

Abstract
Many bacterial pathogens use type three secretion systems (T3SS) to inject virulence factors, named effectors, directly into the cytoplasm of target eukaryotic cells. Most of the T3SS components are conserved among plant and animal pathogens, suggesting a common mechanism of recognition and secretion of effectors. However, no common motif has yet been identified for effectors allowing T3SS recognition. In this work, we performed a biochemical and structural characterization of the Salmonella SopB/SigE chaperone/effector complex by small-angle X-ray scattering (SAXS). Our results showed that the SopB/SigE complex is assembled in dynamic homohexameric-ring-shaped structures with an internal tunnel. In this ring, the chaperone maintains a disordered N-terminal end of SopB molecules, in a good position to be reached and processed by the T3SS. This ring dimensionally fits the ring-organized molecules of the injectisome, including ATPase hexameric rings; this organization suggests that this structural feature is important for ATPase recognition by T3SS. Our work constitutes the first evidence of the oligomerization of an effector, analogous to the organization of the secretion machinery, obtained in solution. As effectors share neither sequence nor structural identity, the quaternary oligomeric structure could constitute a strategy evolved to promote the specificity and efficiency of T3SS recognition.
INTRODUCTION
The type three secretion system (T3SS), also called the injectisome, is used by Salmonella species and many other bacterial pathogens to inject virulence factors directly into the cytoplasm of target eukaryotic cells. Once translocated, these so-called effector proteins hijack a vast array of crucial cellular functions to the benefit of the bacteria (1).
The needle complex is the central core element of the T3SS. It is a large, cylindrical macromolecular complex which spans the multimembrane bacterial envelope and extends into the extracellular environment with a needle filament. Proteins of T3SS share many sequence similarities with those of the flagellar protein export apparatus, and the structure of the injectisome is similar to that of the flagellar hook basal body, strongly suggesting a common mechanism and evolution for these two molecular machineries (2, 3).
From a structural point of view, the needle complex is organized as a series of ring-like structures built from individual subunits which adopt multiple symmetries (4). In Salmonella, PrgH, PrgK, and InvG assemble into concentric ring-like structures at the inner membrane (IM) and outer membrane (OM), creating a so-called basal body which encloses the inner rod structure formed by PrgJ (4–6). This organization defines a central conduit for protein transport from bacterial cytoplasm to eukaryotic cell membrane or cytoplasm. The diameter of the conduit dictates that effectors be translocated in an unfolded conformation (4). The IM rings are associated with the export apparatus built by the SpaP, SpaQ, SpaK, SpaS, and InvA protein families and are connected to an export gate platform and an ATPase hexamer ring stably attached through thin connectors recently identified by cryoelectron tomography (7, 8). The in situ structure of Salmonella injectisome show that the ATPase is far from the gate platform, suggesting that this configuration is in an export-off state and is probably modified after a yet-unknown activation process (8). A recent review observed that the ring formation is a common characteristic of the IM- and OM-associated components of the T3SS and suggested that the assembly of the export apparatus could use the same strategy, as some members have been reported to oligomerize (7, 9–12).
Before translocation, the effectors are stored in the cytoplasm, where they need to be stabilized and maintained in a secretion-competent state, and translocation of some of them requires their binding to specific chaperones (13, 14). To date, there is no consensus sequence that describes a universal type III secretion signal in effectors and chaperones, even if they share similar biochemical properties (7). Despite the lack of sequence identity for chaperones, crystal structures of several of them alone or bound to their cognate effector have revealed a high degree of three-dimensional structural relatedness (15). In the same way, the structural analysis of their interaction with the chaperone-binding domain (CBD) of the effectors (∼100-amino-acid N-terminal region) revealed common binding features: CBD wraps around the homodimeric chaperone, thus being maintained in a nonglobular conformation but retaining several secondary structure elements (15, 16). Structural alignment of these structures revealed a common structural β-motif which is present both in chaperone and in the CBD of effectors of several species and is crucial in the stabilization of the chaperone-substrate complex (17). The conserved effector β-strand interacts with the conserved first β-strand of the chaperone's β-sheet to form protein-protein interaction by β-sheet augmentation, which has already been described for many metabolic pathways (18). In Salmonella, the chaperone SicP alone or in complex with the secreted effector SptP (a virulence factor of Salmonella) is recognized by InvC, the ATPase associated with the T3SS of Salmonella, and this interaction plays a critical role in the substrate recognition by the T3SS machinery as well as in the effector unfolding prior to translocation (19). The InvC protein sequence shares a high degree of sequence identity with FliI flagellar ATPase and with HrcN, the T3SS ATPase of Pseudomonas syringae. HrcN has been shown to adopt a hexameric-ring-shaped structure which fits the base of the needle complex and has its chaperone binding domain directed toward the cytoplasm (20). This suggests that conserved ATPases involved in T3SS adopt the same hexameric active ring-like-organization to bind the export apparatus (20). The role of InvC in T3SS substrate recognition has been investigated by examining its interaction with Salmonella effectors (19, 21). It indicates that the C-terminal domain of InvC recognizes the chaperone SicP alone or bound to SptP through its C-terminal domain and that the hexameric form of the ATPase is required for higher efficiency. Furthermore, InvC induces chaperone release and the unfolding of the effector in an ATP-dependent manner (19). InvC has also be shown to interact with SopD, a crucial effector of Salmonella (21). These works showed the critical role of InvC in substrate recognition and unfolding by the T3SS machine.
The presence of substrate docking sites was also described for members of the YscU families (IM proteins of the export apparatus), containing large cytoplasmic domains which were proposed to be involved in the recognition of effectors (22; also, see reference 7 for a review). Those observations suggest that T3SS contain several substrate-binding sites able to recognize different types of substrates, which contributes to the regulation and organization of the secretion process. This cytoplasmic “sorting platform” has been shown to be sequentially loaded by appropriate secreted proteins to ensure the hierarchy in type III protein secretion (components of the needle complex are secreted before proteins of the export apparatus, which are secreted before the effectors) (23).
The subject of this study, SopB (also called SigD), a T3SS Salmonella effector, is an inositol phosphatase which acts on host cell membrane phospholipids. In eukaryotic cells, the biological activity of SopB is required in many processes affecting the bacterial entry into and persistence in nonphagocytic cells (24–26). In the Salmonella cytoplasm, SopB is maintained in a secretion-competent state by interaction with its cognate chaperone SigE (27). SigE is a class IA chaperone essential for the stability, secretion, and translocation of SopB whose structure has been solved by X-ray crystallography and displays the conserved fold previously described for class IA chaperones, including SicP (28, 29). The SigE/SopB complex formation requires the dimeric state of SigE as well as the presence of a conserved hydrophobic β motif (17, 29). Our recent work (16) showed the interaction of SigE with the chaperone binding domain (CBD) of SopB. In this complex, CBD, mainly folded in α-helices and disordered regions, is stabilized by interaction with the chaperone SigE in an extended conformation, which surrounds both monomers of SigE. This complex is very similar to the SicP/SptP interaction, suggesting a common recognition of this complex by the T3SS apparatus and particularly the ATPase InvC.
