

Conspectus
Enzymes are essential for all living organisms, and their effectiveness as chemical catalysts has driven more than a half century of research seeking to understand the enormous rate enhancements they provide. Nevertheless, a complete understanding of the factors that govern the rate enhancements and selectivities of enzymes remains elusive, due to the extraordinary complexity and cooperativity that are the hallmarks of these biomolecules. We have used a combination of site-directed mutagenesis, pre-steady-state kinetics, X-ray crystallography, nuclear magnetic resonance (NMR), vibrational and fluorescence spectroscopies, resonance energy transfer, and computer simulations to study the implications of conformational motions and electrostatic interactions on enzyme catalysis in the enzyme dihydrofolate reductase (DHFR).
We have demonstrated that modest equilibrium conformational changes are functionally related to the hydride transfer reaction. Results obtained for mutant DHFRs illustrated that reductions in hydride transfer rates are correlated with altered conformational motions, and analysis of the evolutionary history of DHFR indicated that mutations appear to have occurred to preserve both the hydride transfer rate and the associated conformational changes. More recent results suggested that differences in local electrostatic environments contribute to finely tuning the substrate pKa in the initial protonation step. Using a combination of primary and solvent kinetic isotope effects, we demonstrated that the reaction mechanism is consistent across a broad pH range, and computer simulations suggested that deprotonation of the active site Tyr100 may play a crucial role in substrate protonation at high pH.
Site-specific incorporation of vibrational thiocyanate probes into the ecDHFR active site provided an experimental tool for interrogating these microenvironments and for investigating changes in electrostatics along the DHFR catalytic cycle. Complementary molecular dynamics simulations in conjunction with mixed quantum mechanical/molecular mechanical calculations accurately reproduced the vibrational frequency shifts in these probes and provided atomic-level insight into the residues influencing these changes. Our findings indicate that conformational and electrostatic changes are intimately related and functionally essential. This approach can be readily extended to the study of other enzyme systems to identify more general trends in the relationship between conformational fluctuations and electrostatic interactions. These results are relevant to researchers seeking to design novel enzymes as well as those seeking to develop therapeutic agents that function as enzyme inhibitors.
1. Introduction
The exquisite activities, specificities, and selectivities of naturally occurring enzymes continue to inspire studies seeking to more precisely describe the origin of the catalytic prowess of these extraordinary biomolecules. In essence, enzymes lower the free energy barriers of chemical reactions (i.e., increase the probabilities of sampling transition states relative to ground states) by providing electrostatic and geometric interactions that are complementary to the transition state.1 This concept of transition state stabilization generally evokes a static model with singular ground and transition state structures. However, enzymes today are better understood in terms of ensembles of configurations that exist on free energy landscapes2 encompassing innumerable local minima, which are analogous to the rugged free energy surfaces envisioned for protein folding.3 The current understanding of the factors governing enzyme catalysis has been discussed extensively in numerous recent reviews.2,4−6
Electrostatic stabilization of transition states involves polar and hydrogen bonding groups precisely positioned to stabilize the migration of charge during the chemical reaction. The partly hydrophobic character of the interiors of enzymes and the oriented nature of the enzymes’ functional groups within the protein scaffold decrease the energetic penalty of environmental reorganization in the protein relative to bulk solvent.6,7 The importance of electrostatic interactions has led to efforts to isolate and quantify the contribution of electrostatics to enzyme catalysis, as well as to understand the connection between conformational and electrostatic fluctuations.6,8−10
Motion and flexibility within enzymes play important functional roles in substrate binding, product release, and the formation of catalytically competent configurations.2 In enzyme catalysis, small equilibrium conformational changes have been shown to accompany the chemical step in many cases. These changes are essential to stabilize geometrical variations that are necessary for the chemical reaction (e.g., to bring a donor and acceptor closer together or to accommodate a change in hybridization).11,12 According to this perspective, fast thermal equilibrium fluctuations on the femtosecond to picosecond time scale lead to overall conformational changes that occur on the millisecond time scale to reach a state conducive to the relatively fast formation and cleavage of chemical bonds. However, there is no convincing evidence for specific special promoting vibrations13 on the femtosecond to picosecond time scale that are directly coupled to the chemical bond formation and cleavage. The role of protein motions in enzyme catalysis has been demonstrated in a wide variety of enzymes, including cyclophilin A,14 α-amylase,15 adenylate kinase,16 and dihydrofolate reductase (DHFR).
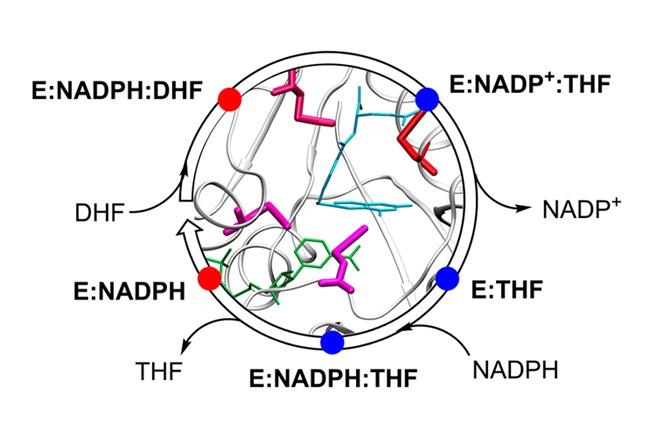
DHFR is a ubiquitous enzyme that catalyzes the reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF) using reduced nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor (Figure 1). DHFR from Escherichia coli (ecDHFR) undergoes a large-scale (∼8 Å) conformational change in a flexible region (residues 9–24) known as the Met20 loop during the course of its catalytic cycle (Figure 2).17,18 In the closed conformation, the Met20 loop interacts with the substrate, while in the occluded conformation, the loop occupies a portion of the cofactor binding pocket and extrudes the nicotinamide moiety of NADP(H) from the active site. Nuclear magnetic resonance (NMR) experiments show the presence of high-energy conformations in each state that correspond to low-energy conformations of neighboring states in the catalytic cycle.19
Figure 1.
