in naïve cells, pSLP76 levels were higher in antigen-exposed cells independent of pCD3ζ levels (Fig. 7A). The near-saturation of the pSLP76 levels can also be seen in the high and flat slope of pCD3ζ-pSLP76 edge at 30 seconds (Fig. 6C). Similarly, pERK amounts were more faithfully transmitted to pS6 in naïve cells, whereas pS6 amounts were basally higher – independent of pERK – in antigen-exposed cells (Fig. 7A). In the case of TCR activation with CD4 co-stimulation, the analysis suggests that the AKT pathway might explain the higher level of pS6 in antigen-exposed cells. The AKT-pS6 edge had higher DREMI in antigen-exposed cells than in naïve cells (Figs. 7B, 7C, Supplementary Fig. 17) in early time points. Moreover, the AKT-pS6 relation was active before stimulation in antigen-exposed cells, providing a constitutive boost in signaling, perhaps through positive feedback between pS6 and pAKT (35).
Figure 7. Comparison of naïve with antigen-exposed T cells on the pAKT pathway.
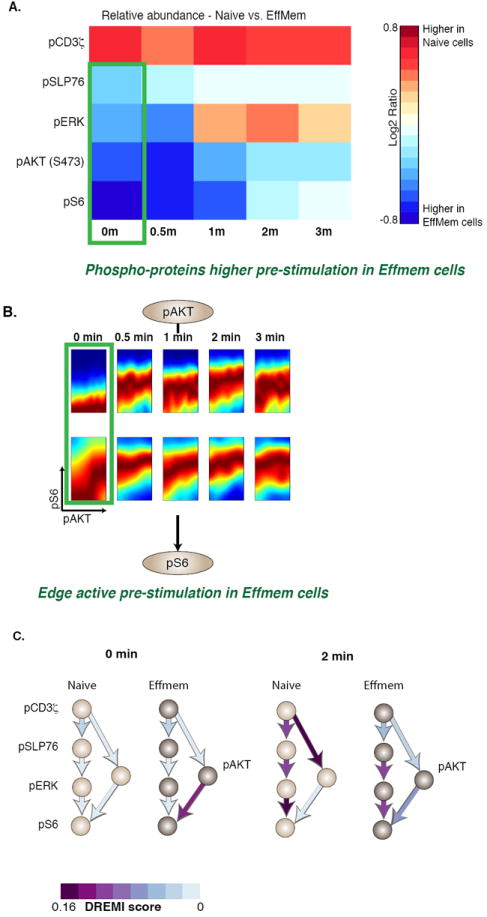
All panels in this figure represent data from CD4+ naïve (CD44low) and effector or memory (antigen-exposed, CD44high) T-lymphocytes from B6 mice with CD3, CD28, and CD4 crosslinking. (A) Comparison of phopho-protein abundances between of naïve CD4+ T cells, with effector or memory CD4+ T cells. Red indicates the phospho-protein levels amounts are higher in naïve CD4+ T cells and blue indicates the phospho-protein amounts are higher in effector or memory CD4+ T cells, demonstrating that differences in the average behavior of nodes (protein species) is not necessarily telling of the differences in edges (relationships between proteins). B) DREVI plots comparing naïve and effector or memory cells on the pAKT-pS6 edge from 0-3 minutes after stimulation. C) Network diagrams depicting DREMI scores in naïve and effector or memory cells, showing the combined pCD3ζ-pSLP76-pERK-pS6 and pCD3ζ-pAKT-pS6 pathways. The pre-stimulation pAKT-pS6 edge is stronger in effector-memory cells at 0 and 2 minutes, while responses downstream of pCD3ζ are stronger in naïve cells at 2 minutes.
Together, these differences suggest that antigen-exposed cells are poised towards a more easily triggered response (12-14, 36). Because the basal levels of the phospho-proteins are higher, a shorter period of signal integration (as indicated by less sustained periods of high-DREMI) would be needed to elicit a cellular response. On the other hand, naïve cells, which are experiencing antigen for the first time, may need to mount a more cautious response, with the signaling cascade transmitting information carefully for longer periods of time to avoid spurious activation (12-14, 36).
ERK-knockout Validation
Because DREMI analysis determined that the influence of pERK on pS6 is stronger in naïve cells, this predicts that knockout of ERK2 should have a larger impact on pS6 levels in naive cells (Fig. 8A) and this difference should be detected at the time the DREMI of this edge increases, i.e., 2 minutes after stimulation. We used the edge-response function and the measured difference in pERK abundance to approximate the expected change in pS6 levels at 2 minutes following knockout with an expectation to observe a change that is approximately 50-60% larger in naïve cells relative to effector/memory cells at 2 minutes (Figs. 8B, 8C). To validate these predictions we collected data from a mouse model (37), which enables tamoxifen-induced knockout of ERK2 within a normally differentiated T cell population. To allow for direct comparison, immune cells from both the B6 and ERK knockout mice were stimulated and measured in the same tube and subsequently identified through congenic markers CD45.1 and CD45.2.
Figure 8. Validation of differences in edge-strength using in an inducible Erk2-knockout mouse.
