

Abstract
Nitrosylcobalamin (NOCbl) is readily formed when Co(II)balamin reacts with nitric oxide (NO) gas. NOCbl has been implicated in the inhibition of various B12-dependent enzymes, as well as in the modulation of blood pressure and of the immunological response. Previous studies revealed that among the known biologically relevant cobalamin species, NOCbl possesses the longest bond between the Co ion and the axially bound 5,6-dimethylbenzimidazole base, which was postulated to result from a strong trans influence exerted by the NO ligand. In this study, various spectroscopic (electronic absorption, circular dichroism, magnetic circular dichroism, and resonance Raman) and computational (density functional theory (DFT) and time-dependent DFT) techniques were used to generate experimentally validated electronic structure descriptions for the “base-on” and “base-off” forms of NOCbl. Further insights into the principal Co–ligand bonding interactions were obtained by carrying out natural bond orbital analyses. Collectively, our results indicate that the formally unoccupied Co 3dz2 orbital engages in a highly covalent bonding interaction with the filled NO π* orbital and that the Co–NO bond is strengthened further by sizable π-backbonding interactions that are not present in any other Co(III)Cbl characterized to date. Because of the substantial NO– to Co(III) charge donation, NOCbl is best described as a hybrid of Co(III)–NO– and Co(II)–NO• resonance structures. In contrast, our analogous computational characterization of a related species, superoxocobalamin, reveals that in this case a Co(III)–O2– description is adequate due to the larger oxidizing power of O2 versus NO. The implications of our results with respect to the unusual structural features and thermochromism of NOCbl and the proposed inhibition mechanisms of B12-dependent enzymes by NOCbl are discussed.
Short abstract
Various spectroscopic and computational techniques were used to generate experimentally validated electronic structure descriptions for the “base-on” and “base-off” forms of nitrosylcobalamin. The principal Co−ligand bonding interactions were examined further by carrying out natural bond orbital analyses. Our results indicate that the Co 3dz2 orbital engages in a highly covalent bonding interaction with one of the NO π* orbitals and that the Co−NO bond is strengthened further by sizable π-backbonding interactions.
Introduction
Cobalamins (Figure 1) consist of a six-coordinate low-spin Co(III) ion that is ligated equatorially by the four nitrogens of a tetrapyrrole macrocycle, known as the corrin ring.1 The “lower” axial position is occupied by a nitrogen from the 5,6-dimethylbenzimidazole (DMB) base that is part of an intramolecular nucleotide loop bound to the corrin ring at C17. At low pH, the coordinating nitrogen of the DMB group becomes protonated, which converts the cobalamins from their “base-on” state to their “base-off” form in which a solvent-derived water molecule now serves as the lower ligand.2 An excellent model of these base-off species at neutral pH is provided by cobinamides, which are naturally occurring cobalamin precursors that lack the terminal DMB group.3
Figure 1.
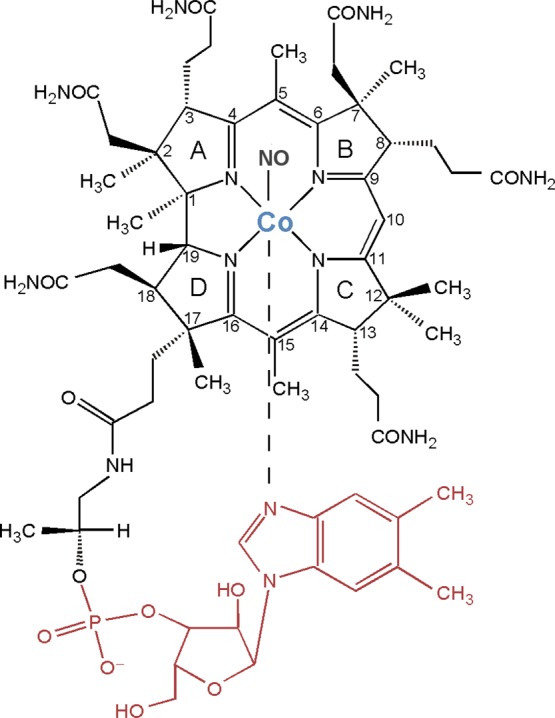
Chemical structure of nitrosylcobalamin (NOCbl), along with the numbering scheme used for the atoms in the corrin ring. Colored in red is the pendant 5,6-dimethylbenzimidazole (DMB) group, which is absent in nitrosylcobinamide (NOCbi+).
The biologically active forms of cobalamin differ with respect to the identity of the variable upper axial ligand, with the best characterized forms being methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl).4 These molecules feature an organometallic bond between the Co(III) ion and either a methyl group or an ATP-derived 5′-deoxyadenosyl group. In humans, MeCbl serves as the cofactor of methionine synthase (MetH), involved in the synthesis of methionine from homocysteine,5 while AdoCbl is required by methylmalonyl-CoA mutase (MMCM) for the isomerization of methylmalonyl-CoA to succinyl-CoA.6 Experimental studies have revealed that both MetH and MMCM are inhibited by nitric oxide (NO).7,8 While these enzymes are chemically unreactive toward NO in their resting states,9 the Co(II)Cbl intermediates formed during catalysis (in the case of MMCM) or cofactor reactivation (MetH) are believed to be susceptible to reactions with this neutral radical species. In support of this assumption, in vitro studies have indicated that NO reacts with Co(II)Cbl on a microsecond time scale to yield nitrosylcobalamin (NOCbl),10,11 while in vivo studies in animals revealed that hydroxycobalamin supplementation can inhibit the physiological response to NO,12 due to efficient NO scavenging by the resulting Co(II)Cbl formed in the cell. Because NOCbl does not support the catalytic activities of MetH and MMCM, high cellular levels of NO are expected to result in a buildup of homocysteine and methylmalonyl-CoA, thereby causing disruption of the homocysteine metabolism and, possibly, the induction of methylmalonic aciduria.6
Despite the wealth of experimental evidence supporting the formation of NOCbl in vivo, the electronic structure of this species remains incompletely understood, in part because NO is a redox-active ligand, thus making an oxidation state assignment for Co ambiguous.13 Using the Enemark–Feltham formalism for metal nitrosyls,14 NOCbl can be described as a {M–NO}8 type species, where the metal–NO fragment is treated as a single unit and is characterized by the total number of metal d and NO π* electrons. From a comparison to {M–NO}8 metalloporphyrin species,15 the Co–NO fragment of NOCbl is expected to adopt a bent geometry, with the NO ligand exerting a strong trans influence. Indeed, the crystal structure of NOCbl obtained at 100 K shows a very long Co–N(DMB) bond (2.32–2.35 Å), a short Co–NO bond (1.91–1.94 Å), and a Co–N–O bond angle of ∼120°.16 These axial bond distances are consistent with structures reported for Co(III)Cbl species featuring a strongly σ-donating upper ligand, thus supporting a Co(III)–NO– description.17 Additionally, the visible region of the electronic absorption (Abs) spectrum of NOCbl in aqueous solution at neutral pH is dominated by a broad asymmetric feature centered at ∼480 nm, which is characteristic of Co(III)Cbl species.11
The dominant contributors to this Abs feature of Co(III)Cbl species are the so-called α/β bands that have been attributed to an electronic transition between corrin π/π*-based molecular orbitals.4 The energies of these bands have been shown to be sensitive to the electron-donating properties of the axial ligands.4 Notably, in the case of NOCbl, the peak position of the α/β bands (∼480 nm) is similar to that of base-off alkylcobalamin species in which the lower axial DMB ligand is replaced by a water molecule (e.g., λmax ≈ 460 nm for MeCbl at pH ≤ 218). These observations suggest that the strongly σ-donating NO– ligand could promote complete dissociation of the DMB group under physiological conditions. However, detailed pH titration and NMR studies by van Eldik and co-workers led to the identification of a pKa value of 5.1 for the protonation and consequent dissociation of the DMB nitrogen, indicating that base-on NOCbl is favored at neutral pH.11 Yet, this pKa value is only ∼0.5 pH units lower than that of the free nucleotide base (pKa of 5.56 in aqueous solution11), suggesting that the Co–N(DMB) bond of NOCbl is very weak. Additional NMR studies by Hassanin et al. led to the suggestion that at neutral pH, ∼33% of NOCbl is present in the base-off form, with the remaining ∼67% being in the base-on form.2
To improve the current understanding of the electronic structures of NOCbl in its base-on and base-off forms, we carried out Abs, circular dichroism (CD), magnetic CD (MCD), and resonance Raman (rR) spectroscopic studies of NOCbl and nitrosylcobinamide (NOCbi+). The spectroscopic data were analyzed within the framework of density functional theory (DFT) and time-dependent-DFT (TDDFT) calculations, employed previously with great success in computational studies of other Co(III)Cbl and Co(III)Cbi species.19−21 To identify the principal Co–ligand bonding interactions, we also carried out natural bond orbital (NBO) analyses. Collectively, our results provide significant new insights into the spectral and electronic properties of NOCbl and NOCbi+. Additionally, by carrying out an analogous computational characterization of superoxocobalamin (O2Cbl), intriguing electronic structure differences between this {Co–O2}9 species and NOCbl were identified.