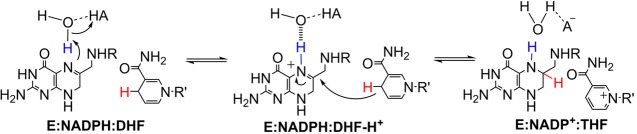
Catalytic mechanism of ecDHFR involving proton transfer from an active site water molecule to N5 of DHF (blue) and subsequent hydride transfer from NADPH to C6 of DHF (red).
Figure 2.
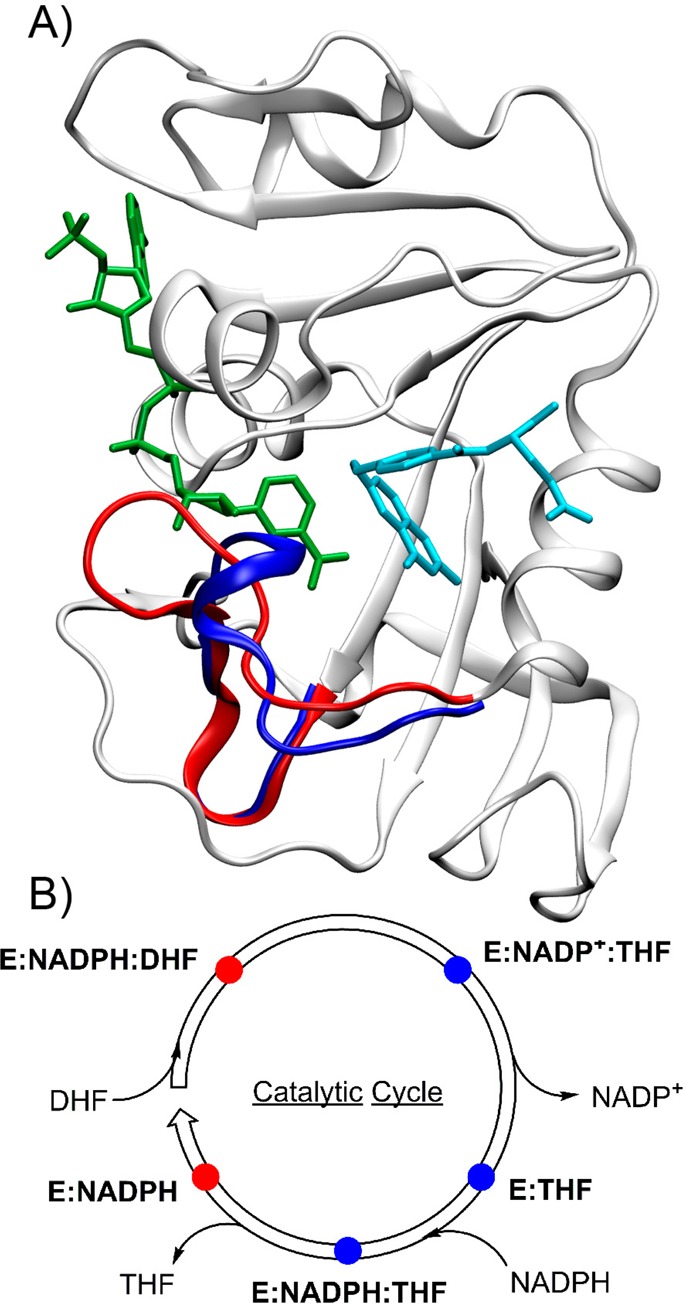
(A) Ribbon structure of ecDHFR with bound NADP+ (green) and folate (sky blue) illustrating the differences between the closed (PDB ID 3QL3; red) and occluded (PDB ID 1RX6; blue) conformations of the Met20 loop. (B) The ecDHFR catalytic cycle distinguishing the closed conformation (red) and occluded conformation (blue). Adapted with permission from ref (10). Copyright 2014 American Chemical Society.
Herein, we describe the collaborative experimental and theoretical research performed in our laboratories on ecDHFR catalysis. The first section summarizes the role of conformational motions on the hydride transfer reaction and the impact of mutations on these motions and catalysis. The second section illustrates the importance of active site residues on finely tuning the pKa’s within the active site, in particular, the pKa of the substrate N5 that is protonated prior to the hydride transfer reaction. The subsequent sections discuss the incorporation of thiocyanate probes into ecDHFR to examine local electrostatic microenvironments within the enzyme and identify electrostatic interactions that directly influence the hydride transfer reaction itself. Taken together, the results of these inquiries highlight the importance of electrostatic interactions and motions in enzyme catalysis.