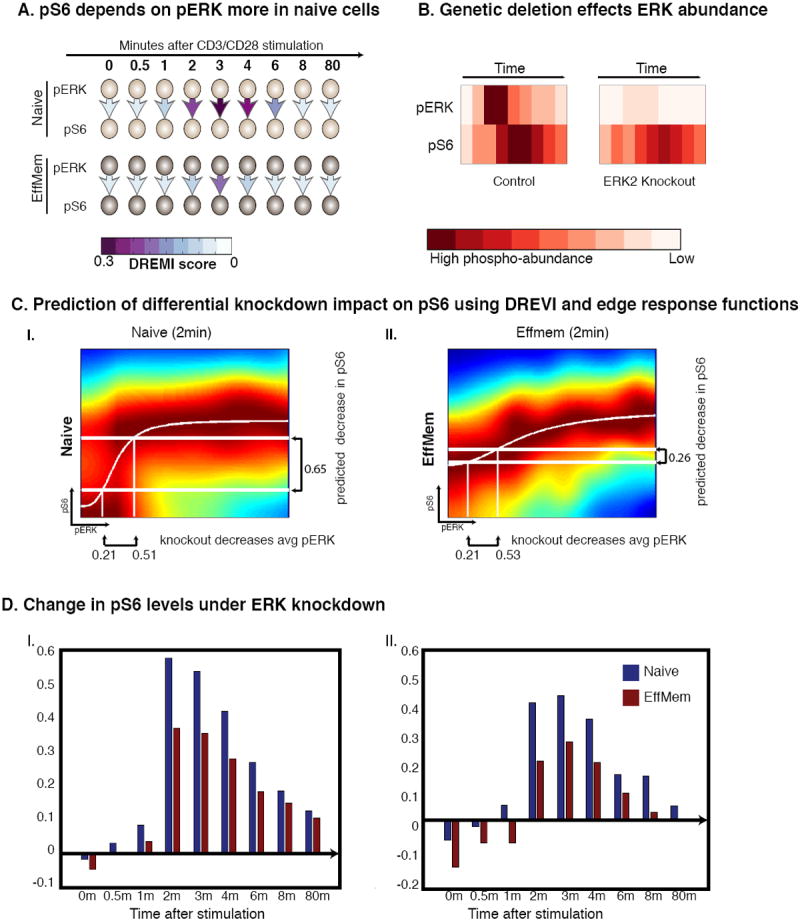
This figure depicts data are from CD4+ naïve T-lymphocytes from both B6 and Erk2-knockout mice with CD3 and CD28 crosslinking. (A) DREMI is used to compare the strength of the pERK-pS6 relationship between naïve and effector or memory T cells in B6 mice. Higher DREMI in naïve T cells predicts a bigger impact of Erk2 knockout on pS6 in naïve cells. (B) Heatmaps show the amounts of pERK and pS6 in the control and knockout mice cells. (C) Edge-response functions for B6 mice at the 2-minute time point predict the outcome of a knockout experiment in (I) naïve cells and (II) effector or memory cells. The x-axes of both figures are annotated with the measured change in mean pERK levels upon knockout, and the y-axis shows the predicted decrease in pS6 levels based on the edge-response function. (D) I-II show two replicates demonstrating the change in pS6 amounts following after ERK2 knockout. The blue bars represent the average difference between pS6 in normal and ERK2-knockout mice cells in naïve T cells. The maroon bars show the same difference in effector or memory cells. The blue bars are significantly higher than the maroon bars, especially after the 2-minute timepoint (as predicted), therefore our edge strength assessment is validated.
To test the hypothesis that naïve T cells showed a stronger relationship between pERK and pS6, based on the relatively higher DREMI in naïve cells, we assessed the impact of the Erk2 depletion on pS6 levels in both naïve and antigen-exposed cells. Indeed, as predicted, changes in pS6 abundance are larger in naïve cells than in antigen-exposed cells. Approximately 50% greater response in naïve cells began at 2 minutes as predicted (Fig. 8D). DREMI also predicts that the influence of pERK on pS6 is stronger in CD4+ T cells than CD8+ T cells (Supplementary Fig. 18), which was also confirmed experimentally (Supplementary Fig. 19), demonstrating that DREMI can correctly predict differences in signaling edge strengths between related T cell subsets.
DISCUSSION
DREVI and DREMI combined with single cell data provide solutions to a challenging problem that has obstructed the analysis of single cell biology: that of quantifying the strength of the underlying complex relationships between proteins from data. Other approaches such as correlation, mutual information, and MIC – which are typically used to quantify the strengths of interactions – are not always reliable in the face of technical noise, natural stochasticity, bi-modality, spurious correlations, and non-linearity inherent in single-cell data. DREMI, on the other hand, consistently quantifies the relative strengths of relationships and how they change following stimulation through time and between different cell types.
We used DREVI and DREMI to functionally and quantitatively analyze edges within intracellular protein networks. DREVI and DREMI directly regard the single-cell population as instantiations of an underlying signal processing circuit, and infer the form and strength of each edge in the circuit. Many DREVI plots have a sigmoid (or partial sigmoid) response curve, suggestive of the signaling kinetics. With absolute quantification of cytometry data, it would be possible to derive biochemical rate constants for multiple enzymatic pairs and multiple subsets, in a single experiment. This would provide an enormous advantage over laborious experiments currently needed to compute such rate constants. Therefore, a versatile, data-driven approach such as that represented here enables the systematic and quantitative comparison of signaling relationships between different stimuli, time points, and cell types.
The analysis in this manuscript focused on mass cytometry data collected from murine T cells measuring 11 phospho-proteins and 9 surface markers. The analysis determined that the TCR signaling network is dynamic – with edges activating and deactivating quickly as the signals are transduced downstream. We used DREMI and DREVI to gain insights into how signaling is fine-tuned between related T cell populations. The differences we identified in TCR signaling between naïve and antigen-exposed T cells suggest that naïve cells more sensitively transmit upstream signaling inputs along a central signaling cascade. This more fine-tuned signaling response might help regulate the cell’s decision to differentiate into either regulatory or helper T cells. By contrast, entrained effector or memory cells seem poised for fast responses upon secondary exposures. To explain this observation, we noted that several alternative pathways had higher activity in antigen-exposed cells, including AKT under CD4 co-stimulation, which likely bolsters phospho-protein levels and shortens input integration time in antigen-exposed cells.