How a highly conserved T3SS apparatus can recognize different effectors in the bacterial cytoplasm is still unclear, but the highly conserved hexameric-ring-shaped ATPase localized at the basis of the needle complex plays a critical role in the specific recognition process of the chaperone-effector complex and the unfolding of the effector prior translocation (19). Furthermore, previous works showed that chaperones and effectors share structural homology, suggesting that these conserved structural motifs could be at the origin of the recognition by T3SS. In the present study, we therefore investigated the structure of the complex between SopB and its cognate chaperone SigE that is the form recognized by T3SS. Combining biochemistry and biophysical approaches, we showed that this complex hexamerizes, forming ring-like structures in solution. These complexes are very dynamic, oscillating between monomeric and oligomeric states. Consequently, small-angle X-ray scattering (SAXS) combined with high-performance liquid chromatography (HPLC) proved to be the most appropriated method for the structural analysis of this complex. This work constitutes the first evidence of a ring-like structure organization of chaperone-effector complexes, analogous to the organization of the secretion machinery, that can promote the specificity and efficiency of substrate recognition. It also suggests a dynamic role for the chaperone in the first steps of translocation.
MATERIALS AND METHODS
Protein cloning, production, and purification.
The Escherichia coli strains XL1 and BL21(DE3) were used for construction of the plasmids and gene expression, respectively. Sequences encoding SigE and all the constructs of SopB were amplified from Salmonella enterica serovar Typhimurium genomic DNA and cloned in the petDuet vector (Novagen), which allows coexpression of multiple target genes in E. coli with BglII-XhoI and BamHI/NotI restriction sites respectively inserting an N-terminal His6 tag on each SopB construct. Four constructs of SopB were used in this work. SopBΔ29, with a deletion of the first N-terminal 29 amino acids, is considered the active fragment, as it was previously shown that this truncated form of SopB is still active and also able to bind SigE in a secretion-competent complex (30). Three N-terminus-truncated forms of SopB have been produced and have been given names with the format SopBΔX, where X indicates the number of deleted amino acid residues. Cells were grown in LB broth supplemented with ampicillin (100 μg/ml) and induced for protein production with isopropyl-β-d-thiogalactopyranoside (IPTG; 0.5 mM) at an optical density at 600 nm (OD600) 0.6 for 3 h at 37°C.
SopB/SigE complexes from harvested cells were resuspended in lysis buffer 1 (phosphate-buffered saline [PBS] buffer with 0.5 M NaCl, 5 μg/ml DNase, and complete protease inhibitor cocktail EDTA-free tablets [one tablet per liter of culture]), lysed by sonication, and centrifuged at 12,000 × g for 60 min at 4°C. Soluble protein extract was purified using nickel-chelating resin (GE Healthcare Life Science) followed by a size exclusion chromatography (SEC) step using a HiLoad 16/60 Superdex 200 (GE Healthcare Life Science) pre-equilibrated in PBS, NaCl (150 mM), glycerol (10%), dithiothreitol (DTT; 2 mM) as described previously (16). This second step of purification using SEC also allowed the analysis of oligomerization properties of the purified proteins (see Fig. 1).
FIG 1.

Purification of SopB/SigE complexes. (A) Elution profiles of SigE/SopBΔ29 (thick line), SigE/SopBΔ112 (dotted line), SigE/SopBΔ144 (dashed line), and SigE/SopBΔ155 (thin line). (Inset) SDS-PAGE of the purified sample of SigE/SopBΔ29. (B) Superimposition of elution profiles of SigE/SopBΔ29 on a size exclusion column (SEC-3, 300 Å; Agilent) after injection of 5 μl (dashed line) or 70 μl (solid line) of the protein sample.
Dispersity and stability of protein samples.
The Zetasizer Nano ZS system (Malvern, United Kingdom) was used on the light scattering measurements for particle size determination as well as to check the polydispersity of the solutions prior to SAXS experiments. The solutions were placed in the thermostat-equipped sample chamber controlled with an accuracy of 0.1°C. A number of measurements were taken (defined automatically by the instrument, normally around 10) to obtain adequate statistics. Measurements were performed immediately after both the SEC step and the concentration step. Dynamic light scattering (DLS) was also employed to estimate the thermostability of protein samples in different buffer solutions. Ten measurements were taken for each temperature (every degree from to 15°C to 64°C). The value of Rh was monitored, and the limit of stability was assumed when the value of Rh increased abruptly due to protein denaturation.
SAXS data collection.
In order to describe the structural organization of the SigE-SopB complex, SopBΔ29/SigE, SopBΔ112/SigE, and SopBΔ144/SigE complex solutions were tested in SAXS experiments, as this technique is a very useful tool for the determination of shape, size, and oligomerization status of the macromolecules in solution. Considering the instability of the solutions and particularly for the SopBΔ144/SigE complex, the configuration of the SWING beamline at Synchrotron SOLEIL (Saint Aubin, France), where an HPLC system is installed upstream of the SAXS capillary, was crucial for our experiments. For SopBΔ29/SigE and SopBΔ112/SigE complexes, this configuration allows us to get rid of some aggregated particles as well as residual monomeric complex molecules. For the SopBΔ144/SigE complex solution in which the oligomeric and monomeric forms coexist, we were able to separate them by HPLC and immediately collect SAXS data for each peak.
All protein sample solutions were centrifuged for 10 min at 10,000 rpm prior to X-ray analysis in order to eliminate all aggregates, and their concentration was measured by UV absorption at 280 nm on a Thermo Scientific NanoDrop 1000 spectrophotometer. For each sample, stock solution was prepared at a final concentration (5 to 10 mg/ml), stored at 4°C, and then directly used for the experiments.