2. Conformational Fluctuations Impact Enzyme Activity
The role of conformational changes in enzyme catalysis is a topic of great interest in enzymology, particularly whether conformational motions are coupled to the chemical step in enzyme catalysis and the quantitative contribution of motions to lowering the free energy barrier of the chemical reaction.20−22 Empirical valence bond (EVB)23 molecular dynamics (MD) simulations of ecDHFR catalysis identified several residues involved in conformational changes and indicated that these changes occur along the collective reaction coordinate associated with the hydride transfer reaction.11,12 Evolutionarily conserved residues including Met42, Tyr100, and Gly121 were observed to undergo modest ≤ 1 Å conformational changes as the reaction progressed from reactant to transition state. These conformational changes bring the donor and acceptor closer together in a favorable orientation and provide an appropriate electrostatic environment, thereby facilitating the hydride movement that occurs near the transition state. Mutations of these residues impacted the catalytic rate constant and altered the conformational changes; furthermore, double mutations of Met42 and Gly121 displayed nonadditive effects, indicating cooperativity between these residues. Thus, even relatively small conformational changes are important to the chemical step in ecDHFR.
Single molecule Förster resonance energy transfer (FRET) experiments suggested that multiple conformations of ecDHFR exist in solution, that these conformations interact differently with the bound ligands, and that they exhibit different hydride transfer rates.24,25 Furthermore, in these experiments and more recent equilibrium FRET experiments,26 some changes in fluorescence were associated with hydride transfer, while other changes were unrelated to hydride transfer. For example, FRET experiments indicated that the G121V mutation reduced the rates of conformational fluctuations, but these rate effects were quantitatively different from those on the hydride transfer rate,25 indicating that the conformational motions are not directly coupled to hydride transfer. The observation that certain conformational changes identified by FRET can be related to the chemical step while others are unrelated to the chemical step highlights the importance of performing experiments designed to distinguish between these two possibilities.26
We recently explored the evolutionary significance of the N23PP mutation from ecDHFR to DHFR from Homo sapiens (hsDHFR),27 which was previously shown to ablate millisecond time scale large-scale conformational changes in the Met20 loop.20 Using bioinformatics, we identified several additional mutations between ecDHFR and hsDHFR that were found to be evolutionarily and functionally significant.27 Mutation in ecDHFR of an asparagine to diproline in the flexible Met20 loop (N23PP) ablates millisecond time scale fluctuations and reduces the hydride transfer rate,20 a three residue insertion near the substrate binding pocket (G51PEKN) has little effect in WT ecDHFR but recovers the WT rate in the N23PP background, and a conservative mutation of an aliphatic to an aromatic hydrophobic residue in the substrate binding pocket (L28F) enhances the hydride transfer rate of N23PP/G51PEKN to near-mammalian levels. EVB-MD simulations of the N23PP and N23PP/G51PEKN mutants suggested that the recovery of the N23PP mutant by the G51PEKN mutation was related to recovering WT-like conformational changes across the chemical step (Figure 3). This interpretation is broadly consistent with the kinetic isotope effect studies of Kohen and co-workers28 and the simulation results of Agarwal and co-workers,29 while Warshel and co-workers suggested that the differences between the N23PP mutant and WT ecDHFR are predominantly electrostatic in nature.30 Recognizing the inextricable connection between conformational and electrostatic changes, these interpretations may be related to the same phenomena rather than mutually exclusive.
Figure 3.
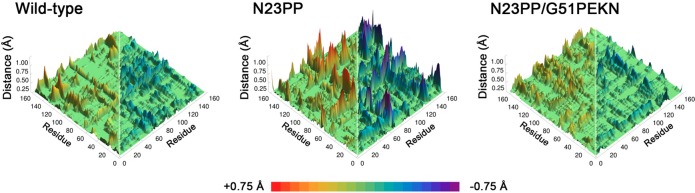
Conformational changes measured by thermally averaged Cα–Cα distance changes from the reactant state to the transition state of the hydride transfer reaction observed in EVB-MD simulations of WT ecDHFR, N23PP ecDHFR, and N23PP/G51PEKN ecDHFR.27 For clarity, only distances that increase are shown on the left side of each plot, while only distances that decrease are shown on the right.
In light of the evidence for the existence of conformational changes in enzymes and their connection to enzyme catalysis, the next step is to determine the source of their contribution to lowering the free energy barrier to catalysis. When polar and charged residues are involved in conformational motions, these structural rearrangements lead to changes in the active site microenvironments that are essential for providing the thermodynamic stabilization of the reacting species. As illustrated in subsequent sections, conformational fluctuations modulate local electrostatic properties in ecDHFR that further contribute to enzymatic activity.
3. Conformational and Electrostatic Influence on Ionization and pKa
Electrostatic influence on the ionization states of catalytic residues has been demonstrated in many enzymes, especially regarding the role of conformational flexibility.31,32 Conformational and electrostatic changes are intimately coupled because structural rearrangements of polar groups within enzymes must affect the local electrostatic microenvironments. Thus, assessing the catalytic contribution of enzymes based solely on electrostatics while discounting the presence of thermal fluctuations only provides a static snapshot along the free energy surface of an enzymatic reaction and fails to capture the ensemble of varying conformational and electrostatic environments that exist in solution.