Methods
Synthesis
Aquacobalamin (H2OCbl+), dicyanocobinamide ((CN)2Cbi), sodium borohydride (NaBH4), potassium formate (HCOOK), sodium nitrite (NaNO2), ascorbic acid, and copper tetrachloride (CuCl4) were purchased from Sigma-Aldrich and used as obtained. Gaseous nitric oxide (NO) was generated by the reaction of NaNO2 with ascorbic acid and aqueous Cu(II) chloride under an argon (Ar) atmosphere.22 NOCbl was prepared by chemical reduction of ∼2 mM H2OCbl+ with a saturated solution of HCOOK under anaerobic conditions to yield Co(II)Cbl, which was subsequently exposed to freshly prepared NO gas for 2 h. To halt the reaction, the solution vials were purged with Ar. NOCbi+ was prepared according to the same procedure, except that in this case ∼2 mM diaquocobinamide ((H2O)2Cbi2+) was used as a precursor. (H2O)2Cbi2+ was synthesized by the addition of NaBH4 to an aqueous solution of (CN)2Cbi, as described in previous reports.23,24 The pH of the sample solutions was 7 unless otherwise indicated.
A comparison of the electronic Abs spectra of the resulting species to published spectra for NOCbl and NOCbi+ confirmed that the reactions went to completion25 (see Supporting Information Figure S1 for complete details). An electron paramagnetic resonance (EPR) characterization of these samples revealed that less than 3% of Co(II)Cbl remained in solution after NO exposure (see Supporting Information, Figure S2). Up to 60% (v/v) degassed glycerol was added under anaerobic conditions to all samples used for low-temperature Abs and MCD experiments to ensure glass formation upon freezing.
Isotopically enriched samples for rR experiments were prepared by the methods described above, except with the use of 15N-labeled NaNO2 (99% pure, Cambridge Isotope Laboratories, Inc.). Frozen pellets were prepared by injecting small aliquots of fluid sample into a liquid N2 bath under an argon atmosphere. Additional samples for rR studies were prepared by the addition of HCl to NOCbl to reach a final pH value of <2. The room-temperature Abs spectra of this low-pH NOCbl species and NOCbi+ were found to be superimposable, indicating that the former species was cleanly converted to its base-off form, as expected on the basis of the pKa value of 5.1 reported for the ligating DMB nitrogen of NOCbl.2
Spectroscopy
Room-temperature CD and low-temperature Abs, CD, and MCD spectra were acquired using a Jasco J-715 spectropolarimeter in conjunction with an Oxford Instruments SpectroMag-4000 8T magnetocryostat. All MCD spectra were obtained by taking the difference between spectra collected with the magnetic field aligned parallel and antiparallel to the light propagation axis to remove contributions from the natural CD and glass strain. Room-temperature Abs spectra were obtained with a Cary 5E UV/vis spectrophotometer.
The rR spectra were obtained upon excitation at 488.0 nm with a Coherent I-305 Ar+ ion laser and ∼40 mW of laser power at the sample. The scattered light was collected using a 135° backscattering arrangement, dispersed by an Acton Research triple monochromator (equipped with 300, 1200, and 2400 grooves/mm gratings), and analyzed with a Princeton Instrument Spec X: 100BR deep depletion, back-thinned CCD camera. Spectra were accumulated at 77 K by placing frozen pellets into a quartz finger dewar filled with liquid N2. Spectra of fluid solution samples, contained in sealed EPR tubes under an argon atmosphere, were obtained with the same setup but by filling the finger dewar with an ice/water mixture. No spectroscopic changes attributable to photolytic or chemical degradation were observed during data collection. All rR spectra were baseline corrected using a piecewise line function to remove the broad nonresonant fluorescence contributions, and the intensities were normalized with respect to the features in the region between 1100 and 1400 cm–1. Peak positions were calibrated against the 984 cm–1 peak of a potassium sulfate standard as well as the ice peak at 228 cm–1 for frozen samples or the water feature at 1637 cm–1 for fluid solution samples.
Computational Studies
Initial coordinates for the model of NOCbl were obtained from the highest-resolution X-ray crystal structure reported by Hassanin et al. in 2010.16 These coordinates were also used as the basis for generating an initial model of NOCbi+, whereby the nucleotide loop was replaced by an H atom at the phosphate position and a water molecule was placed in the lower axial coordination site originally occupied by the DMB base (see Figure 1). Coordinates for the O2Cbl model were obtained from the structure reported by Hohenester et al. and used as is.26 Because of the large number of atoms present in these species, smaller models were prepared for TDDFT excited-state and DFT frequency calculations by removal of atoms that were not expected to contribute to the spectroscopic features observed experimentally. Specifically, the corrin ring substituents were replaced by hydrogen atoms, except for the four methyl groups at the C1, C2, C5, and C15 positions (see Figure 1 for the atom numbering scheme used). Additionally, the two propionamide groups at C2 and C18, along with the nucleotide loop at C7 in the case of NOCbl, were replaced by methyl groups. This truncation scheme was adopted because the methyl groups at the C5 and C15 positions were shown to be crucial for an accurate treatment of the vibrational modes of the corrin ring,27 while the others were found to play a role in preventing excessive flattening of the corrin ring (vide infra). To further increase the likelihood of obtaining realistic corrin fold angles, the entire DMB base was included in all NOCbl models.
Full geometry optimizations of the complete and truncated cofactor models were performed with the Amsterdam Density Functional (ADF) 2012 suite of programs28−30 using the Vosko–Wilk–Nusair (VWN) local-density approximation (LDA)31 and the Perdew, Burke, and Ernzerhof (PBE) gradient corrections for exchange and correlation.32,33 In each case, the triple ζ with polarization (TZP) basis set29,30 was chosen, along with an integration constant of 5.0 and a small frozen core through 1s for C, N, and O and through 2p for Co and P. The optimized models were subjected to frequency calculations to verify that a true energy minimum was found. In each case, only positive frequencies were obtained.
