

Abstract
The bglA gene of Escherichia coli encodes phospho-β-glucosidase A capable of hydrolyzing the plant-derived aromatic β-glucoside arbutin. We report that the sequential accumulation of mutations in bglA can confer the ability to hydrolyze the related aromatic β-glucosides esculin and salicin in two steps. In the first step, esculin hydrolysis is achieved through the acquisition of a four-nucleotide insertion within the promoter of the bglA gene, resulting in enhanced steady-state levels of the bglA transcript. In the second step, hydrolysis of salicin is achieved through the acquisition of a point mutation within the bglA structural gene close to the active site without the loss of the original catabolic activity against arbutin. These studies underscore the ability of microorganisms to evolve additional metabolic capabilities by mutational modification of preexisting genetic systems under selection pressure, thereby expanding their repertoire of utilizable substrates.
INTRODUCTION
The extraordinary versatility and adaptability exhibited by microorganisms are primarily responsible for their success in occupying a broad spectrum of niches. Bacteria have evolved different strategies to cope with a variety of stresses encountered in their natural habitats. When forced to grow in the presence of a novel substrate as the sole carbon source, bacteria are able to evolve the capability to utilize the novel substrate by modifying preexisting genetic systems having related functions (1, 2).
Escherichia coli has several catabolic systems for the utilization of plant-derived aromatic β-glucosides such as arbutin (Arb), salicin (Sal), and esculin (Esc) (Fig. 1) that encode a sugar-specific permease that is a part of the phosphotransferase system (PTS) and a phospho-β-glucosidase necessary for the hydrolysis of the sugar. Interestingly, many of these genetic systems are silent and need a step of mutational activation to be functional. The silent bgl operon of E. coli enables the utilization of arbutin and salicin upon mutational activation (3). A homologue of the bgl operon in Erwinia has been shown to confer the ability to utilize esculin in addition to arbutin and salicin (4). Prompted by this observation, el Hassouni et al. have examined the role of the bgl operon of E. coli in esculin utilization. They have reported that Bgl+ strains of E. coli can hydrolyze esculin and that this ability is lost in mutants in which the transport and hydrolysis functions have been abrogated (4).
FIG 1.

Structure of the aromatic β-glucosides arbutin, salicin, and esculin.
The bgl operon consists primarily of three genes, bglG, bglF, and bglB, encoding a positive regulator that acts as an antiterminator of transcription, the permease that transports and concomitantly phosphorylates arbutin and salicin, and the phospho-β-glucosidase B that hydrolyzes phospho-arbutin and phospho-salicin, respectively. The bgl operon is silent in most wild-type (WT) strains due to the presence of negative elements upstream of the promoter; mutations such as the transposition of insertion elements that abrogate the negative elements result in the activation of the operon. The activated operon confers a Bgl+ phenotype and has been shown to have additional roles beyond the hydrolysis of aromatic β-glucosides (see reference 5 and references therein).
The second silent system involved in β-glucoside utilization in the absence of the bgl operon is the asc operon (6). It includes three genes, ascG, ascF, and ascB. The ascG gene encodes a repressor, ascF encodes a nonfunctional permease, and ascB encodes a phospho-β-glucosidase. In the absence of bglB, activation of the asc operon through a promoter mutation enables salicin utilization, with AscB providing the β-glucosidase function and BglF contributing the permease function (7).
E. coli carries a constitutively expressed bglA gene encoding phospho-β-glucosidase A, which is a part of the glycosyl hydrolase family 1 (8) and can hydrolyze phosphorylated arbutin, para-nitrophenyl-β-glucoside, and phenyl β-glucoside but not phospho-salicin (9). However, wild-type strains are unable to utilize arbutin because of the lack of a permease (9). In the absence of BglB, BglA can hydrolyze arbutin if BglF is active and can provide the transport function (7). The bglA locus of the Gram-positive bacterium Streptococcus mutans, which has 72% similarity with E. coli bglB, has been shown to be involved in esculin utilization (10). The wild-type bglA gene of E. coli, however, is unable to confer the ability to hydrolyze esculin.
In this study, we have explored the possible genetic systems involved in esculin utilization in E. coli and have confirmed that activated bgl and asc operons can confer the ability to utilize esculin. We have also sequentially isolated esculin-positive (Esc+) and salicin-positive (Sal+) mutants from E. coli lacking functional BglB and AscB phospho-β-glucosidases by selecting for growth first on esculin and then on salicin. We show that bglA encoding phospho-β-glucosidase A specific for arbutin is the target of mutations that enable hydrolysis of esculin and salicin in two sequential steps without the loss of its primary catabolic activity against arbutin.