Cumulatively, these techniques can allow the tracking of subtle reconfigurations in signaling that occurs between related cell subtypes. Note that these reconfigurations do not result in an essentially rewired network, but rather a network that is behaviorally reconfigured to produce changes in response. Although we have presented a specific application of our techniques, focused on TCR activation in T cell subsets, we believe that DREVI and DREMI are broadly applicable across biological systems and single cell technologies (38, 39). Our ability to characterize signaling edges and their strengths is of particular value for disease studies, as it has been shown that many disease alleles affect the edge strengths of the signaling network (40). As single-cell data become more abundant, our methods will allow construction of more quantitative models of cellular signaling. Such models can then elucidate how these signaling circuits differ between cell types, as well as characterize essential and therapy-targetable differences between health and disease.
MATERIALS AND METHODS
Mice
Mice were maintained in specific pathogen-free facilities at Harvard Medical School (Institutional Animal Care and Use Committee 99–20, 02954). Age-matched, 4 to 6 week-old male B6g7 mice were used for all experiments. For TCR signaling responses in peripheral T cells from TCR transgenic mice, BDC2.5 and BDC2.5/B6g7. Rag−/− mice were used. This mouse strain was previously described (41).
Cell Stimulation
For analysis of phosphorylation events by mass cytometry, total lymph node cell suspensions from B6g7 were used. Cells were stimulated in medium containing 2 or 6 μg/ml of biotinylated anti-CD3e and anti-CD28 stimulatory antibodies and incubated for 2 min at 37 °C before addition of 8 or 24 μg/ml streptavidin. At various times after cross-linking, the stimulation was stopped by addition of paraformaldehyde (final concentration 2 %) and incubation at room temperature for 20 min. In some experiments biotinylated anti-CD4 antibodies were also included in the stimulation mix. The cells are measured before stimulation and at various time points after stimulation: 30s, 1, 2, 3, 4, 6, 8, 10, 20, 80 mins.
Erk Knockout Experiment
To study the role of ERK in peripheral, T cells we used an inducible model that was developed in (37) with the Erk2 floxed-gene and ER-CRE, which is a tamoxifen-inducible CRE-recombinase. Adult mice are injected with tamoxifen, every day (Sigma) for six consecutive days, to induce the deletion of the floxed gene and create ERk2-deficient cells. The mice were analyzed 2 days later. There are normal numbers of peripheral T cells in these mice, as they are analyzed only 2 days after the tamoxifen-induced deletion. Therefore, these peripheral cells are a result of normal differentiation and transportation from the thymus weeks before the tamoxifen treatment injections (37), but have ERK2 knocked-down for the TCR activation experiment.
Normal B6 and Erk knockout cells express different congenic CD45 surface antigens (Erk-knockout mice express CD45.1, normal B6 express CD45.2), allowing for the stimulation of both cell types in the same tube. B6 and Erk knockout cells are therefore mixed prior to stimulation and identified post-analysis with antibodies against CD45.1 and CD45.2. This strategy has the considerable advantage of eliminating any contribution of experimental variations during the stimulation, staining, and CyTOF processing of the samples (for each time point analyzed).
Measurement of Phospho-signaling Events by Mass Cytometry
In our experiments we used mass cytometry to measure surface and internal proteins. Primary conjugates of the mass cytometry antibodies (see complete list in Supplementary Table 1) were prepared and titrated as described (8) PFA fixed and frozen lymphocyte suspensions were thawed at room temperature and then transferred to ice.
Single samples of 1 million cells were stained in a 50 ul final volume. All samples from a given time series of a stimulation-condition were run in a single barcoded tube. Each barcoded tube contained 20 individual samples and was stained in 300ul. Cells were stained with a cocktail of metal isotope conjugated antibodies (Supplementary Table 1) using a 2-step procedure (surface stains included: CD4, CD8, TCRb, CD5, CD19, CD25, CD44, CD45.1, CD45.2; intracellular stains included: pCD3ζ, pSLP76, pERK1/2, pS6, pCREB, pMAPKAPKII, IkBa, pNFkB, pRB, pFAK, pAKT). The signaling network consisting of all markers is shown in Supplementary Fig. 1. Cell fixation, permeation, and staining were performed as previously described (8). To make all samples maximally comparable, all data was acquired using internal metal isotope bead standards as previously described in (42). Approximately 100,000 events were acquired per sample on a CyTOF I (DVS Sciences) using instrument settings previously described in (42).
Barcoding of samples
To reduce sample-to-sample variability cells corresponding to each stimulation condition and time point were barcoded using all 6 stable palladium isotopes with a method similar to that described by (43). In the barcoding scheme each isotope is used in binary fashion, high for “1” and low for “0”. Hence the barcode itself is a binary string that is can be decoded for each cell to determine the treatment condition for that cell. For this purpose we developed an error-correcting 3-one barcoding scheme that both facilitates ease of debarcoding, and eliminates the majority of doublets. All legal barcodes in our scheme have exactly 3 ones and 3 zeroes, resulting in 20 possible barcodes. This barcoding scheme also enables the detection of most cell doublets because in a well-mixed tube it is unlikely to have a doublet created by two cells from the same condition. Doublets with cells from two different conditions would have an illegal combined barcode (with more than 3 “1”s) that is represented by the logical OR of the individual barcodes. Therefore this scheme eliminates almost all doublet events. Further, each pair of barcodes has a distance of at least 2 and therefore this scheme is also robust to errors during the debarcoding.
Post-processing of Mass Cytometry Data
Individual time series were normalized to the internal bead standards prior to analysis using the method described in (42). In addition, as described in (8) for all analysis of the mass cytometry data, abundance values reported by the mass cytometer were transformed using a scaled arcsinh, with scaling factor 5 to minimize the noise in measurements where the data is close to zero (the limit of detection on the mass cytometry scale). Events recorded by the mass cytometer were gated based on cell length and DNA content (to avoid debris and doublets) as described in (8).