For the computation of spectroscopic properties, the ORCA 2.8 software package developed by Dr. F. Neese was employed.34 While the optimized truncated models were used as-is, the full models were modified using the truncation scheme outlined above and subjected to partial geometry optimizations, whereby only the added H atoms were allowed to move. Electronic transition energies and Abs intensities were computed with the TDDFT method35−37 within the Tamm–Dancoff approximation38,39 and using the PBE functional for exchange and correlation.32,33 The Ahlrichs polarized split valence (SVP) basis set40 was employed for all atoms, except for cobalt, the ligating nitrogens, and the oxygen from NO, for which the triple-ζ valence polarized (TZVP) basis was utilized.41 In each case, 40 excited states were computed by including all one-electron excitations among molecular orbitals within ±3 hartree of the highest occupied molecular orbital/lowest unoccupied molecular orbital (HOMO/LUMO) gap. To increase computational efficiency, the resolution of the identity (RI) approximation in conjunction with the SV/J40,42 auxiliary basis were employed in the evaluation of the Coulomb term for all calculations. The TDDFT results were used as the basis for simulating Abs and CD spectra, assuming that each electronic transition gives rise to a Gaussian-shaped band with a full width at half maximum (fwhm) of v1/2 = 1250 cm–1. All calculated spectra were uniformly red-shifted by 2200 cm–1 to facilitate a visual comparison with the experimental data. Finally, off-resonance Raman spectra for the fully optimized truncated models were computed with ORCA 2.8, by evaluating the numerical frequencies and electronic polarizabilities of all normal modes using DFT and the basis sets and functionals described above. No scaling factor was applied to the computed frequencies.
The ground-state wave functions of the fully optimized models were analyzed further within the natural bond orbital (NBO)43 framework to assess the major bonding interactions, using the NBO5 interface as implemented in ADF via the gennbo and adfnbo programs. The VWN/LDA31 and the PBE gradient corrections for exchange and correlation,32,33 along with the TZP basis set (without frozen core approximation),29 and an integration constant of 5.0 were used to compute the ground-state electron density. Default parameters were chosen for the NBO5 interface. Isosurface plots of all orbitals and electron density difference maps were generated with Pymol using isodensity values of 0.03 au and 0.003 au, respectively.
Results
I. Abs, CD, and MCD Data
The low-temperature (4.5 K) Abs spectrum of NOCbl in frozen solution exhibits partially resolved bands in the lower energy region with an intensity maximum near 20 000 cm–1, corresponding to the so-called α/β bands, along with a series of broad and more intense bands around 31 000 cm–1, historically referred to as the γ region (Figure 2, top). This spectrum is very similar to those exhibited by alkyl-Co(III)Cbl species, such as MeCbl and AdoCbl, which were previously classified as “unique” Abs spectra based primarily on the unusual appearance of the γ region.4 As in those latter spectra, the pair of features at ∼19 000 and 20 000 cm–1 in the Abs spectrum of NOCbl also appear to correspond to the origin and first member of a vibrational progression in a totally symmetric breathing mode of the corrin ring. In support of these assignments, the corresponding features are of the same sign in both the CD and the MCD spectra of NOCbl (Figure 2, middle and bottom). The other bands in the visible region of the NOCbl Abs spectrum are due to at least one or more additional electronic transitions, as indicated by their opposite signs in the CD spectrum. Consistent with its classification as a unique Abs spectrum, several bands that arise from distinct electronic transitions can be identified in the γ region, including two similarly intense features centered at 31 000 cm–1 with a low-energy shoulder at 28 000 cm–1.
Figure 2.
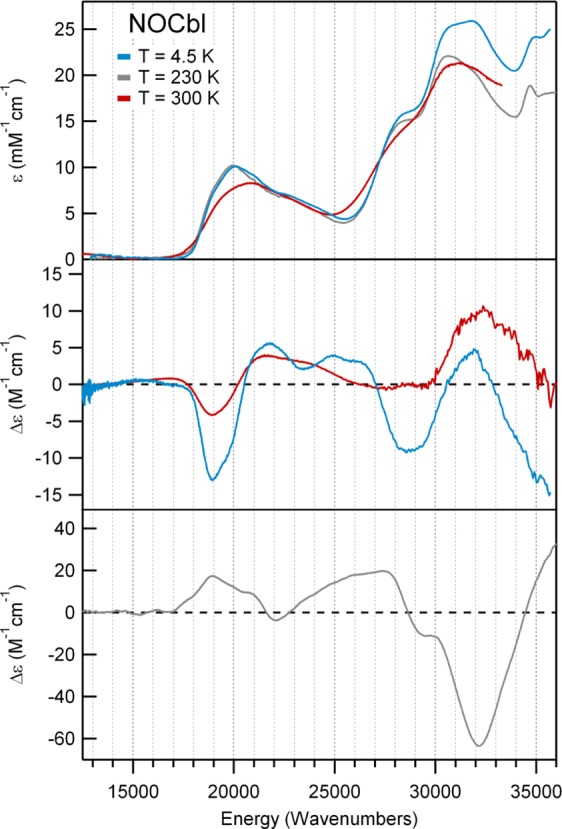
Abs (top), CD (center), and 7 T MCD (bottom) spectra at various temperatures of NOCbl.
Upon substitution of the DMB ligand of NOCbl with a more weakly σ-donating water molecule in NOCbi+, a ∼1000 cm–1 blue shift of the α/β bands is observed (Figures 3, top). Additionally, a positively signed feature at 18 500 cm–1 appears in the CD spectrum of NOCbi+ that has no counterpart in the NOCbl spectrum, while the prominent negative feature at 19 000 cm–1 decreases in intensity and shifts to higher energy (Figure 3, middle). Because the MCD spectra of NOCbl and NOCbi+ are essentially temperature-independent in the 4.5 to 50 K range (Supporting Information, Figures S4 and S5), it can be concluded that both species possess diamagnetic (S = 0) ground states, consistent with a Co(III)/NO– oxidation state assignment. Previous studies of other Co(III) corrinoids have revealed that the spectral changes in the α/β region that occur in response to a DMB → H2O lower ligand substitution reflect a stabilization of the HOMO relative to the LUMO, the extent of which depends on the σ-donating strength of the upper axial ligand.20 As the blue shift of the α/β bands from NOCbl to NOCbi+ (∼1000 cm–1) is considerably smaller than the shift observed from MeCbl to MeCbi+ (∼2500 cm–1),4 NO may appear to be a weaker σ-donating ligand than a methyl group. However, since the α/β bands of NOCbl occur at higher energies than those of MeCbl and all other alkylcobalamins,4 their small blue shifts from NOCbl to NOCbi+ could also be due to the fact that the DMB moiety is only weakly interacting with the Co ion in the former species, as suggested by the unusually long Co–N(DMB) bond observed in the crystal structure of NOCbl.16
Figure 3.