SAXS experiments were conducted on the SWING beamline at Synchrotron SOLEIL (λ = 1.033 Å). The Aviex charge-coupled device detector was positioned to collect data in the Q range of 0.008 to 0.4 Å−1 (Q = 4πsinθ λ−1, where 2θ is the scattering angle). All solutions were mixed in a fixed-temperature (15°C) quartz capillary with a diameter of 1.5 mm and a wall thickness of 10 μm, positioned within a vacuum chamber. The monodispersed samples of proteins were injected onto a size exclusion column (SEC-3, 300 Å; Agilent) using an Agilent HPLC system and eluted directly into the SAXS flowthrough capillary cell at a flow rate of 0.2 ml min−1 (31). A first test was made by injecting 5 μl of protein samples in order to estimate the elution volume. Then 70 μl of protein samples were injected for SAXS measurements. The elution buffer consisted of PBS (pH 7.4), 150 mM NaCl, and 10% glycerol. SAXS data were collected continuously, with a frame duration of 1.0 s and a dead time between frames of 0.5 s. Selected frames corresponding to the main elution peak were averaged using FOXTROT (31), a dedicated homemade application. A large number of frames were collected during the first minutes of the elution, and these were averaged to account for buffer scattering, which was subsequently subtracted from the signal during elution of the protein. Data reduction to absolute units, frame averaging, and subtraction were done using FOXTROT. All subsequent data processing, analysis, and modeling steps were carried out with PRIMUS and other programs of the ATSAS suite (32). We determine the molecular weight of the complex using SAXS MoW, a method to determine the molecular weight of proteins in dilute solution, with an error smaller than ∼10%, by using experimental data of a single SAXS curve measured on a relative scale (33).
Data evaluation.
The experimental SAXS data for all samples were linear in a Guinier plot of the low q region, indicating that the proteins did not undergo aggregation. The radius of gyration (Rg) was derived by the Guinier approximation I(q) = I(0) exp(−q2Rg2/3) for a qRg value of <1.0 using PRIMUS (34). Interference-free SAXS profiles were estimated by extrapolating the measured scattering curves to infinite dilution. The program GNOM (35) was used to compute the pair-distance distribution functions [P(r)]. This approach also features the maximum dimension of the macromolecule (Dmax). Similarities between the scattering curves collected for various hexamer constructs were estimated using the Vr coefficient (36).
Ab initio modeling.
As the scattering curves are results of the contributions of both hexamers and monomers; the overall shapes of the entire assemblies were restored from the experimental data using the program GASBORMX (37), a version of GASBOR which takes into account the partial dissociation of the complex from the P(r) calculation using hexameric symmetry with 25 runs of GASBORMX. These models were averaged to determine common structural features and to select the most typical shapes using the programs DAMAVER (38) and SUPCOMB (39) (see Fig. 2 and 3).
FIG 2.

SAXS data and ab initio models of SigE/SopB complexes. (A) SAXS data for SigE/SopBΔ29 (blue), SigE/SopBΔ112 (green), and SigE/SopBΔ144 in two oligomeric states (red and purple) and the corresponding autocorrelation function [P(r)] calculated with GNOM (inset). These experimental SAXS data from the different complexes are superimposed on the scattering curves calculated from the models (dotted lines) generated by GASBORMX as described in the text. (B) The ab initio models corresponding to the different SigE/SopB complexes are shown with colored beads (same color code as in panel A) and presented in two orientations. (C) Guinier plot of scattering curves. (D) Superimposition of Kratky plot representations of experimental data.
FIG 3.
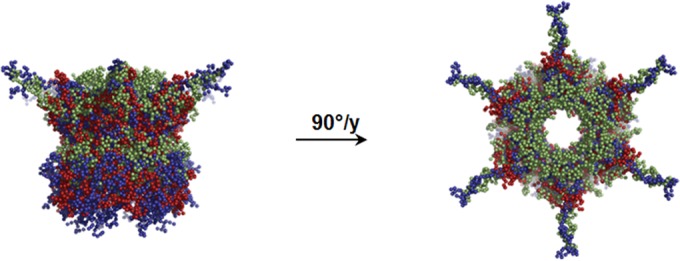
Superimposition of the low-resolution shape of SigE/SopB complexes. Models of complexes SigE/SopB are presented (with the same color code as in Fig. 2) in two different orientations and compared.
Molecular modeling.
In our previous work, we obtained a molecular model of the N-terminal chaperone-binding domain (CBD) of SopB in complex with the chaperone SigE and showed by circular dichroism (CD) experiments that the C-terminal catalytic part of SopB is constituted mainly of alpha helices (16). For this domain we generated a model with I-TASSER (40) and PHYRE2 (41) (see Fig. 4). This model is made by alpha helices organized in an elongated domain, which is in agreement with CD analysis and was further tested with the SAXS data for the monomer SopBΔ144/SigE, giving very good agreement with the data (see Fig. 4).
FIG 4.
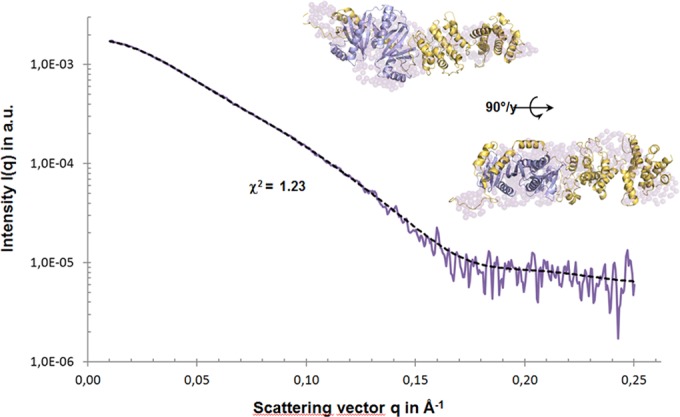
SAXS data analysis of SigE/SopBΔ144 in the monomeric state. The model was achieved by using a complete model of SigE/SopBΔ144 and refined with DADIMODO. The first part comprises a SigE/SopB (residues 144 to 235) complex and the second part a model of SopB from residue 236 to residue 561 by I-TASSER (yellow cartoon). The model superimposed on the low-resolution shape of SigE/SopBΔ144 (surface in purple) produced with GASBORMX is shown in two orientations.
Initial model building for the SopBΔ144/SigE monomeric complex was then performed by homology modeling considering two structural blocks, the first one containing the SigE/CBD model (16) and the second one formed by the C-terminal helical model of SopB described previously. In this way we reduced the possibility of solutions that could mimic the SAXS data. In a final stage of rigid-body refinement, we used DADIMODO, a genetic-algorithm-based rigid-body refinement analysis program (42). The initial structures were first minimized by keeping the main chain of the rigid domain fixed. The parts that were determined to be flexible (C-terminal regions of the CBD) were then subjected to random mutations, whereby phi/psi angles were changed with a maximum amplitude of 45° per step. Continuity of the structure was ensured by subsequent energy minimization. A SAXS χ2 value was then computed for each eligible structure, using CRYSOL with 50 harmonics. The selection protocol, based on the tournament method with progressive increase of the selection, allows the final selection of the best fitting models (42).