MATERIALS AND METHODS
Strains and plasmids.
Bacterial strains and plasmids used in this study are listed in Table 1.
TABLE 1.
List of strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| E. coli strains | ||
| RV | F− lacX74 thi bglR0 bglG+ bglF+ bglB+ (Arb− Sal−) | 17 |
| RV Esc+ | RV bglR (Arb+ Sal+ Esc+) | This work |
| MM1 | RV tnaA::Tn10 (80% linkage to the wild-type bgl operon) | 17 |
| AE328 | MM1 bglR::IS1 (Arb+ Sal+) | 17 |
| SD-2.12 | RV bglR::IS1 ΔbglB (Arb+ Esc− Sal−) | 7 |
| SD-3.12 | SD-2.12 (Arb+ Esc+ Sal+) | 7 |
| SD-3.12 ΔascB | SD-3.12 ascB::kan (Arb+ Esc− Sal−) | 7 |
| SD-3.12 ΔascB Esc+ | SD-3.12 ΔascB bglAE1 (Arb+ Esc+ Sal−) | This work |
| SD-3.12 ΔascB Sal+ | SD-3.12 ΔascB bglAS1 (Arb+ Esc+ Sal+) | This work |
| SD-3.12 ΔascB ΔbglA | SD-3.12 ΔascB bglA677::Tn10 (Arb− Esc− Sal−) | This work |
| Plasmids | ||
| pACDH | Tetr, Plac promoter, pACYC184 origin | 18 |
| pACDH-bglAWT | Wild-type bglA under Plac promoter in pACDH | This work |
| pACDH-bglAEsc+ | bglA from Esc+ mutant under Plac promoter in pACDH | This work |
| pACDH-bglASal+ | bglA from Sal+ mutant under Plac promoter in pACDH | This work |
| pACDH-bglAT583G | bglAT583G under Plac promoter in pACDH | This work |
Media and growth conditions.
Bacterial cultures were grown in L broth at 37 °C. β-Glucoside utilization was detected by growing the strains on MacConkey agar medium supplemented with 1% salicin, 1% arbutin, or 0.4% esculin incubated at 37°C. β-Glucoside-positive strains were scored based on their ability to form bright red colonies, while β-glucoside-negative strains form white/pale yellow colonies. Upon prolonged incubation, newly arising β-glucoside-positive mutants form red outgrowths (papillae). For growth assays on M9 minimal medium, cultures were grown in L broth to early stationary phase, centrifuged, and washed three times with M9 minimal medium without a carbon source. Cells were resuspended in the same medium, and 5 μl of the diluted samples was spotted on M9 minimal medium plates supplemented with 1% salicin, 1% arbutin, or 0.4% esculin as a sole carbon source and incubated at 30 °C for 48 h. Growth on M9 minimal medium with 0.4% glycerol as a sole carbon source served as a positive control.
For RNA extraction, cultures were grown in M9 minimal medium with 0.4% sodium succinate. Antibiotics were added at the following concentrations: ampicillin at 100 μg/ml, kanamycin at 50 μg/ml, and tetracycline at 15 μg/ml. P1 transductions were performed as described by Miller (11).
Construction of plasmids.
For the construction of the plasmids pACDH-bglAWT, pACDH-bglAEsc+ and pACDH-bglASal+ carrying the bglA gene from the wild-type and Esc+ and Sal+ mutant strains, respectively, the bglA coding sequences were amplified using the forward primer 5′-TAGAGCTCAATTGACTCATCTGGAGCCT-3′ and the reverse primer 5′-TAGAATTCCTTCATCGACTTAAAGCTTC-3′. The SacI-EcoRI fragments carrying bglA were cloned into the vector pACDH at SacI and EcoRI restriction sites.
Site-directed mutagenesis of wild-type bglA.
The bglA gene with a T-to-G change at position 583 [bglA(T583G)] was introduced by site-directed mutagenesis using the plasmid pACDH-bglAWT as the template, the forward primer 5′-CTGTTCGGTTACGGCTGCTCCGGCG-3′ and the reverse primer 5′-CGCCGGAGCAGCCGTAACCGAACAG-3′ (where boldface indicates the altered nucleotide to be introduced), and 1 μl of Pfu high-fidelity DNA polymerase (2.5 units/μl) (Thermo Scientific). The PCR program was adopted from a Stratagene QuickChange site-directed mutagenesis protocol.
Sequence analysis.