Gating of cell subsets
Cells were then filtered through a series of gates as depicted in Supplementary Fig. 2 for the purpose of isolating particular subpopulations of T cells such as CD4+ naïve T cells, CD8+ naïve T cells and CD4+ effector/memory T cells. These gates include: 1) a gate that separates T cells (TCRb+ CD19-) from Bcells (TCRb- CD19+), 2) a gate that further separates CD4+ T cells (CD4+) from CD8+ T cells (CD8+), 3) a gate that further separates CD4+ T cells into Tconv (CD25-) and Treg (CD25+) populations, and 4) a gate that further separates CD4+ Tconv T cells and CD8+ T cells into naïve (CD44-) and antigen-exposed effector/memory populations (CD44+). Note that since there is no known CD8+ equivalent of the CD4+CD25+ Treg population, CD8+ cells are not passed through gate 3. The final populations that we isolate and use in our comparisons include: 1) Naïve CD4+ T cells (CD44- CD25- CD4+ CD8- TCRb+ CD19-), 2) Naïve CD8+ T cells (CD44- CD25- CD8+ CD4-TCRb+CD19-) and 3) Effector/memory CD4+ T cells (CD44+ CD25- CD4+ CD8- TCRb+ CD19-). Supplementary Table 2 shows the nodes, cell types, stimulation and time points used in each figure.
Description of computational methods
In the following sections we provide a detailed explanation of how DREVI, DREMI, and the edge-response functions are computed,
Kernel Density and Conditional Density Estimation
A kernel density estimate f̂ (x) is a non-parametric approximation of the probability density function p(x). The standard method for computation of a kernel density estimate is to introduce a narrow Gaussian (or alternative) kernel at each point in the raw data, and calculate the integral of the individual kernel values over a large set of points (21, 44). If (x1, x2, x3, … xn) are independent, identically distributed random samples from an unknown probability density function f, then the kernel density estimator is:
| (Eq. 1) |
Where K is the kernel function. The commonly used Gaussian kernel is given by:
| (Eq. 2) |
Kh refers to the kernel density function scaled by 1/h (where h denotes the bandwidth). Intuitively, one can think of kernel density estimation as placing a small smooth kernel Kh at each sampled point, and integrating over all such kernels to determine a smooth probability density function. The kernel density estimate in two dimensions is given by:
| (Eq. 3) |
This standard method of kernel density estimation suffers from several deficiencies including 1) sensitivity to selected bandwidth and the assumption of normalcy in many bandwidth estimation methods, 2) boundary bias, 3) over or under-smoothing.
Instead we use the heat-diffusion method from (10) for two dimensional kernel density estimation. This method views the kernel density estimate as a solution to the heat-equation, which it evolves for a time-period proportional to the bandwidth. This interpretation of a kernel density estimate derives from the notion of a Weiner process. A Weiner process W is a stochastic Markov process (i.e., the next state can be directly computed by the previous state) with
Initial probability uniformly spread through the d-dimensional data points (x1, x2, x3, … xn).
- The transition probability of going from point xi to xj is given by the Gaussian kernel:
The Gaussian kernel KDE can be interpreted as the probability distribution function for this process at time t is the same as Eq. 2 with bandwidth h:
But since this is an iterative process, the transition satisfies the following differential equation, which is also known as the heat equation.
The initial condition is given by where δ is the dirac delta function (which gives point-masses to all datapoints). The advantage of writing this in PDF form is that if the PDF is known to be 0 outside the range, then boundary conditions enforcing this can be added which results in a theta kernel rather than a Gaussian kernel:
| (Eq. 4) |
The theta kernel is locally adaptive and has the property that it behaves like a Gaussian kernel for small bandwidths and like a uniform kernel for large bandwidths. The evolution of the heat equation decides the shape of the kernel rather than having a fixed Gaussian estimate. This often results in a smoother density function (Supplementary Fig 3). Additionally: 1) it can be solved quickly using fast Fourier transform methods, 2) it can be extended to a more general diffusion equation easily, and finally 3) the bandwidth t can be solved as a fixed point solution to a recursion without a normal assumption on the distribution. The superior performance and speed of computation of the heat-equation based KDE is shown in Supplementary Fig. 3.
To obtain the standard conditional density estimate, the joint density estimate f̂ (x, y) is divided by the marginal density estimate f̂ (x) as follows (45):
| (Eq. 5) |
We utilize the conditional density estimate as a starting point for DREVI and DREMI as explained in the following sections.
Conditional Density-Rescaled Visualization
DREVI is aimed at visualizing the stochastic function representing how X influences Y, by depicting the distribution of Y for each value of X. To visualize 2-dimensional relationships in single-cell data, we begin by computing a 2-dimensional kernel density estimate as described in Eq. 4 (10) on a square mesh grid. The mesh grid of points is denoted G = {(xi, yj), 1<i<n, 1<j<m} and the 2-D kernel density estimate is a matrix D = f̂ (G), where D(i, j) corresponds to f̂ (xi, yj). The mesh grid of points G is selected so that the X-Y ranges are restricted to regions that are well-populated by cells. Most markers (e.g. phospho-proteins) are distributed among the measured cells as heavy-tailed multimodal distributions (Supplementary Fig. 7C). For any pair of markers X and Y, we remove 1% of both tails of these distributions as outliers, removing ~200 cells from a population of ~10000 cells (per condition and time point). The remaining range is generally well populated by cells and is amenable for a robust density estimate, which results in robust visualization and scoring as shown in Supplementary Fig. 7.