The large conformational changes18,19 exhibited by ecDHFR lead to a broad spectrum of active site electrostatic environments that can have substantial effects on the energy landscapes of enzymatic processes. The pKa of the N5 atom of DHF, which is protonated prior to hydride transfer (Figure 1), varies from ∼2.6 in aqueous solution33 up to ∼9 in the enzyme34 depending on the conformation of the Met20 loop. This pKa shift is likely due to changes in the loop conformation that alter the electrostatic environment around the nearby substrate. Because the hydride transfer reaction follows DHF protonation, shifts in the substrate pKa determine the concentration of the reactive species and consequently have substantial effects on the rate of the overall reaction.
Recently, we identified a reactive ecDHFR ternary species that operates under alkaline conditions (pH > 11), in which one or more titratable residues within the enzyme’s active site are likely to be deprotonated.35 Using multiple kinetic isotope effects (KIEs),36 we showed that the reaction mechanism (N5 protonation preceding hydride transfer) remains consistent across the pH range examined (pH 4–12), suggesting elevation of the N5 pKa beyond the previously determined value of ∼9.34 Using free energy perturbation MD simulations,37 we showed that the N5 pKa can be increased by ∼5 units when the nearby Tyr100 is deprotonated.35 In other words, a more negatively charged active site environment with Tyr100 or other residues deprotonated or partially deprotonated, generated under high pH conditions, could increase the N5 pKa from 6.5 in the ternary reactant complex to ∼11 or higher, thus ensuring sufficient DHF protonation. Moreover, thermodynamic evaluation indicates that Tyr100 and Asp27 function synergistically through electrostatic interactions to optimize both the substrate protonation and the subsequent hydride transfer step.
4. Fluctuations in Active Site Electrostatics along the Catalytic Cycle
The importance of electrostatic interactions for enzyme function necessitates methodologies that are capable of monitoring local changes in electrostatics in conjunction with catalysis. One attractive approach is the use of non-natural amino acids containing thiocyanate (SCN) functional groups with infrared (IR) absorption spectroscopy.38 These functional groups are well-suited for studying electrostatics in enzymes due to several properties: their absorption spectra are relatively distinct from that of the enzyme, their stretching modes tend to decouple from other proximal vibrational modes, and their vibrational frequencies are sensitive to the local electrostatic environment. Furthermore, these functional groups generally do not constitute a large perturbation to the local protein structure, allowing the examination of a virtually intact and functional enzyme.
The application of SCN vibrational probes to study enzyme electrostatics was extensively explored by Boxer, Pande, and co-workers.39 The vibrational frequency distribution of the probe within the enzyme is often compared with the vibrational frequency in various solvents and with the Stark tuning rate determined in the solid state. These data can be interpreted in terms of the Onsager reaction field or solvatochromic models,38,40 although the heterogeneous environment of a protein broadly precludes simple characterization via a single dielectric constant. The incorporation of thiocyanate probes is especially powerful when combined with X-ray crystallography or MD simulations of the enzyme that relate the electrostatic data to atomic-level structural details.10,39,41,42 Additional insight into the presence of hydrogen bonding to the nitrile group can be obtained by comparing the 13C NMR chemical shift of the nitrile carbon with the vibrational frequency data.41
Theoretical methods for calculating electric fields and IR vibrational shifts in enzymes have been developed using classical MD39 and mixed quantum mechanical/molecular mechanical (QM/MM) approaches.10,42,43 Classical MD simulations allow the electric field to be quantified within the context of the atomic point charges in the MM force field. Moreover, QM/MM calculations for configurations generated by classical MD enable the calculation of the vibrational frequency shifts of nitrile probes. Specifically, the C–N potential energy curve and associated vibrational energy level splittings are calculated for each configuration. Further decomposition of the electric field based on contributions from each residue illustrates how various protein components shape the enzyme’s electrostatic landscape.
4.1. Probing ecDHFR Microenvironments through Vibrational Spectroscopy
The vibrational spectrum of SCN and other environmentally sensitive vibrational probes can yield quantitative information on the changes in the local electric field around the probes upon ligand binding, protein folding, alteration of the surrounding hydration shell, and protein–protein association.8,38,39,44,45 To determine how the electrostatic microenvironments of ecDHFR respond to the conformational changes occurring along its catalytic cycle, we recently incorporated the Cys-CN probe into four different regions of the enzyme active site (Figure 4; Supporting Information, Table S1). Despite the proximity of the nitrile probes to the reactive center, these mutants retained comparable catalytic activity (∼50–110%) to the WT enzyme, and X-ray crystallography indicated no major structural rearrangements. The minimal catalytic and structural effects induced by the SCN probe suggest that these constructs faithfully represent the electrostatics of the native enzyme.10
Figure 4.

Thiocyanate probes in the ecDHFR active site colored according to their FTIR maximum vibrational frequency (cm–1) for closed (red) and occluded (blue) states along the catalytic cycle. When present, folate or THF is shown in sky blue and NADP(H) is shown in green.
After incorporating the SCN probe into the ecDHFR active site, we examined the changes in the CN vibrational frequency along the catalytic cycle. Figure 4 illustrates the SCN probes colored according to the frequency of maximum FTIR absorption, νmax, detected in each complex.10 Shifts in this frequency reflect changes in the component of the electric field along the CN bond of the probe. Crystal structures18 and MD simulations10 suggest that the L28C-CN, T46C-CN, and L54C-CN probes retain their positions and orientations among the five catalytic complexes. This observation implies that the changes in the electrostatic microenvironments of these probes among the various complexes in the catalytic cycle are due to electrostatic changes arising from ligand binding and rearrangements of the enzyme and solvent. In contrast, the orientation and location of the M20C-CN probe change significantly along the catalytic cycle, relative to a fixed point in the protein, due to alterations in the conformation of the Met20 loop.