DNA sequencing was carried out commercially at Macrogen, South Korea. All DNA and protein alignment studies were carried out using EcoCyc (http://ecocyc.org/), ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2), and UniProt (http://www.uniprot.org).
Structural analysis.
The crystal structure of wild-type BglA was obtained from the Protein Data Bank (PDB) (identification [ID] code 2XHY) (8), and mutant BglA(C195G) was modeled based on the crystal structure using the Rosetta software program (12). For both proteins, the centroid of the binding site of the sugar molecules was approximated using the average of the coordinates of the two catalytic glutamate residues (180 and 377). The docking of salicin to this binding site was performed using the program Autodock, version 4.0 (13).
Saligenin assay.
Cells were grown in M9 minimal medium with 0.4% succinate as a carbon source in the presence and absence of inducer (7 mM salicin). At mid-log phase, 1-ml aliquots of the cultures were harvested, washed in 0.9% saline, and resuspended in 0.1 ml of saline. Salicin (0.1 ml of a 4% solution) was added, and the mixture was incubated at 37 °C for 30 min. The reaction was stopped by the addition of 0.5 ml of 2 M Na2CO3. Production of saligenin by cleavage of phospho-salicin was detected by the addition of 0.5 ml of 0.6% 4-amino-antipyrine followed by 0.25 ml of K3Fe(CN)6 after 15 min at room temperature. A positive reaction, indicated by the appearance of red coloring, was quantified by measuring absorbance at 509 nm. Absorbance at 600 nm was used to normalize for cell density. Enzyme activity was determined using the following formula: activity units = A509 × 103/A600 × 10 × v × t, where v (ml) is the volume of concentrated cells used in the assay and t (min) is the time of incubation (7).
Isolation of total RNA.
Total RNA was isolated using the acid phenol method (14). RNA was quantified by using a Bio-Rad spectrophotometer, and quality was tested by analysis on a 1% agarose-HCHO gel.
Semiquantitative reverse transcription-PCR (RT-PCR).
Total RNA was treated with DNase I (MBI Fermentas) to remove genomic DNA, and 2 μg of it was reverse transcribed using random hexamer primers and 1 μl of Moloney murine leukemia virus (MMuLV) reverse transcriptase (200 U/μl) (MBI Fermentas, Germany) as per the manufacturer's protocol. bglA was amplified using the following primers: the forward primer 5′-ACAACATCGAACCGGTGATC-3′ and the reverse primer 5′-ACATCGTCCGGGTTACAGGA-3′. rrnC was amplified using the forward primer 5′-TTACTGGGCGTAAAGCGCACGC-3′ and the reverse primer 5′-TTCACAACACGAGCTGACGACA-3′. The PCR products were analyzed on a 1.5% agarose gel, and the intensities of the bands were quantified using ImageJ software. Levels of rrnC were used as normalizing controls.
Quantitative RT-PCR (qRT-PCR).
Total RNA was treated with DNase I (MBI Fermentas) to remove genomic DNA, and 2 μg of it was reverse transcribed using random hexamer primers and 1 μl of MMuLV reverse transcriptase (200 U/μl) (MBI Fermentas, Germany). cDNA equivalent to 10 ng of total RNA was used for all of the real-time PCRs. bglA cDNA was amplified using the forward primer 5′-TGGCGGTGAAAGCTGCGCGT-3′ and the reverse primer 5′-ACATCGTCCGGGTTACAGGA-3′. rrnC cDNA was amplified using the forward primer 5′-TTACTGGGCGTAAAGCGCACGC-3′ and the reverse primer 5′-TTCACAACACGAGCTGACGACA-3′. Each 10-μl reaction mixture contained Dynamo SYBR green mix (Finnzymes, Finland), carboxy-X-rhodamine reference dye, the cDNA template, and forward and reverse primers. Amplification reactions were carried out using Applied Biosystems StepOnePlus real-time PCR system (version 2.2.3). Analysis was performed using StepOne (version 2.2.2) software (Applied Biosystems). Reaction mixtures were incubated for 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 30 s at 52 °C, and 1 min at 72 °C for bglA. The annealing condition for rrnC was 30 s at 60 °C. All reactions were performed in triplicates for three biological replicates. The fold change, relative to the calibrator, was calculated by the 2−ΔΔCT (where CT is threshold cycle) method (15) using rrnC as a normalization control. Data were assessed by employing a two-tailed unpaired t test using GraphPad Prism, version 5.0, software (GraphPad software, San Diego, CA). A P value of ≤0.05 was considered to be significant.
Stability of mRNA.