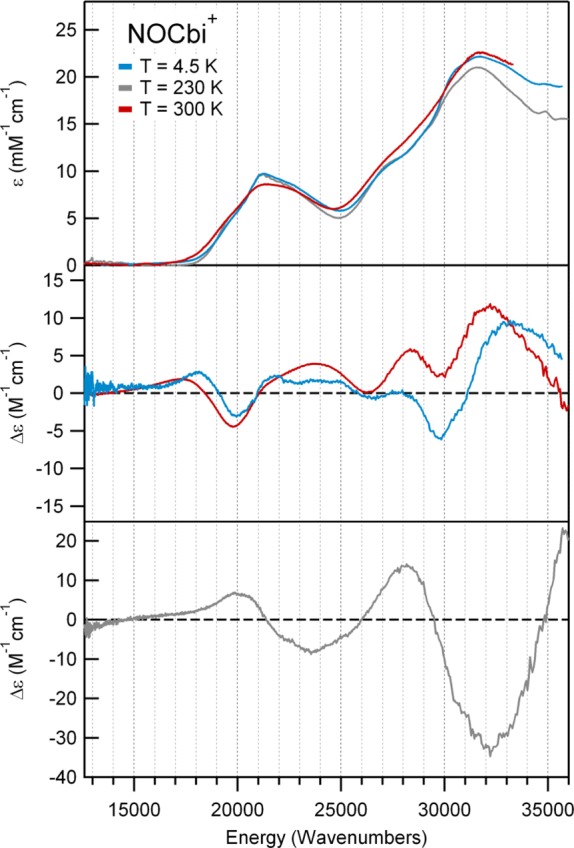
Abs (top), CD (center), and 7 T MCD (bottom) spectra at various temperatures of NOCbi+, a spectroscopic model of base-off NOCbl.
Intriguingly, while the Abs and CD spectra of NOCbi+ obtained at 4.5 and 300 K differ insignificantly with respect to band positions (Figure 3, top and middle), the Abs spectrum of NOCbl collected in fluid solution at 300 K shows the α/β bands blue-shifted by ∼850 cm–1 from their positions at 4.5 K (Figure 2, top). Significant temperature-dependent changes are also observed in the CD spectrum of NOCbl, most notably a drastic decrease in the intensity of the lowest-energy, negatively signed feature at ∼18 000 cm–1 and the appearance of a weak, positively signed feature at even lower energy (Figure 2, middle). As a result, the room-temperature Abs and CD spectra of NOCbl quite closely resemble those of NOCbi+. Collectively, these results suggest that by increasing the temperature from 4.5 to 300 K, the base-on to base-off equilibrium for NOCbl changes, favoring dissociation of the N(DMB) ligand at high temperatures. However, consistent with published NMR results,2 a sizable fraction of NOCbl must remain in the base-on state even under ambient conditions, since the 300 K Abs spectra of NOCbl and NOCbi+ are not superimposable (see Supporting Information, Figure S1). Yet, while the NMR data were interpreted to indicate that at room temperature ∼67% of the NOCbl molecules are in the base-on state, the traces obtained by adding the 4.5 K Abs or CD spectra of NOCbl (scaled by 0.67) and NOCbi+ (scaled by 0.33) differ from the 300 K Abs and CD spectra of NOCbl. This finding suggests that the Co–N(DMB) bond of base-on NOCbl is longer at 300 K than it is at 4.5 K.
II. Resonance Raman Data
The 77 K rR spectrum of NOCbl obtained with laser excitation at 488.0 nm (20,490 cm–1) shows strong enhancement of four features between 1450 and 1650 cm–1. On the basis of our recent study of the vibrational properties of vitamin B12 and its reduced forms,44 all of these features can be attributed to corrin-based vibrational modes. Three of them (termed νS1–νS3) arise from totally symmetric modes (assuming an effective Cs symmetry, with the pseudo mirror plane being oriented along the Co and C10 atoms and perpendicular to the corrin ring plane) and are thus particularly strongly resonance enhanced, while the fourth (νas) is associated with an asymmetric stretching mode. A more detailed analysis of these modes within the framework of our DFT frequency calculations is provided below.
A comparison of the low-energy regions of the rR spectra of NOCbl and its 15NOCbl isotopomer reveals an isotope-sensitive feature at 532 cm–1 (Figure 4, top), a region where the Co–NO stretching and Co–N–O bending modes are expected to occur.10 Subtraction of the 15NOCbl from the NOCbl trace yields a difference spectrum that shows an apparent shift of the 532 cm–1 feature to 496 cm–1 upon 14NO → 15NO substitution. An analogous isotope-sensitive feature is present in the rR spectrum of base-off NOCbl (our model of NOCbi+, Figure 4, bottom traces), though in this case a much better-resolved difference spectrum is obtained. Closer examination of this difference spectrum clearly discloses the presence of a shoulder on the low-energy side of the positive feature, suggesting that two isotope-sensitive modes actually occur in this region. Indeed, a Gaussian deconvolution of the rR spectra in Figure 4 reveals that the vibrational modes of base-on and base-off NOCbl at 515 and 532 cm–1 shift to 500 and 521 cm–1, respectively, upon 14NO → 15NO substitution (see Supporting Information, Figure S9). In a previous rR study of NOCbl, a single isotope-sensitive peak was observed at 514 cm–1 that shifted to 496 cm–1 upon 14NO →15NO substitution. However, the spectral resolution of these published data appears to be relatively low, as only two high-energy (>1,500 cm–1) features associated with corrin-based modes could be identified,10 compared to the four features that are readily apparent in our spectra (Figure 5). Additionally, considering that mode coupling typically leads to lower isotope shifts than expected, the reported isotope shift of 18 cm–1, which largely exceeds the 14 cm–1 decrease in vibrational frequency calculated using a harmonic oscillator model for a localized Co–N stretching mode, seems unreasonably large.
Figure 4.
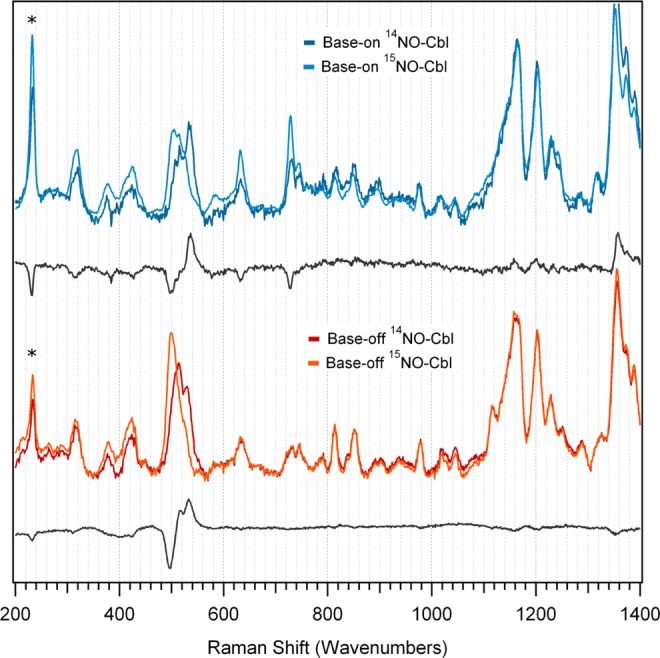
Low-energy region of rR spectra of NOCbl and its 15NO-enriched isotopomer in the base-on (top) and base-off (bottom) conformations, obtained at 77 K with 488 nm (20 491 cm–1) laser excitation. A difference spectrum for each conformation is included below the two data sets to highlight the isotope-sensitive vibrational features. Ice peaks are marked with asterisks.
Figure 5.