The T46C-CN probe is situated proximal to the junction between the two reacting ligands (Figure 4),10,18 DHF and NADPH, allowing us to monitor the microenvironment close to the reactive center. The largest frequency shifts observed for the T46C-CN probe occurred across the two most important steps in the catalytic cycle: (1) upon the conversion between the model Michaelis–Menten E:NADP+:FOL complex and the initial E:NADP+:THF product complex (−4.1 cm–1), which mimics the hydride transfer step, and (2) across the enzyme turnover step, which is the release of THF (5.5 cm–1).
The L54C-CN and L28C-CN probes are situated in the folate binding pocket, located ∼9 Å from the center of the hydride D–A axis (Figure 4). These two probes are sensitive to electrostatic changes upon folate binding, as well as the binding/release of NADP(H) in the cofactor binding pocket. In general, ligand binding induces substantial changes in the active site electrostatic microenvironments for all four positions where the SCN probe was incorporated. For the L54C-CN probe, on one side of the folate binding pocket, the most significant change occurred upon the formation of the ternary E:NADP+:FOL complex (2.4 cm–1), while for the L28C-CN probe, on the opposite side of the pocket, the largest changes occurred during the formation of the Michaelis–Menten complex (−2.9 cm–1) and the enzyme turnover step (2.4 cm–1).10
The M20C-CN probe is located directly on the highly fluctuating Met20 loop, and it migrates between different environments in the closed and occluded conformations. Significant vibrational frequency variation was observed between all steps of the catalytic cycle for the M20C-CN probe, with the largest changes corresponding to steps associated with the transition between the closed and occluded conformation. The large changes in the electric field around the M20C-CN probe are predominantly caused by altered hydrogen-bonding interactions between the nitrile probe and solvent water molecules. This observation is consistent with the proposal that interactions with the hydration shell and bulk solvent are important for conformational changes.46
4.2. Residue-Based Decomposition of Active Site Microenvironments
Complementary QM/MM simulations of the SCN probes in ecDHFR provided atomic-level information regarding the major contributors to the detected local electrostatic changes in each probe location. In particular, we performed QM/MM simulations of the T46C-CN and L54C-CN probes to examine changes in the electric field across the chemical step of the ecDHFR catalytic cycle.10 The calculated νmax of the T46C-CN probe in the modeled Michaelis–Menten complex is 2165.8 cm–1, which is within 1.8 cm–1 of the measured peak in the experiments, and the full width at half-maximum (fwhm) calculated for this probe is 11 cm–1, in good agreement with the experimental value of 14 cm–1. Furthermore, the calculated shifts across the hydride transfer step for the T46C-CN and L54C-CN probes are −3.7 cm–1 and −2.5 cm–1, respectively, compared with the experimentally measured shifts of −4.1 cm–1 and −1.0 cm–1, respectively. The good agreement between the theoretical and experimental values provides validation for the simulation methodology.
We also calculated the electric field at the midpoint of the CN bond due to the MM partial charges in the MD simulations. The average projected electric fields along the T46C-CN nitrile bond for the E:NADP+:FOL and E:NADP+:THF complexes are −15.9 and −19.0 MV/cm, respectively. The component of the electric field along the CN axis for each probe can be decomposed into contributions from individual residues, ligands, ions, and solvent molecules.10 This procedure allows us to assess the importance of the conformational changes of each residue in microenvironment reorganization. For the T46C-CN probe, the NADP+ cofactor and the nearest water molecule are the dominant contributors to the electric field change between the closed and occluded conformations across the hydride transfer step, although Asn18 and Ser49 also make significant contributions. Specifically, the amide nitrogen of Asn18 is ∼1.5 Å closer to the probe in the closed conformation and the hydroxyl group of Ser49 is ∼1.0 Å closer to the nitrile probe in the closed conformation. Moreover, following the closed to occluded transition, the CN probe becomes more accessible to the solvent, altering the hydrogen-bonding interaction of the probe with the nearby water molecule. More subtle effects, such as changes in aromaticity and quadrupole moments, may also play a role.38 In the case of the local environment around L54C-CN, we found that Arg57 exhibits the largest change between the two conformational states. This effect arises because the positively charged region of Arg57 moves away from its orthogonal orientation with respect to the probe in the occluded conformation.
5. Role of Electrostatics in the Catalyzed Hydride Transfer Reaction
On the basis of the experimental validation, we used the same simulation methodology to evaluate the component of the electric field along the hydride D–A axis in the model Michaelis–Menten complex and to decompose this field into contributions from each residue within the enzyme (Figure 5). These calculations yielded valuable information about the reactive center, which is more directly related to catalysis than analogous information about the SCN probes within the active site. Moreover, such information about the reactive center is experimentally inaccessible. We found that the component of the total electric field along the D–A axis in the model ternary reactant complex (E:FOL:NADP+) is −48.9 MV/cm. The negative value indicates that this component of the electric field is highly favorable for transferring a negatively charged hydride from the cofactor to the substrate, and this magnitude could potentially reduce the free energy barrier by several kilocalories per mole.39 Electric field decomposition showed that the substrate and cofactor contribute −32.4 MV/cm to the total field along the D–A axis. The protein contributes −15.4 MV/cm, which is ∼33% of the total field, while the solvent molecules and ions contribute very little (−1.1 MV/cm) to the total field. The quantitative contributions from the ligands to the electric field in the model Michaelis complex may differ from those of the substrate and cofactor during the chemical reaction. Future computational studies will examine the electric field during the chemical reaction catalyzed by DHFR.