To determine the stability of the bglA transcript, the strains SD-3.12 ΔascB (Esc− parent) and SD-3.12 δascB Esc+ (mutant) were grown to early log phase (optical density at 600 nm [OD600] of ∼0.3) and transcription initiation was arrested using 200 μg/ml of rifampin. Total RNA was isolated from shaking cultures at different time points after the addition of rifampin and treated with DNase I to remove genomic DNA. Two micrograms of RNA was reverse transcribed using random hexamer primers and MMuLV reverse transcriptase. cDNA equivalent to 10 ng of total RNA was used for all PCRs. The PCR products were analyzed on 1.5% agarose gels, and the intensities of the bands were quantified using ImageJ software. The rrnC transcript was used as a normalizing control. The half-life of the bglA transcript in the wild-type and mutant strains was determined by plotting the normalized levels of the transcript remaining at each time point relative to transcript level from untreated cultures (0 min) against time.
Determination of the bglA TSS.
RNA was extracted from the Esc− parent and Esc+ mutant strains. First-strand cDNA was synthesized from equal amounts of RNA using 1 μl of Transcriptor reverse transcriptase (25 U/μl) per reaction volume and the first reverse primer specific to bglA (SP1, 5′-CTACCGTATTGCTGCACCAGATGC-3′), according to the protocol of the Roche Second-Generation 5′/3′ RACE (rapid amplification of cDNA ends) kit. The cDNA was purified using a Fermentas High Pure Product Purification kit. Addition of a homopolymeric A tail to the 3′ end of first-strand cDNA was carried out using dATP and 1 μl of terminal transferase (80 units/μl) per reaction volume. The dA-tailed cDNA was amplified using a second nested reverse primer, SP2, located upstream of SP1 (SP2, 5′-GCTTGATGTCTTCCTTATAGTGACC-3′), an oligo(dT) anchor forward primer (5′-GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTTV-3′, where V is A, C, or G), and high-fidelity Pfu DNA polymerase. The PCR products were sequenced to determine the transcription start site (TSS).
RESULTS
Isolation of Esc+ mutants.
Wild-type strains of E. coli are unable to utilize the aromatic β-glucoside esculin as a sole carbon source. Esculin-positive (Esc+) mutants could be obtained from papillae appearing on MacConkey-esculin medium within 24 to 48 h. Introduction of the wild-type (silent) copy of the bgl operon into the Esc+ mutants by P1 transduction resulted in the loss of the Esc+ phenotype, suggesting the involvement of the activated bgl operon in conferring the Esc+ phenotype. The strain SD-2.12, harboring an activated bgl operon but carrying a deletion of the bglB gene that encodes the enzyme phospho-β-glucosidase B, also showed an Esc− phenotype, confirming the involvement of the bglB gene in esculin utilization. The strain could, however, regain the ability to utilize esculin via activation of the silent asc operon, which was confirmed by the loss of the Esc+ phenotype upon introduction of a disruption of the ascB locus in the mutants. The strain SD-3.12 ΔascB carrying a deletion of both bglB and ascB showed an Esc− phenotype. The strain, however, was able to hydrolyze arbutin via the constitutively expressed bglA gene encoding phospho-β-glucosidase A, with transport function provided by bglF.
In an attempt to observe whether the bglB ascB double mutant can gain the ability to utilize esculin, the strain SD-3.12 ΔascB was streaked on MacConkey-esculin medium. Esculin-positive mutants appeared after 5 days as papillae. These Esc+ mutants retained the arbutin-positive (Arb+) phenotype. Deletion of the bglA locus in the mutants resulted in an Esc− Arb− phenotype, indicating that both phenotypes are related to the bglA locus.
The ability of the Esc+ mutant to hydrolyze esculin could be due to enhanced transcription/translation of bglA in this mutant. Alternatively, the mutant may carry an alteration within the bglA coding sequence. Introduction of a multicopy plasmid carrying the wild-type bglA gene in the strain SD-3.12 ΔascB resulted in an Esc+ phenotype (Table 2), suggesting that the phenotype is related to enhanced expression of bglA in the mutant. This is consistent with the absence of any mutation within the bglA coding region, as indicated by nucleotide sequencing of the bglA locus from the Esc+ mutant (Fig. 2).
TABLE 2.