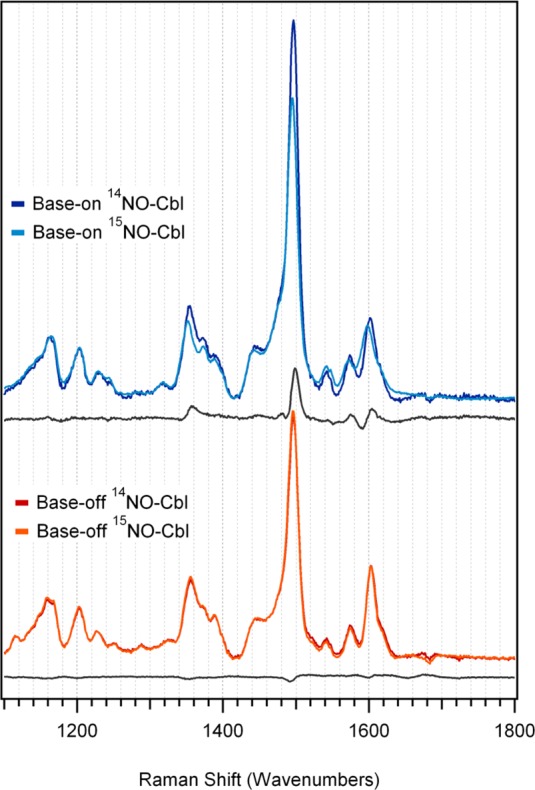
High-energy region of rR spectra of NOCbl and its 15NO-enriched isotopomer in the base-on and base-off conformations, obtained at 77 K with 488 nm (20 491 cm–1) laser excitation. A difference spectrum for each conformation is included below the two data sets to highlight the isotope-sensitive vibrational features.
The high similarity between the low-energy regions of the rR spectra of base-on and base-off NOCbl (Figure 4) indicates that the Co–NO bonding interaction is largely unperturbed by the DMB → H2O lower-ligand substitution. Consistent with this conclusion, the rR spectra of fluid solution samples of base-on and base-off NOCbl are very similar to each other as well as to the corresponding 77 K spectra (see Supporting Information, Figures S7 and S8), despite the thermochromism exhibited by NOCbl.
III. Computational Studies
DFT has been used by us with great success for the study of corrinoids in their Co(III), Co(II), and Co(I) oxidation states.19 While different functionals have been shown to provide variable descriptions of spectroscopic properties,45 careful evaluations of the computational results on the basis of experimental data have afforded a detailed understanding of the chemical and spectroscopic properties of a large number of different corrinoid species.19,4,23,21,27,46 From a recent study of MeCbl, Kozlowski and co-workers have suggested that pure GGA functionals, rather than hybrid functionals, provide DFT results more consistent with correlated wave function-based methods.47 Furthermore, work by our group20 and others48 has revealed that complete cofactor models should be used to obtain accurate geometric structures of cobalamins, while truncated models can be used to compute various spectroscopic parameters. In light of these findings, we performed geometry optimizations of complete NOCbl and NOCbi+ species using the pure PBE functional. The resulting structures were then suitably truncated to predict the Abs and CD spectra of these species using TDDFT, as well as to analyze their vibrational properties (see Methods for details). For comparison, we also carried out full geometry optimizations of these truncated models and calculated their Abs and CD spectra.
i. Geometries
The most significant differences between the X-ray crystal structure and our DFT-optimized model of NOCbl include the Co–N(DMB) bond distance and the folding of the corrin ring. While the crystal structure of NOCbl shows an unusually long Co–N(DMB) bond of 2.35 Å, our computation predicts this bond to be elongated by an additional 0.16 Å, to 2.51 Å (see Table 1). A further elongation of this bond by 0.1 Å, along with a tilting of the DMB ring plane relative to the Co–N(DMB) bond vector, occurred during the geometry optimization of our truncated model lacking the propionamide side chains. Although DFT geometry optimizations of cobalamin models consistently overestimate the Co–N(DMB) bond distance by ∼0.1 Å,20,49 the elongated Co–N(DMB) bonds in our models may also stem from the neglect of intermolecular interactions that modulate the length of this bond in the solid state and in solution. Regardless, these findings support the notion that the Co–N(DMB) bond of NOCbl is very weak and can thus be stretched, or potentially even broken, quite readily.
Table 1. Relevant Structural Parameters of NOCbl and NOCbi+, as Derived Experimentally or Obtained Computationally.
| species | model | Co–Xupper (Å) | Co–Xlower (Å) | N–O (Å) | ∠Co–N–O (0) | θ(LA) (0) | Φ(SA) (0) |
|---|---|---|---|---|---|---|---|
| NOCbl | crystala | 1.91 | 2.35 | 1.14 | 119.4 | 12.4 | 7.5 |
| full | 1.87 | 2.51 | 1.19 | 119.8 | 9.2 | 8.9 | |
| truncated | 1.87 | 2.61 | 1.18 | 119.3 | 3.3 | 5.8 | |
| NOCbi+ | full | 1.85 | 3.58 | 1.18 | 120.9 | 9.2 | 10.43 |
| truncated | 1.85 | 2.98 | 1.18 | 120.2 | 3.6 | 4.5 |
From ref (16).
Distortions of the corrin ring can be assessed on the basis of the long-axis and short-axis fold angles, θ(LA) and Φ(SA), respectively. θ(LA) is defined here as the angle between the plane containing the NA, C4, C5, C6, and NB atoms and the plane containing the NC, C14, C15, C16, and ND atoms, while Φ(SA) corresponds to the angle between the planes containing the ND, C19, C1, and NA atoms and the NB, C9, C10, C11, and NC atoms (see Figure 1 for the atom numbering scheme used). As such, θ(LA) correlates with the amount of “butterfly fold” of the corrin ring and is thus particularly sensitive to the positioning or removal of the bulky DMB ligand. Alternatively, Φ(SA) reflects the extent of “ring ruffling” due to steric constraints imposed on the propionamide and methyl groups on the A and D rings. As shown in Table 1, the θ(LA) values derived from the crystal structure and DFT-optimized complete model of NOCbl vary quite substantially, as expected from the sizable variation in the corresponding Co–N(DMB) bond distances. In contrast, the Φ(SA) values are nearly identical, suggesting that the corrin ring conformation observed experimentally is minimally affected by crystal-packing effects and/or intermolecular H-bonding interactions. The fact that DFT predicts both fold angles to decrease quite substantially upon removal of the propionamide side chains is consistent with our previous finding that distortions along the θ(LA) and Φ(SA) coordinates require very little energy.50
Despite these modest discrepancies between the experimentally determined and DFT-optimized NOCbl structures, the metric parameters for the Co–NO unit agree very well. Specifically, the Co–N(O) distances and Co–N–O bond angles are identical to within 0.04 Å and <1°, respectively. Notably, the Co–N(O) bond is significantly shorter than the upper axial ligand–Co bond distances reported for all other Co(III)Cbl species,51,52 suggesting that this bond is exceptionally strong. Although the computation appears to overestimate the N–O bond distance by 0.05 Å, note that the value of 1.14 Å determined experimentally is surprisingly small, given that (i) the N–O bond distance in nitric oxide is 1.15 Å53 and (ii) the NO ligand in NOCbl acquires a partial negative charge and thus increased π-antibonding electron density (vide infra).