Figure 5.
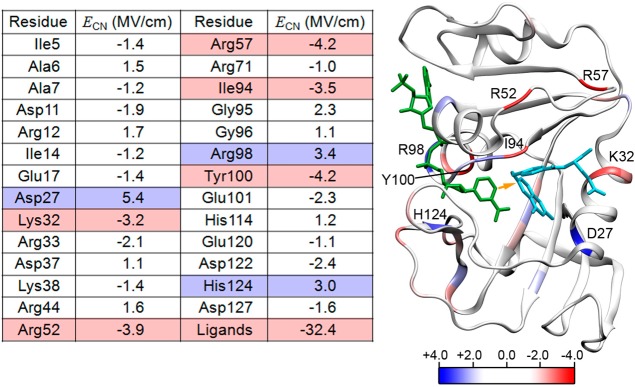
Ribbon structure of ecDHFR colored according to residue-based contributions to the calculated electric field along the hydride transfer D–A axis (depicted as an orange arrow) for selected residues from MD simulations of the model Michaelis complex E:FOL:NADP+. Positive values of the electric field disfavor hydride transfer, while negative values facilitate hydride transfer. NADP+ is colored green, and folate is colored sky blue.
Further residue-based decomposition of the protein electric field (−15.4 MV/cm) showed that there is a strong degree of overlap between the residues that contribute significantly to the field along the D–A axis and the network of coupled motions that has been identified for DHFR catalysis.10,12 This network of coupled motions corresponds to equilibrium conformational changes that occur as the reaction evolves from the reactant to the transition state along the collective reaction coordinate associated with hydride transfer. Such motions facilitate the chemical reaction by bringing the substrate and cofactor closer together in a favorable orientation and providing a suitable electrostatic environment for hydride transfer. The residues Ile14, Tyr100, and Asp122 contribute −1.2, −4.2, and −2.4 MV/cm, respectively, and they were all implicated in the network of coupled motions.12 Additional residues that significantly contribute to the electric field along the D–A axis in a manner that facilitates hydride transfer are Lys32 (−3.2 MV/cm), Arg52 (−3.9 MV/cm), and Arg57 (−4.2 MV/cm). The residues that contribute to the field along the D–A axis in a manner that disfavors hydride transfer include Asp27 (5.4 MV/cm), Arg98 (3.4 MV/cm), and His124 (3.0 MV/cm). All of these residues represent potential targets to alter the electrostatic contribution to catalysis. Interestingly (see section 3), two of the major contributors (Asp27 and Tyr100) were found to be important catalytic residues that operate synergistically to affect various aspects of the enzymatic reaction, such as the pKa of N5 on DHF, proton transfer from a solvent molecule to DHF, and hydride transfer from NADPH, via electrostatic interactions.35
6. Conclusion, Implications, and Future Prospects
The results from recent studies of DHFR are consistent with the view that a series of rearrangements brings the substrates into a conformation that realizes the highly favorable electrostatic field for facilitating the hydride transfer reaction.2,4,10 Protein motions are particularly important in enzymes with a high degree of conformational flexibility due to a greater number of accessible structural and electrostatic states. An integrated computational–experimental approach provides a very powerful tool for probing the detailed active site microenvironments, active site hydration states, and electrostatic contributions along the catalytic cycle.
With scientific disciplines presently shifting in ways that favor the translational value of research, fundamental enzymology queries into enzyme function and catalysis still present important potential for discovery. Enzymes are highly flexible entities, especially under biological conditions with significant thermal energy available, and it is logical that conformational fluctuations will generate an ensemble of species covering a wide range of electrostatic variation. Since the electrostatic environment can have a dramatic effect on enzyme catalysis, a more dynamic perspective must be adopted when considering the source of catalytic efficacy. This approach could be helpful in guiding the development of novel small molecules or peptides for inhibiting protein activity47,48 as well as the design of catalysts with activities comparable to those of naturally occurring enzymes.49
Acknowledgments
This work was supported by US National Institutes of Health (NIH) Grants GM092946 (P.H., C.T.L., and S.J.B.) and GM056207 (S.H.-S.).
Biographies
Philip Hanoian obtained his B.A. from Wesleyan University (2007) and Ph.D. from Penn State University (2014). He is currently a postdoctoral fellow at Penn State University.
C. Tony Liu received his B.S. (2006) and Ph.D. (2011) in Chemistry from Queen’s University in Kingston, Ontario, Canada. He is currently a postdoctoral fellow at Penn State University.
Sharon Hammes-Schiffer received her B.A. from Princeton (1988) and her Ph.D. from Stanford (1993). She is currently the Swanlund Professor of Chemistry at the University of Illinois at Urbana–Champaign.
Stephen J. Benkovic received his B.S. in Chemistry and B.A. in English Literature from Lehigh University (1960) and his Ph.D. in Organic Chemistry from Cornell University (1963). He is currently an Evan Pugh Professor of Chemistry and Eberly Chair in Chemistry at Penn State University.