Aromatic β-glucoside phenotypes of transformants with plasmids carrying different alleles of bglA
| Transformant | Phenotypea |
||
|---|---|---|---|
| Arbutin | Esculin | Salicin | |
| SD-3.12 ΔascB(pACDH) | + | − | − |
| SD-3.12 ΔascB(pACDH-bglAWT) | + | + | − |
| SD-3.12 ΔascB(pACDH-bglAEsc+) | + | + | − |
| SD-3.12 ΔascB(pACDH-bglASal+) | + | + | + |
| SD-3.12 ΔascB(pACDH-bglAT583G) | + | + | + |
The phenotype could be observed even without induction by isopropyl-β-d-thiogalactopyranoside.
FIG 2.

Alignments of DNA and protein sequences for bglA from the wild-type and Esc+ and Sal+ mutant strains. (A) The T-to-G transversion at position 583 within the bglA gene in the Sal+ mutant. (B) The corresponding amino acid change at position 195 of the bglA ORF in the Sal+ mutant. Arrows show the two active-site glutamate residues at position 180, which is a potential proton donor, and at 377, which is a hypothetical nucleophile site, based on sequence similarity (8).
Mapping of the start site of transcription indicated that bglA transcription starts at the identical nucleotide in both the wild type and the Esc+ mutant corresponding to the start site reported by RegulonDB (16) 23 nucleotides upstream of the translational start site of the bglA open reading frame (ORF) (Fig. 3). An insertion of 4 nucleotides was detected upstream of the TSS within the spacer region of the putative promoter of the bglA gene. The insertion results in a new −35 sequence with a 17-nucleotide optimal spacer as opposed to the 16-nucleotide suboptimal spacer present in the parent strain (Fig. 3). This is expected to result in enhanced transcription of bglA in the mutant.
FIG 3.

Nucleotide sequence of the region upstream of bglA showing the putative promoter elements. The A residue at +1 indicates the transcription start site (TSS) corresponding to genome coordinate 3041661 reported in RegulonDB (http://regulondb.ccg.unam.mx/index.jsp). The triangle shows the position of the 4-nucleotide insertion within the spacer region between the predicted −10 and −35 elements in the Esc+ and Sal+ mutants. The insertion results in a new putative −35 element, improving the suboptimal spacer region of the wild-type promoter. The putative bglA translational start site is shown in green.
The possibility of enhanced expression of bglA in the mutant was confirmed by measuring the levels of the bglA transcript in the wild-type and mutant strains using quantitative RT-PCR. The steady-state levels of the bglA transcripts were substantially higher in the Esc+ and Sal+ mutants than in the Esc− parental strain (Fig. 4). Measurement of the stability of the bglA transcript showed that the half-life of the transcript is 2.5-fold higher in the mutant than in the wild type (Fig. 5). Therefore, the higher steady-state level of the bglA transcript in the mutant is the result of enhanced transcription as well as increased stability of the transcript.
FIG 4.

Steady-state expression of bglA mRNA in SD-3.12 ΔascB (Esc−), SD-3.12 δascB Esc+, and SD-3.12 δascB Sal+ strains measured by qRT-PCR. The y axis represents the fold change of bglA expression relative to the Esc− calibrator. The rrnC (16S rRNA) transcript was used as a control for normalization (ns, nonsignificant).
FIG 5.

The relative levels of bglA mRNA plotted against time in the Esc− parent strain SD-3.12 ΔascB and in the Esc+ mutant. The levels of bglA mRNA at different time points after rifampin treatment were analyzed by semiquantitative RT-PCR. The percentage of mRNA remaining relative to mRNA from untreated cultures (0 min) at each time point was plotted on the y axis versus time on the x axis.
Isolation of Sal+ mutants.
The Esc+ mutants described above as well as the original strain SD-3.12 ΔascB carrying multiple copies of bglA remained Sal− (Table 2). However, Sal+ papillae appeared within 3 to 4 days of incubation on MacConkey-salicin medium in the case of the Esc+ mutants but not in the case of the parental strain. Interestingly, the Sal+ mutants retained the ability for degradation of both arbutin and esculin.
Hydrolysis of salicin in the Sal+ mutants is through bglA since deletion of bglA in the mutant resulted in loss of the Sal+ phenotype. Transformation of the parent strain with a plasmid carrying the bglA gene from the Sal+ mutant resulted in an Sal+ phenotype (Table 2), indicating that the mutant carries an alteration within bglA. This was confirmed by nucleotide sequencing that revealed a single point mutation, thymine to guanine transversion at position 583 of the bglA gene (T583G), resulting in an amino acid change of cystine to glycine at position 195 (C195G) of the BglA ORF close to the active site (Fig. 2). Upon introduction of the T583G mutation in the wild-type bglA gene by site-directed mutagenesis, transformants of the parent strain carrying the mutated bglA on a multicopy plasmid showed an Sal+ phenotype (Table 2), indicating that the C195G mutation is necessary and sufficient to confer an Sal+ phenotype when present in multiple copies.