After the 2-dimensional kernel density estimate is computed, each column of D (corresponding to a fixed X-value, X = xi) is renormalized by the marginal density estimate of X = xi as shown in Eq. 5, f̂ (x) for that column to obtain the conditional density matrix
| (Eq. 6) |
This is an estimate of P(Y∣X) on the same mesh grid. This matrix is rescaled for the purposes of visualization as follows:
| (Eq. 7) |
We denote the matrix of rescaled kernel density estimates as
| (Eq. 8) |
The matrix of Eq. 8 is visualized as a heatmap using the “jet” colorscale, where densest regions, corresponding to the highest rescaled conditional density are maroon/red and the sparsest regions are blue. This way, the X-Y relationship is revealed even in regions populated by fewer cells, because each X-slice is renormalized individually. The computation of DREVI is demonstrated Fig. 1.
Conditional Density-Resampled Mutual Information (DREMI)
We developed a method for scoring relationships termed conditional-Density Resampled Estimate of Mutual information (DREMI). Like mutual information this will depend on how well the value of X can predict the value of Y. However unlike mutual information, we are not interested in how the entropy of Y decreases with X in a given sample on average. Instead, we are interested in looking at the underlying stochastic function F(X) = Y, with the aim of learning the strength of this relationship over the entire dynamic range of X, independently of a specific sample.
The key mathematical insight in DREMI is that we provide a score for the conditional distribution of P(Y∣X) rather than joint distribution P(X, Y). DREMI resamples from the conditional density estimate as described above, taking care to generate an uniform number of samples from each column of the matrix Δ*, so that the relationship between X-and-Y is scored through the full dynamic range of X that is well-sampled by the data.
DREMI begins with the rescaled conditional density matrix Δ* described in Eq. 8. To filter out noise, we round values of Δ* (xi, yj) < ε to zero and eliminate the corresponding points from the grid G. We generate n new points from the density distribution in each column i of Δ*, denoted . We generate a total of n × m samples S = {S1, S2, … Sm}. Since points in each column of have a similar value X=xi, we are generating an equal number of samples from the entire range of X (up to the fine-mesh grid level of granularity).
Finally, we define a discrete partition of the X-and Y-axes and compute mutual information I(X:Y) as in Eq. 10 using the samples in S. We generally use 8 equi-partitions of the X and Y-axes to compute the mutual information on these samples, but our software can use any number of equi-partitions based on a user-defined parameter. Our partitions are formed by pooling all data from conditions being compared for a particular variable, taking the maximal range of the X and Y and partitioning that space into 8 equal units along each axis. We normalize DREMI by the log base 2 of the number of partitions to provide a score that lies between 0 and 1.
To derive the mathematical expression for DREMI, we start with standard mutual information, computed as the differential entropy between Y and Y∣X:
| (Eq. 9) |
Alternatively mutual information can be regarded as a measure of the distance between the joint distribution P(X, Y) and the marginal distributions P(X) and P(Y). An equivalent formula for computing mutual information is given by(26):
| (Eq. 10) |
The first three steps of the DREMI algorithm involve estimating the conditional density by normalizing the joint kernel density estimate and filtering out noise. These steps result in a smoothed and de-noised conditional density estimate that will fill gaps in the raw data. The final step involves estimating mutual information from this computed conditional density. In Supplementary Fig. 20, we show intuitively how sampling from the conditional density estimate is equivalent to taking weighted samples from the joint distribution. Each point (xi, yj) from the joint distribution has sample weight proportional to .
Given this, DREMI can be written in terms of the differential entropy formulation of mutual information (Equation 8) as follows:
| (Eq. 11) |
Where Hc (Y) and Hc (Y∣X) are reweighted entropies where each point (xi, yj) in the joint distribution is weighted by given as follows:
| (Eq. 12) |
| (Eq. 13) |
Substituting Eq. 13 and Eq. 12 into Eq. 11 gives:
| (Equation 14) |
Note that the final form in Eq. 14 is equivalent to Eq. 10, with each point reweighted by . Hence, Ic maintains many properties of mutual information including the fact that Ic (Y∣X) = 0 if p(x, y) = p(x)p(y).
Robustness Analysis
We demonstrate that DREVI and DREMI are robust methods. Supplementary Fig. 7 shows that the downsampling of the data by 20% results in virtually identical DREVI plots and very similar DREMI scores (standard deviation of .02 or less) for several edges. The reason underlying this robustness is our use of density estimation with a locally adaptive smoothing that results from the evolution of a differential equation whose solution is equivalent to the kernel density estimate (10). The density estimation method interpolates and fills gaps within the data by integrating using a smooth kernel.
Fitting Edge-response functions
For strong relationships, P(Y∣X) is highly peaked for any value of X. Moreover, the peak of P(Y∣X) for adjacent X-slices tend to follow along a smooth and continuous curve, the edge-response function, which we fit to the data. We derive the edge-response function by fitting points sampled from the noise-filtered conditional density Δ* to one of three models using regularized regression: linear, sigmoidal, or double sigmoidal (27). If none of the models obtain significant fits (based on p-values), we fit to a free-form curve (See Fig. 1C, 3, 5C for examples).
We define the edge-response function as the function f(X) that best fits the data. We derive the edge-response function by performing non-linear, mixed-model regression (46) of the high valued regions of the conditional-density estimate. The models we use are:
linear: f1(x) = ax + b
sigmoidal: f (x) = (L − U)/1 + (xn/Kd)) + U
- double-sigmoidal:
free-form curve
We minimize over both the models and parameters in 1)-3). If no model results in sufficiently high significance of fit, we use LOESS regression (47) to fit to a non-parametric curve.
Performance of DREMI on Synthetic Data
We used synthetic data to evaluate how DREMI behaves under increasingly noisy conditions (Supplementary Fig. 6). We generated the synthetic data based on characteristics of real pairwise relations observed in single-cell cytometry data:
X is concentrated in a narrow band within the entire X-range.
The high density region of the conditional distribution reveals a consensus of response which is smooth and can be fit to a curve.
The noise in Y is variable.
To fulfill these conditions we generated data as follows:
For condition 1) The X is a random variable that follows the distribution normal distribution with standard deviation 0.25, i.e., N(2, .25).