Supporting Information Available
Frequencies of maximum absorbance for SCN probes in each of the five states along the DHFR cycle. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Protein Motion in Catalysis”.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Pauling L. Molecular Architecture and Biological Reactions. Chem. Eng. News 1946, 24, 1375–1377. [Google Scholar]
- Hammes G. G.; Benkovic S. J.; Hammes-Schiffer S. Flexibility, Diversity, and Cooperativity: Pillars of Enzyme Catalysis. Biochemistry 2011, 50, 10422–10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill K. A.; MacCallum J. L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Catalytic Efficiency of Enzymes: A Theoretical Analysis. Biochemistry 2013, 52, 2012–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D.; Natarajan A. Fundamental Challenges in Mechanistic Enzymology: Progress toward Understanding the Rate Enhancements of Enzymes. Biochemistry 2013, 52, 2050–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H. B.; Olsson M. H. M. Electrostatic Basis for Enzyme Catalysis. Chem. Rev. 2006, 106, 3210–3235. [DOI] [PubMed] [Google Scholar]
- Jencks W. P. Binding Energy, Specificity, And Enzymic Catalysis: The Circe Effect. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219–410. [DOI] [PubMed] [Google Scholar]
- Jha S. K.; Ji M.; Gaffney K. J.; Boxer S. G. Direct Measurement of the Protein Response to an Electrostatic Perturbation That Mimics the Catalytic Cycle in Ketosteroid Isomerase. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16612–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwans J. P.; Sunden F.; Gonzalez A.; Tsai Y.; Herschlag D. Evaluating the Catalytic Contribution from the Oxyanion Hole in Ketosteroid Isomerase. J. Am. Chem. Soc. 2011, 133, 20052–20055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. T.; Layfield J. P.; Stewart R. J. III; French J. B.; Hanoian P.; Asbury J. B.; Hammes-Schiffer S.; Benkovic S. J. Probing the Electrostatics of Active Site Microenvironments along the Catalytic Cycle for Escherichia coli Dihydrofolate Reductase. J. Am. Chem. Soc. 2014, 136, 10349–10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Hammes-Schiffer S. Nuclear Quantum Effects and Enzyme Dynamics in Dihydrofolate Reductase Catalysis. J. Phys. Chem. B 2002, 106, 3283–3293. [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Rajagopalan P. T. R.; Benkovic S. J.; Hammes-Schiffer S. Network of Coupled Promoting Motions in Enzyme Catalysis. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S. D.; Schramm V. L. Enzymatic Transition States and Dynamic Motion in Barrier Crossing. Nat. Chem. Biol. 2009, 5, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmesser E. Z.; Millet O.; Labeikovsky W.; Korzhnev D. M.; Wolf-Watz M.; Bosco D. A.; Skalicky J. J.; Kay L. E.; Kern D. Intrinsic Dynamics of an Enzyme Underlies Catalysis. Nature 2005, 438, 117–121. [DOI] [PubMed] [Google Scholar]
- Kosugi T.; Hayashi S. Crucial Role of Protein Flexibility in Formation of a Stable Reaction Transition State in an α-Amylase Catalysis. J. Am. Chem. Soc. 2012, 134, 7045–7055. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Lei M.; Thai V.; Kerns S. J.; Karplus M.; Kern D. A Hierarchy of Timescales in Protein Dynamics Is Linked to Enzyme Catalysis. Nature 2007, 450, 913–916. [DOI] [PubMed] [Google Scholar]
- Fierke C. A.; Johnson K. A.; Benkovic S. J. Construction and Evaluation of the Kinetic Scheme Associated with Dihydrofolate Reductase from Escherichia coli. Biochemistry 1987, 26, 4085–4092. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. Loop and Subdomain Movements in the Mechanism of Escherichia coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 1997, 36, 586–603. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. Millisecond Timescale Fluctuations in Dihydrofolate Reductase Are Exquisitely Sensitive to the Bound Ligands. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Lee J.; Ekiert D. C.; Gam J.; Wilson I. A.; Dyson H. J.; Benkovic S. J.; Wright P. E. A Dynamic Knockout Reveals That Conformational Fluctuations Influence the Chemical Step of Enzyme Catalysis. Science 2011, 332, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y.; Cembran A.; Ma S.; Gao J. Connecting Protein Conformational Dynamics with Catalytic Function as Illustrated in Dihydrofolate Reductase. Biochemistry 2013, 52, 2036–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Warshel A. The Empirical Valence Bond Model: Theory and Applications. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 30–45. [Google Scholar]
- Zhang Z. Q.; Rajagopalan P. T. R.; Selzer T.; Benkovic S. J.; Hammes G. G. Single-Molecule and Transient Kinetics Investigation of the Interaction of Dihydrofolate Reductase with NADPH and Dihydrofolate. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antikainen N. M.; Smiley R. D.; Benkovic S. J.; Hammes G. G. Conformation Coupled Enzyme Catalysis: Single-Molecule and Transient Kinetics Investigation of Dihydrofolate Reductase. Biochemistry 2005, 44, 16835–16843. [DOI] [PubMed] [Google Scholar]