The Sal+ phenotype of the mutant seen on indicator medium was confirmed by a saligenin assay that measures specifically the phospho-β-glucosidase activity of the enzyme against salicin (7). In the presence of 7 mM salicin (necessary for inducing bglF expression to provide permease function), the Sal+ mutant strain as well as transformants of the parent strain carrying the bglASal+ allele and the bglAT583G allele generated by site-directed mutagenesis showed significantly enhanced activity compared to the respective control strains, consistent with the phenotype seen on indicator plates (Fig. 6).
FIG 6.
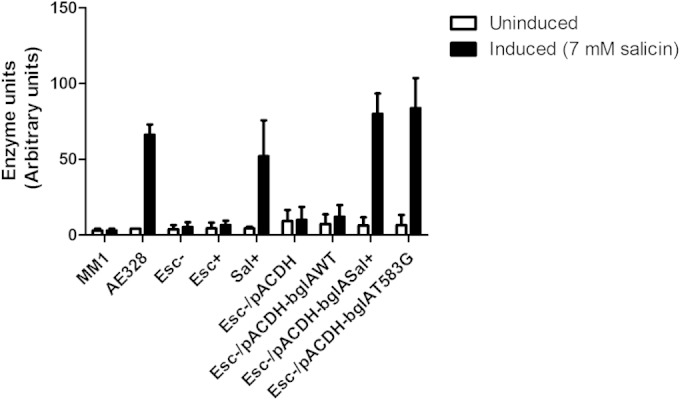
Saligenin assay carried out on SD-3.12 ΔascB (Esc−), SD-3.12 δascB Esc+, and SD-3.12 δascB Sal+ strains and SD-3.12 ΔascB transformants carrying pACDH (vector control), pACDH-bglAWT, pACDH-bglASal+, and pACDH-bglAT583G. Strains MM1 (bgl wild type) and AE328 (Bgl+) carrying an activated bgl operon served as the reference strains. Cells were grown in M9 minimal succinate medium in the presence and absence of inducer (7 mM salicin). Error bars represent the standard deviations obtained from a minimum of three independent biological replicates.
DISCUSSION
We have explored the role of known aromatic β-glucoside catabolic systems in esculin utilization in E. coli. The primary system involved in esculin catabolism is the bgl operon. Upon activation of the operon, BglF is presumed to transport esculin inside the cell, and phospho-β-glucosidase B encoded by bglB is presumed to cleave the phosphorylated esculin.
Previous work in the lab (7) has shown that in an E. coli strain with a disrupted bglB gene, activation of the silent asc operon is involved in salicin degradation. We observed that the E. coli bglB strain can form papillae on MacConkey-esculin medium and gain the ability for esculin degradation through activation of the asc operon. This was confirmed by the observation that disruption of ascB results in an Esc− phenotype in the mutants, indicating that the activated ascB is responsible for esculin hydrolysis. When both bglB and ascB are absent, the strain SD-3.12 ΔascB can mutate to become esculin positive on MacConkey-esculin medium. In these mutants, hydrolysis of esculin is through the bglA gene encoding phospho-β-glucosidase A.
Sequencing of the bglA locus from wild-type and Esc+ mutants showed that the bglA ORF is unaltered, but an insertion of 4 bases was detected in the spacer region of the putative bglA promoter in the mutant. The mutation generates a new −35 sequence with an optimal spacing of 17 nucleotides as opposed to the 16-nucleotide spacer present in the wild-type strain. The enhanced steady-state level of the bglA transcript in the mutant detected by quantitative RT-PCR is consistent with the enhanced transcription expected from the altered promoter. Interestingly, though transcription is initiated at the same nucleotide in both cases, the decay rate of the bglA transcript is higher in the wild-type strain than in the mutant. This is likely to be related to the increased transcription initiation in the mutant leading to saturating concentrations of the transcript that have a protective effect.
The observation that enhanced levels of the wild-type bglA transcript can confer an Esc+ phenotype suggests that wild-type BglA is able to cleave esculin with a low efficiency, but this activity is insufficient to enable growth on esculin as a sole carbon source. When expression levels are enhanced, the cumulative activity of the enzyme becomes sufficient to enable growth on esculin.