For condition 2) The mean Y is functionally related to X, i.e., E(Y∣X=x) = f(x). We use linear, sigmoidal and quadratic functional relationships in the synthetic data.
For condition 3) Y has varying degrees of Gaussian noise added on top of its mean value, i.e. Y = f(X) + N(0, s), for parameter s.
Each synthetic dataset contained around 20,000 points. We generated synthetic data following the above conditions for 5 different values of noise: s = {0.25, 0.5, 0.75, 1, 1.25}. Supplementary Fig. 6 shows the results of computing DREMI on this synthetic data. Results show that DREMI scores decrease roughly linearly with the standard deviation of the Gaussian noise, regardless of the underlying functional shape of the relationship.
In additional to the analysis described in the main text we also evaluate how varying the noise-filtering threshold influences the results. Supplementary Fig. 6 also shows how the noise-filtering threshold ε can be used to eliminate experimental noise. Supplementary Fig. 6B depicts a horizontal line that equalizes the scores of all of the noise levels. Thus the value of ε can be tuned to compensate for experimental or measurement noise.
There are certain settings that provide naturally noisier signals than others. For instance certain antibodies can have weak binding affinities with their target proteins, others can non-specifically bind to a variety of proteins. In such cases a higher level of the outlier-elimination threshold may be useful. In other situations such as the pSLP76-pERK edge, the response of the protein itself is stochastic and therefore the stochasticity itself has meaning. In such cases the outlier-elimination threshold can be set higher to capture the natural stochasticity in the data. Other global contexts, such as measurements on primary cells as opposed to cell-lines, lead to higher noise and weaker response. In these cases it may be useful to set the noise-filtering threshold to a particular global value. We use a globally fixed noise-filtering threshold of 0.85 for all figures and DREMI values computed for this manuscript, as the data all come from B6 murine T cells. As shown in Supplementary Fig. 6, within any set noise-filtering threshold, the DREMI score decreases linearly with the standard deviation of noise, and as such is able to relatively rank DREMI scores for comparison purposes.
Comparing DREMI to other methods
We compared the performance of DREMI against other commonly used dependency measures: 1) MIC (32), 2) adaptively partitioned MI (48) and, 3) Pearson correlation (see Fig. 5). Since no gold-standard exists to compare strength of signal transfer along an edge, we created a framework to conduct the comparisons based on the dynamics of signaling. We used DREVI to visually determine the peak timing of the dynamic signaling response and in cases where peak timing is known our visual inspection matched published literature(2). We then assess whether each of the four metrics can recover the peak timing and also order the time points correctly according to strength of signal transfer. We systematically computed all of the metrics along the time-series in each of the six edges (Fig 5I and Supplementary Fig 10). DREMI outperformed all other metrics.
To show that DREMI is able to quantify and rank relationships in settings other than TCR-stimulated mice cells, we also compared DREMI on three known signaling edges on previously published B cell data (8) from the bone marrow of healthy human donors following BCR (B cell Receptor) stimulation (Supplementary Fig. 11). This data only has a single time point post BCR stimulation, therefore instead of ranking edge strength along time points, we rank edge strength across increasingly mature B cell populations. The five populations, in order of maturity, are 1) Pre-BI, 2) Pre-BII, 3) Immature B cells, 4) Mature CD38+ B cells and 5) Mature CD38- B cells. The cells are gated into these populations based on phenotypic B cell markers such as CD20, CD38, CD45RA, CD19 and CD123 as described in (8). We used prior biological knowledge to determine the cell population expected to have strongest signaling, determined before computing the metrics. DREMI outperformed all other metrics on this data.
An important goal of MIC is “equitability”. Intuitively, this means that different types of edge shapes are given similar scores if they are similarly “fuzzy”(33). Evaluating MICs performance, this does not seem to be a suitable assumption for molecular relations. Adaptive partitioning puts more weight into the densest part of the distribution. While this might make sense, in essence, it makes a large number of microscopic partitions over the small range where most of the density lays. Thus the partitions often represent random measurement noise rather than biological signal. Pearson correlation prefers linear dependencies to other types of dependencies, whereas non-linear dependencies are frequently present in signaling relationships.
In Fig. 5II we compare DREMI on synthetic data, where the generating function is known. Here we generate a weak linear relationship with low slope compared to the range of the data: Y = aX+ N(0, 0.1), to compare against strong non-linear relationships
A quadratic relationship with a high degree of noise: Y = -AX2 + N(0, 0.75).
A sigmoidal relationship X-Y with both Gaussian noise and uniform noise in the range [0, Y], for any value of Y: Y = sigmoid(X) + N(0,.75) + U(0, sigmoid(X)), where U is a uniform random function.
In both cases, the noisy-yet-strong relationships scored lower than the weak linear relationship with low noise for all metrics other than DREMI.
Evaluating Edge-Response Functions on Raw verses Resampled Conditional Data
In Supplementary Fig. 8 we show that the data resampled from the conditional density is more suitable for curve fitting than the raw data. In the first two columns, we use linear, sigmoidal or double sigmoidal least-squares regression on the raw mass cytometry data. In the third column, we resample from the dense regions of the conditional density and perform the same types of regression on this resampled data. Finally, in the fourth column we perform regression on the conditional mean (computed from the conditional density).
In each case, the best fit on the raw data has higher RMSE (root-mean-squared-error) than the best-fit data that is resampled from the conditional density (measured on the raw data). In addition, the linear fit always results in a better fit on the raw data, while the conditional data results in a variety of shapes linear, sigmoidal and double sigmoidal. The raw data tends to be concentrated in a small region of the X-Y plane, resulting in a weak penalty for deviations in most regions of the X-Y plane, and a heavy penalty for deviation only in the region of data-concentration. Therefore, a line that passes through the concentrated region of the raw data generally results in the best fit.