- Liu C. T.; Wang L.; Goodey N. M.; Hanoian P.; Benkovic S. J. Temporally Overlapped but Uncoupled Motions in Dihydrofolate Reductase Catalysis. Biochemistry 2013, 52, 5332–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. T.; Hanoian P.; French J. B.; Pringle T. H.; Hammes-Schiffer S.; Benkovic S. J. Functional Significance of Evolving Protein Sequence in Dihydrofolate Reductase from Bacteria to Humans. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10159–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis K.; Stojkovic V.; Kohen A. Preservation of Protein Dynamics in Dihydrofolate Reductase Evolution. J. Biol. Chem. 2013, 288, 35961–35968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan A.; Agarwal P. K. Evolutionarily Conserved Linkage between Enzyme Fold, Flexibility, and Catalysis. PLoS Biol. 2011, 9, e1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczyk A. J.; Cao J.; Kamerlin S. C. L.; Warshel A. Catalysis by Dihydrofolate Reductase and Other Enzymes Arises from Electrostatic Preorganization, Not Conformational Motions. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14115–14120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R.; Head M. S.; Moult J.; Gilson M. K. pKa Shifts in Small Molecules and HIV Protease: Electrostatics and Conformation. J. Am. Chem. Soc. 1998, 120, 6138–6146. [Google Scholar]
- Harris T. K.; Turner G. J. Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life 2002, 53, 85–98. [DOI] [PubMed] [Google Scholar]
- Maharaj G.; Selinsky B. S.; Appleman J. R.; Perlman M.; London R. E.; Blakley R. L. Dissociation Constants for Dihydrofolic Acid and Dihydrobiopterin and Implications for Mechanistic Models for Dihydrofolate Reductase. Biochemistry 1990, 29, 4554–4560. [DOI] [PubMed] [Google Scholar]
- Khavrutskii I. V.; Price D. J.; Lee J.; Brooks C. L. 3rd Conformational Change of the Methionine 20 Loop of Escherichia coli Dihydrofolate Reductase Modulates pKa of the Bound Dihydrofolate. Protein Sci. 2007, 16, 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. T.; Francis K.; Layfield J. P.; Huang X.; Hammes-Schiffer S.; Kohen A.; Benkovic S. J. Escherichia coli Dihydrofolate Reductase Catalyzed Proton and Hydride Transfers: Temporal Order and the Roles of Asp27 and Tyr100. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 18231–18236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermes J. D.; Roeske C. A.; Oleary M. H.; Cleland W. W. Use of Multiple Isotope Effects to Determine Enzyme Mechanisms and Intrinsic Isotope Effects - Malic Enzyme and Glucose-6-Phosphate-Dehydrogenase. Biochemistry 1982, 21, 5106–5114. [DOI] [PubMed] [Google Scholar]
- Hansen N.; van Gunsteren W. F. Practical Aspects of Free-Energy Calculations: A Review. J. Chem. Theory Comput. 2014, 10, 2632–2647. [DOI] [PubMed] [Google Scholar]
- Kim H.; Cho M. Infrared Probes for Studying the Structure and Dynamics of Biomolecules. Chem. Rev. 2013, 113, 5817–5847. [DOI] [PubMed] [Google Scholar]
- Suydam I. T.; Snow C. D.; Pande V. S.; Boxer S. G. Electric Fields at the Active Site of an Enzyme: Direct Comparison of Experiment with Theory. Science 2006, 313, 200–204. [DOI] [PubMed] [Google Scholar]
- Reichardt C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar]
- Fafarman A. T.; Sigala P. A.; Schwans J. P.; Fenn T. D.; Herschlag D.; Boxer S. G. Quantitative, Directional Measurement of Electric Field Heterogeneity in the Active Site of Ketosteroid Isomerase. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E299–E308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield J. P.; Hammes-Schiffer S. Calculation of Vibrational Shifts of Nitrile Probes in the Active Site of Ketosteroid Isomerase upon Ligand Binding. J. Am. Chem. Soc. 2013, 135, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist B. A.; Furse K. E.; Corcelli S. A. Nitrile Groups As Vibrational Probes of Biomolecular Structure and Dynamics: An Overview. Phys. Chem. Chem. Phys. 2009, 11, 8119–8132. [DOI] [PubMed] [Google Scholar]
- Taft R. W.; Kamlet M. J. Linear Solvation Energy Relationships. 8. Solvent Effects on NMR Spectral Shifts and Coupling-Constants. Org. Magn. Reson. 1980, 14, 485–493. [Google Scholar]
- Waegele M. M.; Culik R. M.; Gai F. Site-Specific Spectroscopic Reporters of the Local Electric Field, Hydration, Structure, and Dynamics of Biomolecules. J. Phys. Chem. Lett. 2011, 2, 2598–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H.; Chen G.; Berendzen J.; Fenimore P. W.; Jansson H.; McMahon B. H.; Stroe I. R.; Swenson J.; Young R. D. A Unified Model of Protein Dynamics. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G. M.; Craik C. S. Trapping Moving Targets with Small Molecules. Science 2009, 324, 213–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J. W. Communication Breakdown: Protein Dynamics and Drug Design. Structure 2009, 17, 319–320. [DOI] [PubMed] [Google Scholar]
- Kiss G.; Çelebi-Ölçüm N.; Moretti R.; Baker D.; Houk K. N. Computational Enzyme Design. Angew. Chem., Int. Ed. 2013, 52, 5700–5725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.