Owing to the overall good agreement between the X-ray crystal structure and DFT-optimized model of NOCbl, a closer examination of the energy-minimized NOCbi+ model is warranted. As expected, replacing the bulky DMB base with a water molecule in the lower axial position causes sizable changes to both of the corrin fold angles (Table 1). More importantly, the optimized Co–O(H2) distance in our complete NOCbi+ model is 3.58 Å, suggesting that the Co ion resides in an effective five-coordinate ligand environment. Although this distance shortens by 0.60 Å upon removal of the propionamide side chains, the “lone pairs” on oxygen in our truncated model are not actually oriented properly to engage in a bonding interaction with the Co ion. A closer inspection of the complete NOCbi+ model reveals that several of the propionamide side chains form a cage around the H2O molecule. The fact that removing these side chains causes a large rearrangement of the water molecule suggests a role of the propionamide groups in modulating the ligation of the lower ligand by imposing steric constraints and/or providing a solvent-protected pocket for the ligand.
Finally, as expected in light of the strong Co–NO bonding interaction in NOCbl, the Co–NO unit is largely unaffected by the DMB → H2O lower-ligand substitution (Table 2, see NBO analysis for more details), with the most notable change being a small (0.02 Å) decrease in the Co–N(O) bond distance. A similar shortening was predicted previously for the Co–C(methyl) bond upon conversion of MeCbl to MeCbi+.4 Hence, despite the long Co–N(DMB) bonds in NOCbl and MeCbl, the base does play a small role in modulating the upper axial bonding interactions in these species.
Table 2. Summary of the Results Obtained from a Natural Population Analysis (NPA) for the NOCbl, NOCbi+, and O2Cbl Truncated Models.
| natural
charge |
natural bond order |
|||||
|---|---|---|---|---|---|---|
| species | Co | Xupper | Xlower | Co–Xupper | Co–Xlower | Xupper–O |
| NOCbl | 0.82 | 0.15 | –0.47 | 0.82 | 0.07 | 1.98 |
| NOCbi+ | 0.82 | 0.16 | –0.96 | 0.84 | 0.02 | 1.98 |
| O2Cbl | 0.84 | –0.14 | –0.42 | 0.47 | 0.25 | 0.75 |
ii. TDDFT Results
The computed Abs spectrum for the NOCbl model derived from the complete structure correctly predicts the major features observed experimentally, including the presence of two clusters of intense features near 20 000 cm–1 and 31 000 cm–1, corresponding to the α/β region and the γ region, respectively (Figure 6, top). The computed CD spectrum is also in reasonable agreement with our experimental spectrum, both exhibiting a derivative-shaped feature associated with two oppositely signed transitions centered at 20 000 cm–1 and a series of features with alternating signs in the γ region (Supporting Information, Figure S10, top). While the computed Abs and CD spectra for the optimized truncated NOCbl model are similar to those obtained with the modified complete model, the α/β bands are blue-shifted by 900 cm–1, and the lowest-energy, negatively signed feature in the CD spectrum is considerably weaker, which agrees less well with our experimental data (Supporting Information, Figure S11).
Figure 6.

Abs spectra of NOCbl (top) and NOCbi+ (bottom) collected at 4.5 K (dotted lines) superimposed on the TD-DFT results. The TD-DFT computed transitions (vertical sticks) were convoluted with Gaussian bands with a constant fwhm of 1250 cm–1 to obtain the predicted spectra plotted in dark blue for NOCbl and in light blue for NOCbi+. In each case, the EDDM for the α-band transition is shown on the left, where regions of loss and gain of electron density are shown in gray and gold, respectively. The calculated spectra were uniformly red-shifted by 2200 cm–1 to facilitate a direct comparison with the experimental results.
Upon DMB → H2O lower-ligand substitution, the α/β bands in the TDDFT-computed Abs and CD spectra blue shift by ∼3500 cm–1, and the negative CD feature at 20 000 cm–1 decreases in intensity, causing an apparent blue shift of the derivative-shaped feature to 21 000 cm–1 (Figure 6, bottom, Supporting Information, Figure S10, bottom). As expected on the basis of the negligible Co–O(H2) bonding interactions in the two different NOCbi+ models, the Abs and CD spectra predicted for these species are almost identical (Supporting Information, Figure S12). Overall, the computed Abs spectra for both NOCbi+ models agree quite well with the experimental spectrum across the entire spectral range investigated, except for the presence of a small feature at ∼15 000 cm–1 that is not observed experimentally. In contrast, the computed CD spectra only modestly reproduce the experimental spectrum, which is not unexpected because magnetic-dipole allowed transitions pose a significant challenge for TDDFT computations.54
iii. Vibrational Frequencies
While considerable differences are observed between the experimental Abs, CD, and MCD spectra of NOCbl and NOCbi+ (cf. Figures 2 and 3), the rR spectra obtained for these species show minimal differences with respect to the frequencies of the major corrin and Co–NO-based modes (vide supra). For this reason, and for computational practicality, the truncated NOCbl and NOCbi+ models were employed in our DFT-assisted vibrational analysis. The DFT-computed off-resonance Raman spectra for these models exhibit four relatively intense features in the 1450–1650 cm–1 region (Supporting Information, Figure S13 and S14). A graphical representation of the corresponding normal modes of NOCbi+ (which are very similar to those of NOCbl) in terms of atomic displacement vectors is provided in Figure 7. The three modes that retain the approximate Cs symmetry of NOCbi+ are expected to be most strongly resonance-enhanced, because only totally symmetric modes can couple to electronic transitions and thus gain rR intensity via the Franck–Condon mechanism.55,56 Even though DFT methods typically overestimate vibrational frequencies, the agreement between the experimental and computed frequencies for these modes is excellent, thus permitting a straightforward assignment of the relevant vibrational features. Specifically, the 1497 cm–1 feature observed experimentally is assigned to the formally totally symmetric νs1 mode predicted at 1506 cm–1, which involves nuclear motion primarily along the short axis of the corrin ring. Alternatively, the 1541 and 1603 cm–1 features in the experimental rR spectra are attributed to the νs2 and νs3 modes predicted at 1554 and 1613 cm–1, respectively. These modes mainly entail C–N stretching motion along the long axis of the corrin ring and symmetric methine stretching motion, respectively. The remaining feature observed at 1572 cm–1 is then assigned to the antisymmetric νas mode predicted at 1585 cm–1. While the large depolarization ratio (0.71) computed for νas suggests that this mode should not be resonance-enhanced, it is predicted to carry significant off-resonance Raman intensity and to entail large displacements of corrin ring atoms, thus facilitating intensity enhancement via Herzberg–Teller coupling.55,56 Overall, these assignments are consistent with our vibrational analysis recently carried out for CNCbl and its reduced derivatives.44
Figure 7.