Supplementary Material
Acknowledgments
We thank B. Bodenmiller, N. Friedman, N. HaCohen, I. Pe’er, A. Regev, and S. Reiner for valuable comments. We respectfully appreciate the provision of murine samples and key mechanistic discussions with C. Benoist and S. Hedrick. This research was supported by the National Science Foundation CAREER award through grant number MCB-1149728 and National Centers for Biomedical Computing Grant 1U54CA121852-01A1 and the NIH S10 Shared instrumentation grant NIH S10 SIG S10RR027582-01. D.P. holds a Packard Fellowship for Science and Engineering. S.C.B. is supported by the Damon Runyon Cancer Research Foundation Fellowship (DRG-2017-09). E. S. is supported by NIH K01 award 1K01DK095008. G.P.N. is supported by the Rachford and Carlota A. Harris Endowed Professorship and grants from U19 AI057229, P01 CA034233, HHSN272200700038C, 1R01CA130826, CIRM DR1-01477 and RB2-01592, NCI RFA CA 09-011, NHLBIHV-10-05(2), European Commission HEALTH.2010.1.2-1, and the Bill and Melinda Gates Foundation (GF12141-137101).
Footnotes
Author Contributions SK, GPN, DP conceived the study. SK, DP designed and developed DREVI, DREMI and additional computational methods in this manuscript. SK wrote all computer programs used in this manuscript. MHS, SCB developed reagents. MM stimulated the mice and collected the biological samples. MHS, MM, SCB designed murine experiment and performed cognate data acquisition. ES developed the inducible ERK-knockdown mouse. SK, OL performed statistical analysis. SK, DP performed the biological analysis and interpretation. SK, GPN, and DP wrote the manuscript.
References
- 1.Iezzi G, Karjalainen K, Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8:89–95. doi: 10.1016/s1074-7613(00)80461-6. published online EpubJan. [DOI] [PubMed] [Google Scholar]
- 2.Lanzavecchia A, Lezzi G, Viola A. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 1999;96:1–4. doi: 10.1016/s0092-8674(00)80952-6. published online EpubJan 8. [DOI] [PubMed] [Google Scholar]
- 3.Miskov-Zivanov N, Turner MS, Kane LP, Morel PA, Faeder JR. The duration of T cell stimulation is a critical determinant of cell fate and plasticity. Sci Signal. 2013;6:ra97. doi: 10.1126/scisignal.2004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol. 2009;183:4895–4903. doi: 10.4049/jimmunol.0901459. published online EpubOct 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005;3:e356. doi: 10.1371/journal.pbio.0030356. published online EpubNov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antebi YE, Reich-Zeliger S, Hart Y, Mayo A, Eizenberg I, Rimer J, Putheti P, Pe’er D, Friedman N. Mapping differentiation under mixed culture conditions reveals a tunable continuum of T cell fates. PLoS Biol. 2013;11:e1001616. doi: 10.1371/journal.pbio.1001616. published online EpubJul. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. published online EpubAug 15. [DOI] [PubMed] [Google Scholar]
- 8.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. published online EpubMay 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeGroot MH, Schervish MJ. Probability and statistics. 4. Addison-Wesley; Boston: 2012. p. xiv, 893. [Google Scholar]
- 10.Botev ZI, Grotowski JF, Kroese DP. Kernel Density Estimation Via Diffusion. Annals of Statistics. 2010;38:2916–2957. doi: 10.1214/10-Aos799. published online EpubOct. [DOI] [Google Scholar]
- 11.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berard M, Tough DF. Qualitative differences between naive and memory T cells. Immunology. 2002;106:127–138. doi: 10.1046/j.1365-2567.2002.01447.x. published online EpubJun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandok MR, Farber DL. Signaling control of memory T cell generation and function. Semin Immunol. 2004;16:285–293. doi: 10.1016/j.smim.2004.08.009. published online EpubOct. [DOI] [PubMed] [Google Scholar]
- 14.MacLeod MK, Kappler JW, Marrack P. Memory CD4 T cells: generation, reactivation and re-assignment. Immunology. 2010;130:10–15. doi: 10.1111/j.1365-2567.2010.03260.x. published online EpubMay. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308:523–529. doi: 10.1126/Science.1105809. published online EpubApr 22. [DOI] [PubMed] [Google Scholar]
- 16.Mingueneau M, Krishnaswamy S, Spitzer MH, Bendall SC, Stone EL, Hedrick SM, Pe’er D, Mathis D, Nolan GP, Benoist C. Single-cell Mass Cytometry of TCR signaling: Amplification of small initial differences results in low ERK activation in NOD mice. Proceedings of the National Academy of Sciences. 2014 doi: 10.1073/pnas.1419337111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baaten BJ, Li CR, Bradley LM. Multifaceted regulation of T cells by CD44. Commun Integr Biol. 2010;3:508–512. doi: 10.4161/cib.3.6.13495. published online EpubNov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baaten BJ, Li CR, Deiro MF, Lin MM, Linton PJ, Bradley LM. CD44 regulates survival and memory development in Th1 cells. Immunity. 2010;32:104–115. doi: 10.1016/j.immuni.2009.10.011. published online EpubJan 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. 2013;13:257–269. doi: 10.1038/Nri3403. published online EpubApr. [DOI] [PubMed] [Google Scholar]
- 21.Rosenblatt M. Remarks on Some Nonparametric Estimates of a Density-Function. Annals of Mathematical Statistics. 1956;27:832–837. doi: 10.1214/Aoms/1177728190. [DOI] [Google Scholar]
- 22.Hall P, Racine J, Li Q. Cross-validation and the estimation of conditional probability densities. Journal of the American Statistical Association. 2004;99:1015–1026. doi: 10.1098/01621450400000548. published online EpubDec. [DOI] [Google Scholar]
- 23.Darbellay GA, Vajda I. Estimation of the information by an adaptive partitioning of the observation space. Ieee Transactions on Information Theory. 1999;45:1315–1321. doi: 10.1109/18.761290. published online EpubMay. [DOI] [Google Scholar]