This model of the SopBΔ144/SigE monomeric form was used as a template to rebuild the hexameric state by using SASREFMX (43, 44), a new version of SASREF allowing the analysis of samples with polydispersity in order to perform molecular modeling of hexamers introducing a contribution of the monomers. The fraction volumes of hexamer/monomer estimated and used in this step by SASREFMX are given in Table 1. For the molecular modeling of SopBΔ29/SigE and SopBΔ112/SigE, we also used SASREFMX but with a more complete model of SopBΔ29 and SopBΔ112 built in the same manner as SopBΔ144 (see Fig. 5).
TABLE 1.
SAXS data analysis of the truncated SopB/SigE complexes
| Complex | Rg Guinier (Å) | Rg P(r) (Å) | Dmax (Å) | Porod vol (Å) | Hexamer/monomer ratio |
|---|---|---|---|---|---|
| SigE-SopB/D29 (hexamer) | 64.6 | 65 | 245 | 830 × 103 | 0.82 |
| SigE-SopB/D112 (hexamer) | 58.2 | 57.4 | 220 | 600 × 103 | 0.63 |
| SigE-SopB/D144 (hexamer) | 56 | 56.7 | 195 | 582 × 103 | 0.52 |
| SigE-SopB/D144 (monomer) | 37 | 38,8 | 145 | 110 × 103 |
The Rg and internal maximum distance (Dmax) values were calculated from Guinier extrapolation and P(r) calculation, and the molecular volume from Porod curve. The ratio hexamer/monomer is calculated with SASREFMX.
FIG 5.

Atomic models of truncated SopB/SigE complexes. (A) For each atomic model, the chaperone dimer SigE is represented with clear blue beads and the different parts of SopB with blue spheres for SopBΔ29, green spheres for SopBΔ112, and red spheres for SopBΔ144. The different models are presented in two orientations, and the monomeric part is highlighted with a cartoon model (blue for SigE and yellow for SopB). (B) Superimposition of experimental and calculated SAXS data from the different complexes. The model was achieved by using a complete model of SigE-SopB and achieved with SASREFMX applying P6 symmetry. Dots connected with colored lines show the experimental SAXS data (red for SigESopBΔ144, green for SigESopBΔ112, and blue for SigESopBΔ29), and black dotted lines show the corresponding curves obtained from the atomic model of each construct.
SAXS data have been deposited in the SAXS database BIOISIS (http://www.bioisis.net).
RESULTS
The SopB/SigE complex forms hexamers.
To ascertain the structural organization of the functional SopB/SigE complex, we coexpressed, produced, and purified SopBΔ29, which is considered the active fragment, as it was previously shown to be active and able to bind SigE in a secretion-competent complex (30), and SigE (Fig. 1). The complex was purified as described previously (16). The second step of purification using SEC reveals an oligomeric form for the complex (major peak) (Fig. 1A) and a residual minor peak corresponding to a very unstable form of the SigE/SopB complex. The elution volume of the major peak is consistent with a hexameric form of the complex which is stable and monodispersed at 4°C for 2 or 3 days.
The effect of protein concentration on hexamers was observed by SEC during SAXS measurements. At a high protein concentration (70 μl of protein sample [see Materials and Methods for details]), the SEC profile showed a major peak at the elution volume corresponding to the hexamer. When a smaller amount of protein was injected onto the column (5 μl of protein sample), this peak's intensity decreased to the benefit of a second peak, which corresponded to the monomeric unstable form of the complex (Fig. 1B).
Regions of SopB involved in the interaction with SigE and/or the oligomerization.
In order to clarify the role of the N-terminal domain of SopB in the hexamerization, we prepared several SopB constructs with different deletions at the N terminus chosen with regard to function of secondary structure and disordered predictions—SopBΔ112, SopBΔ144, and SopBΔ155 (the number in each designation represents the number of amino acid residues deleted from the N terminus)—and coexpressed them with SigE (see Materials and Methods).
The SopBΔ112/SigE complex presented biochemical properties and a stability profile very similar to those of SopBΔ29/SigE. This complex was produced and purified using the protocol used for SopBΔ29/SigE (16). The elution profile showed a major peak corresponding to the hexameric form and a minor peak corresponding to the monomeric complex (Fig. 1A), showing that the region from position 29 to 112 of SopB is not crucial for both SigE binding and hexamerization.
The SEC profile for the SopBΔ144/SigE complex revealed two peaks corresponding to the hexameric and the monomeric forms of the chaperone/effector (Fig. 1A) complex, but in that case there was a rapid equilibrium between the two oligomerization states, and we could not isolate the monomeric form which hexamerizes upon the increase in concentration. At low concentrations, the proteins started degrading. The hexameric form was rather stable and monodispersed and could be concentrated to 10 mg/ml for further structural analysis. The monomeric form isolated by SEC was more stable than the monomeric forms of SopBΔ29 constructs described above, but it had a tendency to hexamerize upon concentration. This result suggests that SopBΔ144 is still able to bind SigE but its ability to maintain the oligomers stability is reduced.
The SEC profile after expression of the SopBΔ155/SigE (Fig. 1A) complex presented a major peak corresponding to a very unstable solution of SopBΔ155 alone, which was no longer able to bind the chaperone or form hexamers. This construct could not be used for further structural analysis due to its rapid aggregation, but it showed that, as discussed in our recent work (16), the region from 144 to 155 is crucial for the binding of SigE and the region from 112 to 155 is involved in oligomerization.
Together, these observations suggested that the monomeric form of the complex is rather unstable in solution but it can be stabilized by forming hexamers at high concentration. For this phenomenon as well as for chaperone binding, the region from 144 to 155 appeared to be crucial.
SigE-SopB hexamers organize in ring-like structures.
Crystallization trials performed at several different temperatures and protein concentrations did not lead to any crystals, and for most of them we observed precipitates after 48 h.
As the complexes were stable and monodispersed in solution for few hours, we investigated the complex structure by small-angle X-ray scattering (SAXS) analysis.
SAXS data collection.
SAXS curves obtained using SopBΔ29/SigE, SopBΔ112/SigE, and SopBΔ144/SigE are shown in Fig. 2. The biophysical parameters Rg, maximal internal distance (Dmax), and molecular volume were computed, respectively, from Guinier extrapolation, P(r), and Porod volume calculations (Table 1).