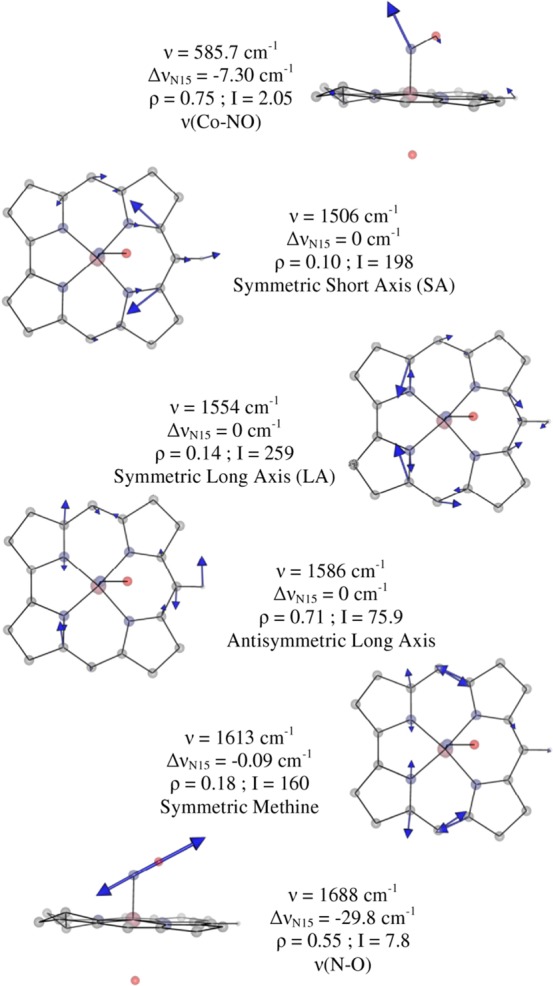
Eigenvector representations of the relevant normal modes for the truncated NOCbi+ model, as obtained with DFT. The computed frequency (ν), isotope shift upon 14NO → 15NO substitution (ΔνN15), depolarization ratio (ρ), off-resonance Raman intensity (I), and assignment are shown for each mode.
Our calculations for NOCbi+ also predict two modes at 569 and 585 cm–1 with 14NO → 15NO isotope shifts of ΔνN15 = −7.3 and −6.0 cm–1, respectively. These predictions agree reasonably well with the frequencies (515 and 532 cm–1) and isotope shifts (ΔνN15 = −15 and −11 cm–1) observed experimentally. Both of these modes involve large atomic displacements along the Co–N(O) stretching and Co–N–O bending coordinates and are coupled out of phase and in phase, respectively, with corrin ring breathing modes. As such, the distinction between bending and stretching modes becomes ambiguous in this case. Only one additional 14NO → 15NO isotope-sensitive mode is computed at 1688 cm–1, corresponding to a relatively pure N–O stretch. While this mode is predicted to be strongly IR active, it carries negligible intensity in the computed off-resonance Raman spectrum (Supporting Information, Figures S13 and S14, respectively). This observation, as well as the lack of mechanical coupling between the N–O and corrin ring stretches and the weak electronic coupling between the NO moiety and corrin π system (see below) predicted computationally are consistent with the absence of a feature attributable to the N–O stretch in our experimental rR spectra of NOCbl and NOCbi+.
iv. DFT-Computed Molecular Orbital Diagrams
The good agreement between the experimental and computed Abs, CD, and vibrational data presented above indicates that DFT successfully models the salient bonding interactions present in NOCbl and NOCbi+. For both species, the relative energies of the Co 3d-based MOs reflect the strongly σ- and weakly π-donating nature of the tetradentate corrin ligand. Among these orbitals (see Figure 8), the Co 3dx2–y2-based MO (#158 in the case of NOCbl) is essentially nonbonding and thus lowest in energy, whereas the Co 3dxy-based MO (distributed over MOs #167 and #168 due to mixing with another, energetically proximate MO) is strongly σ-antibonding with respect to the Co–N(corrin) bonds and therefore highest in energy (note that the x and y axes are rotated by 45° about the z axis from their usual orientations due to the Co–N(corrin) π-bonding interactions). Intriguingly, the HOMO and LUMO of NOCbl derive mostly from Co 3d and NO π*orbitals, in contrast to the case of alkylcobalamins (e.g., MeCbl) where these MOs are primarily corrin π/π*-based.4 Specifically, the HOMO of NOCbl contains 28% Co 3dz2 and 40% NO π∥* orbital contributions (to differentiate between the two NO π* orbitals, the one with its lobes roughly parallel to the Co–N(O) bond axis will be denoted as π∥* and the other as π⊥*), making it strongly σ-bonding with respect to the Co–N(O) bond (see Figure 8 for MO plots and Supporting Information, Table S1 for compositions). Alternatively, the LUMO corresponds to the π-antibonding combination of the Co 3dyz and the NO π⊥* orbitals (15% and 78%, respectively), with the bonding counterpart being considerably lower in energy (MO #162).
Figure 8.
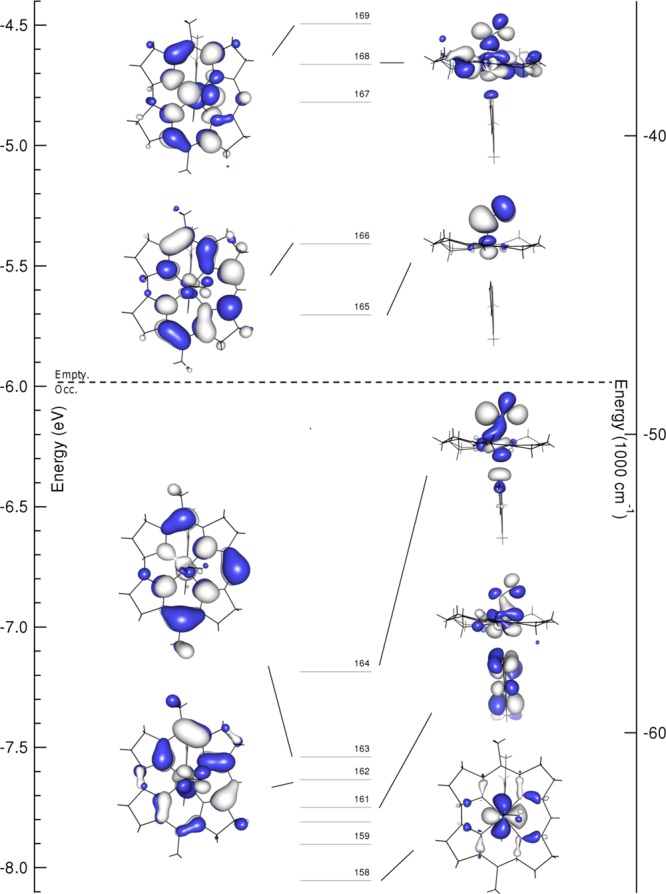
Isosurface plots of the relevant MOs of NOCbl. The dashed line separates occupied from empty orbitals.
Upon replacement of the axial DMB ligand of NOCbl with a more weakly σ-donating water molecule in NOCbi+, MOs with large Co 3dz2 orbital contributions (MOs #163, #164, #167, and #168 of NOCbl, corresponding to MOs #128, #130, #133, and #134 of NOCbi+, see Figure 9) are stabilized by ∼0.1 eV relative to the Co 3dx2–y2-based MO, which was chosen as the reference point because its composition remains essentially unchanged from NOCbl to NOCbi+ (see Supporting Information, Tables S1 and S2). In particular, the Co 3dz2/NO π∥*-based HOMO of NOCbi+ (#130) loses the σ-antibonding interaction with the lower axial ligand and gains contributions from the corrin π7 MO. This mixing causes a large stabilization of the corrin π7-based MO #128 relative to the corrin π8-based MO #132 of NOCbi+ (corresponding to MOs #163 and #166 of NOCbl).
Figure 9.