Sequencing of the bglA locus from the Sal+ mutant showed the presence of a transversion of thymine to guanine at position 583 of the bglA gene (T583G), resulting in an amino acid change from cysteine to glycine at position 195 of the BglA ORF (C195G) close to the active site (Fig. 2). Introduction of the bglA locus from the wild type and Esc+ and Sal+ mutants in the parent strain SD-3.12 ΔascB (Esc− Sal−) showed that the bglA allele from all three strains in multiple copies conferred an Esc+ phenotype. However, only the bglA allele from the Sal+ mutant in multiple copies resulted in an Sal+ phenotype (Table 2). These results show that in the first-step Esc+ mutant, overexpression of wild-type bglA is sufficient for the Esc+ phenotype while the point mutation in the structural gene is necessary for the Sal+ phenotype. Measurement of phospho-β-glucosidase activity indicated that presence of the bglAT583G allele leads to a significant increase in enzyme activity, consistent with the phenotype of the mutant observed on indicator plates.
As seen in the structure of the wild-type BglA (8), C195 is exposed to the substrate-binding site (Fig. 7). Binding energies of salicin to wild-type BglA and BglA(C195G), estimated using Autodock, version 4.0 (13), indicated that the mutant protein showed marginally better binding to salicin than wild-type BglA (data not shown). This might be attributed to the loss of stearic hindrance for the binding of salicin when C195 is replaced by the smaller amino acid glycine in the mutant protein.
FIG 7.
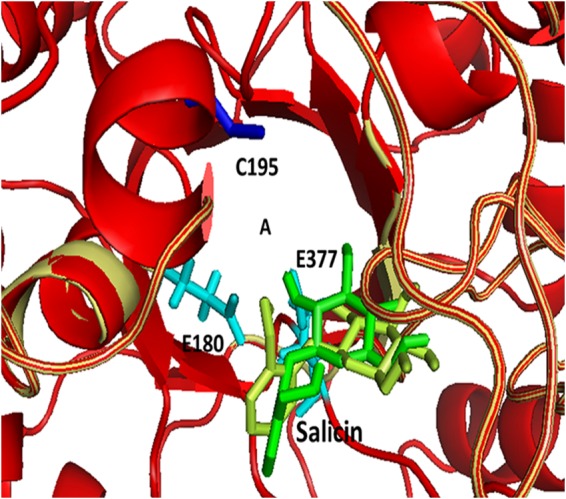
The overlaid structures of wild-type (yellow) and mutant (red) BglA proteins. The structure of the wild-type protein is based on PDB ID 2XHY (8), and the structure of mutant BglA was modeled using the Rosetta software with 2XHY as the template. The letter A indicates the approximate substrate binding pocket. The position of C195 in the wild-type structure is highlighted in blue, and the active-site glutamate residues E180 and E377 are shown in cyan. Salicin docked in the binding pocket is shown in green for the wild-type structure and in lemon for the mutant.
These observations corroborate the idea that microorganisms, when confronted by a novel substrate, have the capacity to evolve additional metabolic capabilities by mutational modification of preexisting genetic systems under selection pressure. While a mutation that leads to overexpression of a hydrolytic enzyme is sufficient to enable the metabolism of one of the novel substrates, in the case of the second substrate, in addition to overexpression, modification of the active site is necessary to mediate catalysis. The former was achieved by enhancing the steady-state levels of the structural gene mRNA, and the latter was made possible by a structural gene mutation (Fig. 8).
FIG 8.

Schematic of esculin and salicin utilization in E. coli in the absence of bglB and ascB. In the parent strain, the level of bglA expression is sufficient only for arbutin degradation. In the first-step Esc+ mutant, insertion of four nucleotides within the bglA promoter (jagged arrow) results in higher levels of BglA (filled ovals), which are necessary for both esculin and salicin degradation. In the next-step Sal+ mutant, a point mutation within the bglA ORF (filled star) results in salicin degradation. The mutant protein is shown as ovals with white stars.
Interestingly, the Sal+ mutant carrying a modified residue in the active site retained the ability to hydrolyze all three aromatic β-glucosides, indicating that the evolution of a novel metabolic function need not necessarily involve a trade-off of the original catalytic activity of the enzyme. In an environment where related substrates are available in cycles, promoting promiscuity of the catabolic enzyme will be a preferred strategy. Though these studies have been carried out using catabolic systems involved in β-glucoside utilization, these mechanisms are likely to be a part of the normal arsenal that microorganisms carry in their struggle for survival in the complex environments in which they live.
ACKNOWLEDGMENTS
We thank Amrisha Bhosle, Department of Biochemistry, Indian Institute of Science, for help in the modeling and docking studies and the anonymous referees for making several constructive suggestions.