The scattering curves obtained for SopBΔ29/SigE, SopBΔ112/SigE, and SopBΔ144/SigE complexes were typical curves of globular/folded multidomain proteins with a high degree of radial symmetry (Fig. 2A). The similarity parameter Vr shows that the curves corresponding to the forms SopBΔ112/SigE and SopBΔ144/SigE are close (Vr = 0.58) and are significantly different from the SopBΔ29/SigE curve (Vr = 7.73 and 8.31, respectively). The dimensionless Rg-based Kratky plot shows that the curves corresponding to SopBΔ29/SigE, SopBΔ112/SigE, and SopBΔ144/SigE are folded into the same maximum, where q · Rg is 1.75 and I(q)/I (0) · (qRg)2 is 1.1, indicating that the three hexameric complexes present a compact and globular structure, in contrast to the SAXS curve of the monomer SopBΔ144/SigE, which exhibits a profile with an elongated or partially unfolded shape (Fig. 2D).
The molecular weight of the complexes, calculated with SAXS MoW, confirmed the hexameric forms (considering the monomer as a single molecule of SopB in complex with a dimeric form of SigE as described previously [16]). Although the three forms were able to form hexameric complexes, the proportion of the monomeric form increased with the size of the deleted N-terminal parts of SopB (Table 1). Furthermore, even when a SEC step was used to separate the two forms, we observed a significant proportion of monomers in the data for the hexameric form, especially for the SopBΔ112/SigE and SopBΔ144/SigE complexes, suggesting a dynamic equilibrium of different forms in solution. This hypothesis was confirmed by the analysis of the Porod volume on each of SAXS curves, where the molecular volume of the hexameric form of SopBΔ144/SigE (582 × 103 Å3) was lower than the volume of monomeric (110 × 103 Å3) form multiplied by 6.
Ab initio modeling.
The ab initio modeling of low-resolution shapes for SopBΔ29/SigE, SopBΔ112/SigE, and SopBΔ144/SigE was performed as described in Materials and Methods. The most typical forms, which had the least deviation from other envelopes (Fig. 2B), clearly showed heights of 80, 65, and 55 Å, respectively, and a constant 115-Å-diameter bell-like ring-shaped organization. The top views of the hexamer in Fig. 2B show an organization of the six monomers delimiting together a central tunnel 45 Å in diameter. The lateral views of the three complexes reveal a similar organization in two domains separated by a narrow region. Each monomer possessed an extended domain which emerged from the cylinder, giving the overall appearance of the complexes of a 6-branched star shape, looking along the axis of the tunnel. The superimposition of the three shapes (Fig. 3) confirms the common architecture of the three constructions, with significant differences in the height of the tunnel and the length of the extended parts emerging from the structure.
As from our previous data (16) we reported that the N terminus of SopB in complex with SigE is disordered and not involved in the complex formation, at this step we hypothesize that this emerging chain present in the SopBΔ29/SigE could be the N terminus of the effector maintained in the unfolded state required for the very first steps of translocation.
The scattering curve obtained for SopBΔ112/SigE had the same characteristics as the curve obtained for SopBΔ29/SigE (Fig. 2A), confirming SEC results and revealing a multidomain arrangement with smaller dimensions (Table 1). We observed the same bell-like ring-shaped hexameric organization as for SopBΔ29/SigE, in which the emerging chains were shorter (Fig. 2B and 3), confirming that those chains correspond to the disordered N termini of SopB. However, this truncated form of SopB showed other modifications. First, an extended N terminus of SopB still protruded outside the complex regardless of its length, indicating that SigE was still able to bind along the truncated CBD and maintain the N terminus in an extended conformation. In this complex, the height of the core ring decreased in comparison to that of SopBΔ29/SigE (Fig. 3). This suggested that when the N terminus of SopB is truncated, SigE is still able to bind the effector's CBD but at a position located further toward the C terminus at the expense of the following folded region, thus shortening the height of the complex.
Molecular modeling of different SopB/SigE complexes.
The complex SopBΔ144/SigE is of particular interest for the investigation of the structural organization of the SopB/SigE oligomeric forms. The presence of a stable monomers of SopBΔ144/SigE provides SAXS data on the elementary building block of the hexamers.
Combining SAXS data with our previous data on SopB and the SopB/CBD complex (16), we performed atomic modeling of effector/chaperone monomeric complex based on multiple scattering data sets using DADIMODO (42). No structure of the catalytic domain of SopB was available in the protein structure database. However, by combining multiple approaches of molecular modeling, such as I-TASSER (44) and PHYRE2 (5), we could obtain an atomic model of SopB to be used as the template in DADIMODO. The SAXS-compatible models of the SopBΔ144/SigE monomeric form (Fig. 4) showed that SigE separates two domains of SopB, the unfolded N end and the folded C terminus. An ab initio approach with GASBOR was also performed, and the low-resolution shape superimposed on the structure exhibited significant agreement in size and form with the calculated atomic model of the complex SopBΔ144/SigE.
SopBΔ29/SigE, SopBΔ112/SigE, and SopBΔ144/SigE hexameric structures.
The SopBΔ29/SigE, SopBΔ112/SigE, and SopBΔ144/SigE hexameric models obtained with SASREFMX showed good agreement with SAXS data (Fig. 5). The comparative analysis of the different models allowed us to precisely determine the organization of the different partners in the complex as well as the position of SigE in the oligomers. The bell-shaped hexamers resulted from the superposition of two ring-like structures: the enlarged face's ring of the bell is made by the SopB/CBD part of the complex, whereas the C terminus of SopB is organized in a ring constituting the narrow part of the bell (Fig. 5). The models of the truncated versions of SopB confirmed the ring-like hexameric organization of SigE/SopB complexes. In those models, the C terminus of SopB decreased to the benefit of the secreted N terminus part, suggesting that SigE can bind the CBD at different positions.
In this configuration, SigE maintained the CBD in an extended conformation and presented the unfolded N terminus of SopB, which is secreted first to the secretion apparatus. This organization is compatible with an interaction with InvC, the T3SS-associated ATPase of Salmonella, for many reasons. First, InvC is able to bind the chaperone SicP alone or bound to the effector SptP (19), and in our model of the chaperone/effector hexameric complex the SopB-CBD/SigE part (whose structure has been shown to be very similar to the SptP-CBD/SicP structure [16]) forms an independent ring located on one side of the complex and is accessible for interaction with the ATPase. The active form of T3SS ATPases is hexameric, and their structure, predicted to be very close to the structure of EscN (45), fits our models, where each monomer of ATPase faces one monomer of SigE/SopB-CBD (Fig. 6A and B).
FIG 6.