- 24.Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nature Protocols. 2006;1:663–672. doi: 10.1038/Nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- 25.Jang IS, Margolin A, Califano A. hARACNe: improving the accuracy of regulatory model reverse engineering via higher-order data processing inequality tests. Interface Focus. 2013;3 doi: 10.1098/Rsfs.2013.0011. published online EpubAug 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cover TM, Thomas JA. In: Elements of information theory. 2. Hoboken NJ, editor. Wiley-Interscience; 2006. pp. xxiii–748. [Google Scholar]
- 27.Hastie T, Tibshirani R, Friedman JH. Springer series in statistics. Springer; New York: 2001. The elements of statistical learning : data mining, inference, and prediction : with 200 full-color illustrations; p. xvi.p. 533. [Google Scholar]
- 28.Artyomov MN, Lis M, Devadas S, Davis MM, Chakraborty AK. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16916–16921. doi: 10.1073/Pnas.1010568107. published online EpubSep 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaslaver A, Mayo AE, Rosenberg R, Bashkin P, Sberro H, Tsalyuk M, Surette MG, Alon U. Just-in-time transcription program in metabolic pathways. Nature Genetics. 2004;36:486–491. doi: 10.1038/Ng1348. published online EpubMay. [DOI] [PubMed] [Google Scholar]
- 30.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. published online EpubJan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, Goronzy JJ. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. 2012;18:1518–1524. doi: 10.1038/nm.2963. published online EpubOct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reshef DN, Reshef YA, Finucane HK, Grossman SR, McVean G, Turnbaugh PJ, Lander ES, Mitzenmacher M, Sabeti PC. Detecting novel associations in large data sets. Science. 2011;334:1518–1524. doi: 10.1126/science.1205438. published online EpubDec 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinney JB, Atwal GS. Equitability, mutual information and the maximal information coefficient. Proceedings of the National Academy of Sciences. 2014 doi: 10.1073/pnas.1309933111. published online EpubFebruary 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB. Systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310:1646–1653. doi: 10.1126/Science.1116598. published online EpubDec 9. [DOI] [PubMed] [Google Scholar]
- 35.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol. 2013;31:675–704. doi: 10.1146/annurev-immunol-032712-095946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adachi K, Davis MM. T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T cells. Proc Natl Acad Sci U S A. 2011;108:1549–1554. doi: 10.1073/pnas.1017340108. published online EpubJan 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang CF, D’Souza WN, Ch’en IL, Pages G, Pouyssegur J, Hedrick SM. Polar opposites: Erk direction of CD4 T cell subsets. J Immunol. 2012;189:721–731. doi: 10.4049/jimmunol.1103015. published online EpubJul 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schuffler PJ, Grolimund D, Buhmann JM, Brandt S, Varga Z, Wild PJ, Gunther D, Bodenmillerthat B. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417. doi: 10.1038/Nmeth.2869. published online EpubApr. [DOI] [PubMed] [Google Scholar]
- 39.Angelo M, Bendall SC, Finck R, Hale M, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, Natkunam Y, Nolan GP. Quantitative Imaging of Ten Markers in Human Breast Tumors Using Multiplexed Ion Beam Imaging (MIBI) and Metal-Tagged Antibodies. Lab Invest. 2014;94:518A–518A. published online EpubFeb. [Google Scholar]
- 40.Zhong Q, Simonis N, Li QR, Charloteaux B, Heuze F, Klitgord N, Tam S, Yu H, Venkatesan K, Mou D, Swearingen V, Yildirim MA, Yan H, Dricot A, Szeto D, Lin C, Hao T, Fan C, Milstein S, Dupuy D, Brasseur R, Hill DE, Cusick ME, Vidal M. Edgetic perturbation models of human inherited disorders. Mol Syst Biol. 2009;5:321. doi: 10.1038/msb.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mingueneau M, Jiang W, Feuerer M, Mathis D, Benoist C. Thymic negative selection is functional in NOD mice. J Exp Med. 2012;209:623–637. doi: 10.1084/jem.20112593. published online EpubMar 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe’er D, Nolan GP, Bendall SC. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–494. doi: 10.1002/cyto.a.22271. published online EpubMay. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodenmiller B, Zunder ER, Finck R, Chen TJ, Savig ES, Bruggner RV, Simonds EF, Bendall SC, Sachs K, Krutzik PO, Nolan GP. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30:858–867. doi: 10.1038/nbt.2317. published online EpubSep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parzen E. Estimation of a Probability Density-Function and Mode. Annals of Mathematical Statistics. 1962;33:1065. doi: 10.1214/Aoms/1177704472. [DOI] [Google Scholar]
- 45.Rosenblatt M. In: Multivariate Analysis II. PR K, editor. Academic Press; New York: 1969. pp. 25–31. [Google Scholar]
- 46.Koller D, Friedman N. Probabilistic graphical models : principles and techniques. MIT Press; Cambridge, MA: 2009. Adaptive computation and machine learning; pp. xxi–1231. [Google Scholar]
- 47.Cleveland WS, Devlin SJ. Locally Weighted Regression - an Approach to Regression-Analysis by Local Fitting. Journal of the American Statistical Association. 1988;83:596–610. doi: 10.2307/2289282. published online EpubSep. [DOI] [Google Scholar]
- 48.Steuer R, Kurths J, Daub CO, Weise J, Selbig J. The mutual information: detecting and evaluating dependencies between variables. Bioinformatics. 2002;18(Suppl 2):S231–240. doi: 10.1093/bioinformatics/18.suppl_2.s231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.