This work was supported by grants from the Department of Biotechnology, Government of India. Infrastructural support for the work was provided by the University Grants Commission and the Department of Science and Technology.
REFERENCES
- 1.Wright BE. 2004. Stress-directed adaptive mutations and evolution. Mol Microbiol 52:643–650. doi: 10.1111/j.1365-2958.2004.04012.x. [DOI] [PubMed] [Google Scholar]
- 2.Patrick WM, Quandt EM, Swartzlander DB, Matsumura I. 2007. Multicopy suppression underpins metabolic evolvability. Mol Biol Evol 24:2716–2722. doi: 10.1093/molbev/msm204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prasad I, Schaefler S. 1974. Regulation of the beta-glucoside system in Escherchia coli K-12. J Bacteriol 120:638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.el Hassouni M, Chippaux M, Barras F. 1990. Analysis of the Erwinia chrysanthemi arb genes, which mediate metabolism of aromatic beta-glucosides. J Bacteriol 172:6261–6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harwani D, Zangoui P, Mahadevan S. 2012. The β-glucoside (bgl) operon of Escherichia coli is involved in the regulation of oppA, encoding an oligopeptide transporter. J Bacteriol 194:90–99. doi: 10.1128/JB.05837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall BG, Xu L. 1992. Nucleotide sequence, function, activation, and evolution of the cryptic asc operon of Escherichia coli K12. Mol Biol Evol 9:688–706. [DOI] [PubMed] [Google Scholar]
- 7.Desai SK, Nandimath K, Mahadevan S. 2010. Diverse pathways for salicin utilization in Shigella sonnei and Escherichia coli carrying an impaired bgl operon. Arch Microbiol 192:821–833. doi: 10.1007/s00203-010-0610-8. [DOI] [PubMed] [Google Scholar]
- 8.Totir M, Echols N, Nanao M, Gee CL, Moskaleva A, Gradia S, Iavarone AT, Berger JM, May AP, Zubieta C, Alber T. 2012. Macro-to-micro structural proteomics: native source proteins for high-throughput crystallization. PLoS One 7:e32498. doi: 10.1371/journal.pone.0032498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prasad I, Young B, Schaefler S. 1973. Genetic determination of the constitutive biosynthesis of phospho-beta-glucosidase A in Escherichia coli K-12. J Bacteriol 114:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cote CK, Cvitkovitch D, Bleiweis AS, Honeyman AL. 2000. A novel beta-glucoside-specific PTS locus from Streptococcus mutans that is not inhibited by glucose. Microbiology 146:1555–1563. [DOI] [PubMed] [Google Scholar]
- 11.Miller JH. 1992. A short course in bacterial genetics. A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 12.Rohl CA, Strauss CE, Misura KM, Baker D. 2004. Protein structure prediction using Rosetta. Methods Enzymol 383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 13.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh J, Mukerji M, Mahadevan S. 1995. Transcriptional activation of the Escherichia coli bgl operon: negative regulation by DNA structural elements near the promoter. Mol Microbiol 17:1085–1092. doi: 10.1111/j.1365-2958.1995.mmi_17061085.x. [DOI] [PubMed] [Google Scholar]
- 15.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protocol 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 16.Salgado H, Peralta-Gil M, Gama-Castro S, Santos-Zavaleta A, Muniz Rascado L, Garcia-Sotelo JS, Weiss V, Solano-Lira H, Martinez-Flores I, Medina-Rivera A, Salgado-Osorio G, Alquicira-Hernandez S, Alquicira-Hernandez K, Lopez-Fuentes A, Porron-Sotelo L, Huerta AM, Bonavides-Martinez C, Balderas-Martinez YI, Pannier L, Olvera M, Labastida A, Jimenez-Jacinto V, Vega-Alvarado L, Del Moral-Chavez V, Hernandez-Alvarez A, Morett E, Collado-Vides J. 2013. RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res 41:D203–D213. doi: 10.1093/nar/gks1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moorthy S, Mahadevan S. 2002. Differential spectrum of mutations that activate the Escherichia coli bgl operon in an rpoS genetic background. J Bacteriol 184:4033–4038. doi: 10.1128/JB.184.14.4033-4038.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao AR, Varshney U. 2002. Characterization of Mycobacterium tuberculosis ribosome recycling factor (RRF) and a mutant lacking six amino acids from the C-terminal end reveals that the C-terminal residues are important for its occupancy on the ribosome. Microbiology 148:3913–3920. [DOI] [PubMed] [Google Scholar]