Recognition and secretion mechanism of SopB. (A) Alignment of the model of SigE/SopBΔ29 (blue and yellow) with the superposition of the cryo-electron microscopy density map of isolated (EMD-1875; red) and in situ (EMD-2521; gray) needle complexes in which models of MxiA, a homolog of InvA (magenta; PDB ID 4A5P), and the hexameric ring model of FliI ATPase, a homolog of InvC (green) are included, as described in reference 8. (B) Superimposition of the SigE/SopBΔ29 and ATPase models along the axis of the central tunnel of their ring with the same color code as in panel A. (C) The proposed mechanism explains the injectisome recognition by the SigE/SopB complex and the translocation initiation of this complex through the T3SS (see the text for more details).
It is therefore likely that the ring-like hexameric organization is a structural feature important for substrate recognition and processing by ATPase in bacterial cytoplasm.
DISCUSSION
To date, the mechanism of substrate recognition by the T3SS is still a matter for discussion. T3SS apparatus as well as flagellar systems possess conserved motifs among animal and plant pathogens. However, these conserved motifs are recognized by very different effectors. Althought they are different, these effectors share several biochemical properties, and their N termini have been shown to be important for their secretion. This region harbors both the N-terminal T3SS signal, which is structurally disordered, providing structural flexibility, which might facilitate recognition by the T3SS apparatus (16, 46), and the chaperone-binding domain, which is maintained in an elongated configuration by chaperones, suggesting that unfolded regions required for translocation are immediately available to play a critical role in recognition.
It is assumed that T3SS is a hierarchical process: components of the secretion apparatus are secreted before effectors, and some effectors are secreted at different phases of the infection. Several mechanisms have been proposed for substrate recognition by the T3SS apparatus, including substrate interaction with different components emerging from the IM rings and ATPase (7, 23, 47), but the hierarchical process of secretion likely implies several control mechanisms at different levels.
Other regions of T3SS substrates were shown also to contribute to the secretion of the effector protein SipB from Salmonella (48; this study). The CBD of SopB has been shown to harbor several crucial functions: it is required for chaperone binding, ATPase unfolding activity, hexamerization, and SopB localization in eukaryotic cells. In SptP, the CBD alone contains all the signals required for heterologous protein translocation by T3SS, suggesting that C-terminal domains in effectors are not involved in the translocation process (19). We previously showed that SigE bound to the 168 first amino acids of SopB is not able to hexamerize (16); however, this work shows that SopB with a truncation of its 155 N-terminal amino acids is not able to bind the chaperone or to hexamerize.
Together, these results may indicate that the SopB C-terminal domain is able to hexamerize only if SigE is bound to the CBD. In that case, binding of SigE leads not only to the stabilization of SopB in the cytoplasm but also to the formation of stable oligomers which recognize and fit the injectisome. Before translocation, SopB has to be stabilized by its specific chaperone, SigE, to be maintained in a soluble form in the bacterial cytoplasm, and the interaction of SigE with SopB likely occurs during SopB translation (16). This work suggests that the SopB/SigE complex should be able to oligomerize in the bacterial cytoplasm when the concentration increases before translocation, making the complex rather stable, whereas the monomeric form of the complex is rapidly degraded. The efficiency of infection requires that a large quantity of virulence factors be injected very rapidly into the host cell, and it has been shown that the effector must accumulate in the bacterial cytoplasm prior to its secretion (49). It is likely, then, that prior to translocation, SigE stabilizes SopB during its translation, and when the concentration increases, the complex may be stabilized by forming hexamers. The stabilization of the complex by hexamerization could constitute a first level of the hierarchical secretion regulation process.
Structural analysis of the hexamers showed a ring-like structure organization similar to the organization of the T3SS components. This is the first time that this ring-like hexameric organization has been observed for effectors of the T3SS. We showed that in this organization, N termini of SopB are disordered and emerge from one side of the ring being thus accessible for translocation. Chaperones constitute the arms of the star-like side of the ring. We showed that reducing the N-terminus of SopB to 144 amino acids implies several modifications of the complex even if SigE is still able to bind SopB and hexamerization occurs. In reduced-N-terminus complexes, we observed that the N terminus of SopB, even if shorter, is still maintained in an accessible and disordered conformation and that the height of the C-terminal domain decreases.
If we consider that truncated versions of SopB could simulate, for SigE, molecules already engaged in the T3SS apparatus at the first step of translocation, these results suggest that during the very first steps of the process, the chaperone remains bound to SopB and can slide along the chain, probably to provide a tightened chain to the T3SS. After secretion of the ∼144 first residues, SigE, no longer able to bind the effector, is released. This leads to the dissociation of the hexamer, and SopB can then be picked up by another component of the T3SS (probably InvA) to achieve the complete translocation. Recent work using cryoelectron tomography on the Salmonella injectisome in situ showed a dense region 24 nm from the needle complex in which the hexameric structure of FliI ATPase could be fitted (8). Examination of this globular region of the EM density map (EM-2520) shows that there is an extra density map fitting the dimensions of the hexamer of SigE/CBD. This observation needs to be confirmed by further binding experiments, but it might indicate that hexameric ATPase interacts with a ring-like structure in the cytoplasm of the bacteria with dimensions analogous to those of the hexameric SigE/CDB-SopB complex investigated in this work.
Model of mechanism of SopB secretion by T3SS.
In conclusion, we showed for the first time that the complex chaperone/effector forms hexameric ring like structures with dimensions fitting the ATPase rings and/or the T3SS apparatus (Fig. 6A and B). We propose that the recognition of SopB by the T3SS relies on the presence of unfolded N-terminal region of the effector maintained in a good configuration by the ring motif for T3SS recognition. This mechanism is consistent with the hierarchical control of effector secretion, which has been shown to be related to different effector translocation rates. In this model, the recognition of SopB by T3SS would depend on the transcription rate of SopB/SigE genes, which directly control the cytoplasmic protein concentration and then the hexameric structure association.
We propose a putative mechanism of SopB translocation by T3SS in which the hexamerization process and ring structure-based recognition are important (Fig. 6C). In Salmonella, SigE and SopB are coexpressed and SigE is then able to stabilize SopB, possibly during the translation (step1 in Fig. 6B). When the concentration of this complex increases before translocation, it is organized in hexameric ring-like structures (step 2) that are likely the structures recognized by the ATPase InvC, located in the cytoplasm of the bacteria (step 3). Identifying the in situ position of ATPase, Kawamoto et al. (8) suggested that during the translocation process the ATPase can move closer to the export gate. Consistent with our results, the chaperone-effector complex is probably recruited by ATPase (step 4) that starts to unfold the accessible N terminus of the effector as SigE tightens this part by moving along the CBD of SopB. The length of the partially folded 150-residue protein is roughly 30 nm (data not shown), and it is fascinating to speculate that the CBD of effectors can bridge the ATPase to the sorting platform of T3SS at the beginning of translocation. At the end of CBD, SopB can then be picked up by InvA (the Salmonella FhlA family member) as described in reference 50; as a consequence, SigE is released, and the hexamer dissociates (step 5). After secretion in host cells, the unstable N-terminal part of SopB can be stabilized by interacting with other partners, such as Cdc42 (step 6).
SopB/SigE is the first example of this ring organization, which was not easy to capture due to its intrinsic dynamic character and concentration dependency. This discovery opens the possibility that some effectors, despite their variability, are organized in a ring-shape structure involved in secretion similar to the ring-like auto-assembling structure of needle complex components. Much further work will be required to test this model and to show the role of the ring structures in molecular recognition and interaction between T3SS protein members, but this finding opens up a large new field of structural investigations that can shed light onto the first steps of the T3SS mechanism.
ACKNOWLEDGMENTS
This work was supported mainly by the CNRS (Centre National de la Recherche Scientifique, USR3078 CNRS/USTL, France) and also by the Région Nord Pas-de-Calais and the CPER CIA.
Access to SOLEIL was granted by Beam Allocation Groups “Conformational properties of proteins in solution.” SAXS data were collected on beamline SWING at Synchrotron SOLEIL (Saint-Aubin, France).
REFERENCES
- 1.Galan JE, Collmer A. 1999. Type III secretion machines: bacterial devices for protein delivery into host cells. Science 284:1322–1328. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- 2.Minamino T, Imada K, Namba K. 2008. Mechanisms of type III protein export for bacterial flagellar assembly. Mol Biosyst 4:1105–1115. doi: 10.1039/b808065h. [DOI] [PubMed] [Google Scholar]
- 3.Erhardt M, Namba K, Hughes KT. 2010. Bacterial nanomachines: the flagellum and type III injectisome. Cold Spring Harb Perspect Biol 2:a000299. doi: 10.1101/cshperspect.a000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marlovits TC, Kubori T, Sukhan A, Thomas DR, Galan JE, Unger VM. 2004. Structural insights into the assembly of the type III secretion needle complex. Science 306:1040–1042. doi: 10.1126/science.1102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marlovits TC, Kubori T, Lara-Tejero M, Thomas D, Unger VM, Galan JE. 2006. Assembly of the inner rod determines needle length in the type III secretion injectisome. Nature 441:637–640. doi: 10.1038/nature04822. [DOI] [PubMed] [Google Scholar]
- 6.Schraidt O, Marlovits TC. 2011. Three-dimensional model of Salmonella's needle complex at subnanometer resolution. Science 331:1192–1195. doi: 10.1126/science.1199358. [DOI] [PubMed] [Google Scholar]
- 7.Buttner D. 2012. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol Mol Biol Rev 76:262–310. doi: 10.1128/MMBR.05017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawamoto A, Morimoto YV, Miyata T, Minamino T, Hughes KT, Kato T, Namba K. 2013. Common and distinct structural features of Salmonella injectisome and flagellar basal body. Sci Rep 3:3369. doi: 10.1038/srep03369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Sourjik V. 2011. Assembly and stability of flagellar motor in Escherichia coli. Mol Microbiol 80:886–899. doi: 10.1111/j.1365-2958.2011.07557.x. [DOI] [PubMed] [Google Scholar]
- 10.Diepold A, Wiesand U, Cornelis GR. 2011. The assembly of the export apparatus (YscR,S,T,U,V) of the Yersinia type III secretion apparatus occurs independently of other structural components and involves the formation of an YscV oligomer. Mol Microbiol 82:502–514. doi: 10.1111/j.1365-2958.2011.07830.x. [DOI] [PubMed] [Google Scholar]
- 11.Lilic M, Quezada CM, Stebbins CE. 2010. A conserved domain in type III secretion links the cytoplasmic domain of InvA to elements of the basal body. Acta Crystallogr D Biol Crystallogr 66:709–713. doi: 10.1107/S0907444910010796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Worrall LJ, Vuckovic M, Strynadka NC. 2010. Crystal structure of the C-terminal domain of the Salmonella type III secretion system export apparatus protein InvA. Protein Sci 19:1091–1096. doi: 10.1002/pro.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wattiau P, Cornelis GR. 1993. SycE, a chaperone-like protein of Yersinia enterocolitica involved in Ohe secretion of YopE. Mol Microbiol 8:123–131. doi: 10.1111/j.1365-2958.1993.tb01209.x. [DOI] [PubMed] [Google Scholar]
- 14.Stebbins CE, Galan JE. 2003. Priming virulence factors for delivery into the host. Nat Rev Mol Cell Biol 4:738–743. doi: 10.1038/nrm1201. [DOI] [PubMed] [Google Scholar]
- 15.Stebbins CE, Galan JE. 2001. Maintenance of an unfolded polypeptide by a cognate chaperone in bacterial type III secretion. Nature 414:77–81. doi: 10.1038/35102073. [DOI] [PubMed] [Google Scholar]
- 16.Roblin P, Lebrun P, Rucktooa P, Dewitte F, Lens Z, Receveur-Brechot V, Raussens V, Villeret V, Bompard C. 2013. The structural organization of the N-terminus domain of SopB, a virulence factor of Salmonella, depends on the nature of its protein partners. Biochim Biophys Acta 1834:2564–2572. doi: 10.1016/j.bbapap.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Lilic M, Vujanac M, Stebbins CE. 2006. A common structural motif in the binding of virulence factors to bacterial secretion chaperones. Mol Cell 21:653–664. doi: 10.1016/j.molcel.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 18.Remaut H, Waksman G. 2006. Protein-protein interaction through beta-strand addition. Trends Biochem Sci 31:436–444. doi: 10.1016/j.tibs.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Akeda Y, Galan JE. 2005. Chaperone release and unfolding of substrates in type III secretion. Nature 437:911–915. doi: 10.1038/nature03992. [DOI] [PubMed] [Google Scholar]
- 20.Muller SA, Pozidis C, Stone R, Meesters C, Chami M, Engel A, Economou A, Stahlberg H. 2006. Double hexameric ring assembly of the type III protein translocase ATPase HrcN. Mol Microbiol 61:119–125. doi: 10.1111/j.1365-2958.2006.05219.x. [DOI] [PubMed] [Google Scholar]
- 21.Boonyom R, Karavolos MH, Bulmer DM, Khan CM. 2010. Salmonella pathogenicity island 1 (SPI-1) type III secretion of SopD involves N- and C-terminal signals and direct binding to the InvC ATPase. Microbiology 156:1805–1814. doi: 10.1099/mic.0.038117-0. [DOI] [PubMed] [Google Scholar]
- 22.Lorenz C, Schulz S, Wolsch T, Rossier O, Bonas U, Buttner D. 2008. HpaC controls substrate specificity of the Xanthomonas type III secretion system. PLoS Pathog 4:e1000094. doi: 10.1371/journal.ppat.1000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lara-Tejero M, Kato J, Wagner S, Liu X, Galan JE. 2011. A sorting platform determines the order of protein secretion in bacterial type III systems. Science 331:1188–1191. doi: 10.1126/science.1201476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raffatellu M, Wilson RP, Chessa D, Andrews-Polymenis H, Tran QT, Lawhon S, Khare S, Adams LG, Baumler AJ. 2005. SipA, SopA, SopB, SopD, and SopE2 contribute to Salmonella enterica serotype Typhimurium invasion of epithelial cells. Infect Immun 73:146–154. doi: 10.1128/IAI.73.1.146-154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steele-Mortimer O, Knodler LA, Marcus SL, Scheid MP, Goh B, Pfeifer CG, Duronio V, Finlay BB. 2000. Activation of Akt/protein kinase B in epithelial cells by the Salmonella typhimurium effector sigD. J Biol Chem 275:37718–37724. doi: 10.1074/jbc.M008187200. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez LD, Hueffer K, Wenk MR, Galan JE. 2004. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304:1805–1807. doi: 10.1126/science.1098188. [DOI] [PubMed] [Google Scholar]
- 27.Darwin KH, Robinson LS, Miller VL. 2001. SigE is a chaperone for the Salmonella enterica serovar Typhimurium invasion protein SigD. J Bacteriol 183:1452–1454. doi: 10.1128/JB.183.4.1452-1454.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo Y, Bertero MG, Frey EA, Pfuetzner RA, Wenk MR, Creagh L, Marcus SL, Lim D, Sicheri F, Kay C, Haynes C, Finlay BB, Strynadka NC. 2001. Structural and biochemical characterization of the type III secretion chaperones CesT and SigE. Nat Struct Biol 8:1031–1036. doi: 10.1038/nsb717. [DOI] [PubMed] [Google Scholar]
- 29.Knodler LA, Bertero M, Yip C, Strynadka NC, Steele-Mortimer O. 2006. Structure-based mutagenesis of SigE verifies the importance of hydrophobic and electrostatic residues in type III chaperone function. Mol Microbiol 62:928–940. doi: 10.1111/j.1365-2958.2006.05418.x. [DOI] [PubMed] [Google Scholar]
- 30.Marcus SL, Knodler LA, Finlay BB. 2002. Salmonella enterica serovar Typhimurium effector SigD/SopB is membrane-associated and ubiquitinated inside host cells. Cell Microbiol 4:435–446. doi: 10.1046/j.1462-5822.2002.00202.x. [DOI] [PubMed] [Google Scholar]
- 31.David G, Perez J. 2009. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small angle X-ray scattering experiments at the synchrotron SOLEIL SWING beamline. J Appl Crystallogr 42:892–900. doi: 10.1107/S0021889809029288. [DOI] [Google Scholar]
- 32.Konarev PV, Volkov VV, Petoukhov MV, Svergun DI. 2006. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Crystallogr 39:277–286. doi: 10.1107/S0021889806004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. 2010. determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J Appl Crystallogr 43:101–109. doi: 10.1107/S0021889809043076. [DOI] [Google Scholar]
- 34.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 35.Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25:495–503. doi: 10.1107/S0021889892001663. [DOI] [Google Scholar]
- 36.Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, Tainer JA. 2013. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nat Methods 10:453–454. doi: 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svergun DI, Petoukhov MV, Koch MH. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys J 80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volkov VV, Svergun DI. 2003. Uniqueness of ab initio shape determination in small angle scattering. J Appl Crystallogr 36:860–864. doi: 10.1107/S0021889803000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kozin MB, Svergun DI. 2001. Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34:33–41. doi: 10.1107/S0021889800014126. [DOI] [Google Scholar]
- 40.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 42.Evrard G, Mareuil F, Bontems F, Sizun C, Perez J. 2011. DADIMODO: a program for refining the structure of multidomain proteins and complexes against small-angle scattering data and NMR-derived restraints. J Appl Crystallogr 44:1264–1271. doi: 10.1107/S0021889811035758. [DOI] [Google Scholar]
- 43.Petoukhov MV, Svergun DI. 2005. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J 89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petoukhov MV, Billas IM, Takacs M, Graewert MA, Moras D, Svergun DI. 2013. Reconstruction of quaternary structure from X-ray scattering by equilibrium mixtures of biological macromolecules. Biochemistry 52:6844–6855. doi: 10.1021/bi400731u. [DOI] [PubMed] [Google Scholar]
- 45.Zarivach R, Vuckovic M, Deng W, Finlay BB, Strynadka NC. 2007. Structural analysis of a prototypical ATPase from the type III secretion system. Nat Struct Mol Biol 14:131–137. doi: 10.1038/nsmb1196. [DOI] [PubMed] [Google Scholar]
- 46.Buchko GW, Niemann G, Baker ES, Belov ME, Smith RD, Heffron F, Adkins JN, McDermott JE. 2010. A multi-pronged search for a common structural motif in the secretion signal of Salmonella enterica serovar Typhimurium type III effector proteins. Mol Biosyst 6:2448–2458. doi: 10.1039/c0mb00097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cherradi Y, Schiavolin L, Moussa S, Meghraoui A, Meksem A, Biskri L, Azarkan M, Allaoui A, Botteaux A. 2013. Interplay between predicted inner-rod and gatekeeper in controlling substrate specificity of the type III secretion system. Mol Microbiol 87:1183–1199. doi: 10.1111/mmi.12158. [DOI] [PubMed] [Google Scholar]
- 48.Kim BH, Kim HG, Kim JS, Jang JI, Park YK. 2007. Analysis of functional domains present in the N-terminus of the SipB protein. Microbiology 153:2998–3008. doi: 10.1099/mic.0.2007/007872-0. [DOI] [PubMed] [Google Scholar]
- 49.Trcek J, Wilharm G, Jacobi CA, Heesemann J. 2002. Yersinia enterocolitica YopQ: strain-dependent cytosolic accumulation and post-translational secretion. Microbiology 148:1457–1465. [DOI] [PubMed] [Google Scholar]
- 50.Abrusci P, Vergara-Irigaray M, Johnson S, Beeby MD, Hendrixson DR, Roversi P, Friede ME, Deane JE, Jensen GJ, Tang CM, Lea SM. 2013. Architecture of the major component of the type III secretion system export apparatus. Nat Struct Mol Biol 20:99–104. doi: 10.1038/nsmb.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]