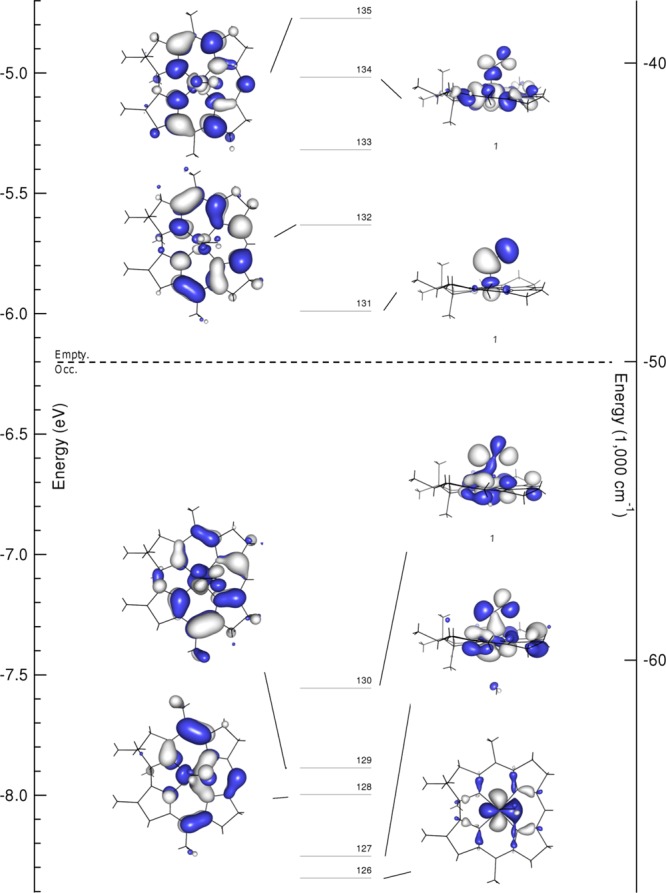
Isosurface plots of the relevant MOs of NOCbi+. The dashed line separates occupied from empty orbitals.
v. TDDFT-Assisted Spectral Assignments
Given the satisfactory agreement between the experimental and computed spectroscopic data of NOCbl and NOCbi+, it is reasonable to assign the key Abs features of these species within the framework of the DFT-based MO descriptions as provided by the TDDFT results. Because in the TDDFT formalism electronic transitions are described as linear combinations of one-electron excitations between occupied and virtual MOs, it becomes difficult to identify the nature of a given transition in cases where multiple excitations make significant contributions. One approach to overcome this limitation involves computing electron difference density maps (EDDMs), which provide a visual representation of the changes in electron density accompanying an electronic transition. On the basis of such EDDMs (Figure 6), the electronic transitions producing the dominant contributions to the TDDFT-computed Abs spectra can be assigned as shown in Table 3 for NOCbl and Table 4 for NOCbi+. In the case of NOCbl, the five lowest-energy transitions carrying significant Abs intensity give rise to one broad feature in region i of the computed Abs spectrum (Figure 6, top), with two transitions (involving excited states 12 and 15) having the largest oscillator strengths. On the basis of its EDDM, transition 12 primarily entails a one-electron excitation from the corrin π7-based MO #163 to the corrin π8-based MO #166, characteristic of the transition that is generally responsible for the α/β bands. The largest changes in electron density occur within the C9–C10–C11 fragment of the corrin ring, consistent with the strong rR enhancement of the νs1 mode observed experimentally (Figure 5).
Table 3. TDDFT-Calculated Energies (in cm–1), Polarizations, Oscillator Strengths, and Percent Contributions from One-Electron Excitations for the Major Electronic Transitions of NOCbl (Band Assignments Are Shown in Figure 6). Donor and Acceptor MOs Are Labeled According to Their Predominant Co/Corrin-π Character, with Percent Co Contributions Shown in Parentheses.
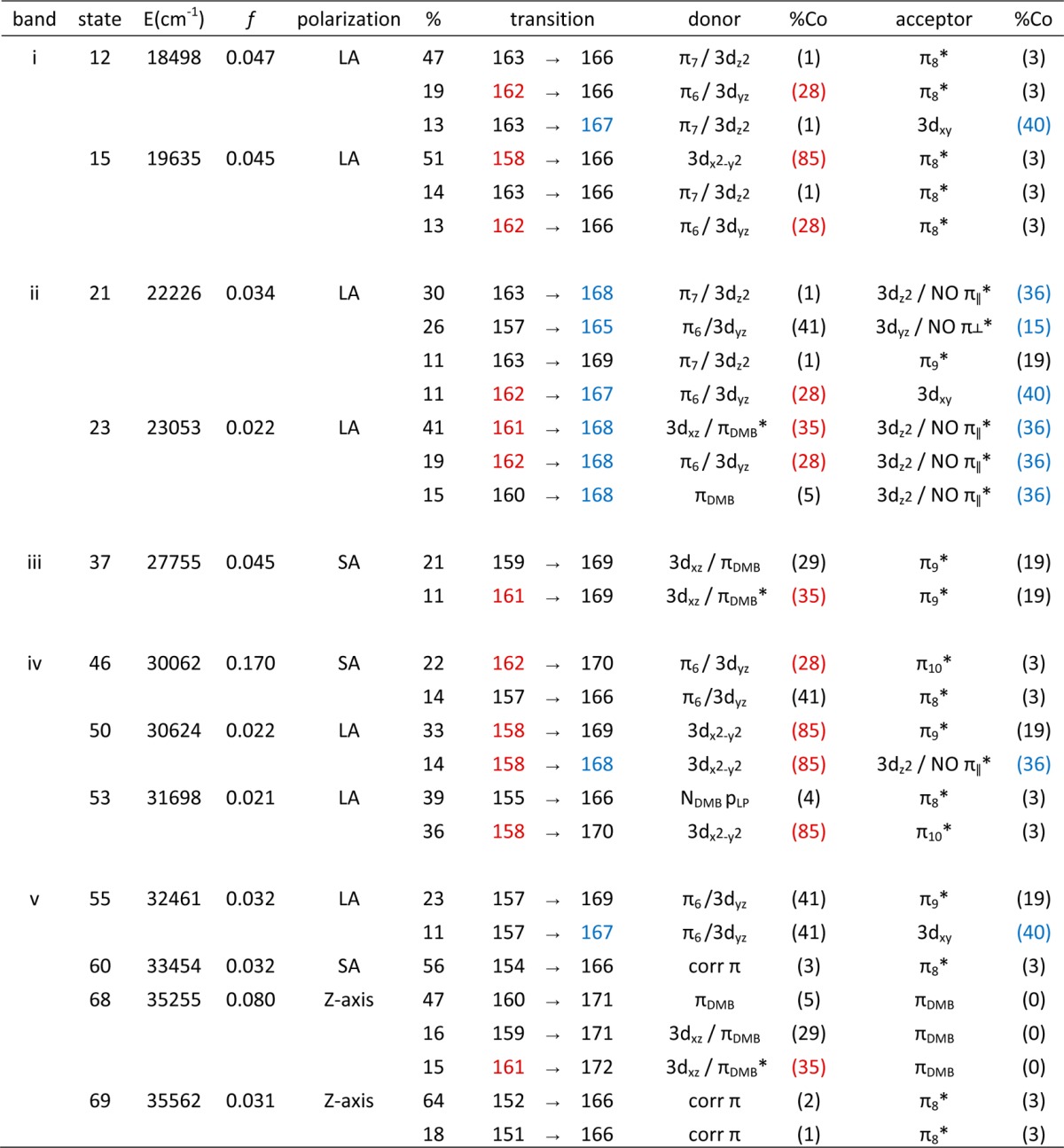
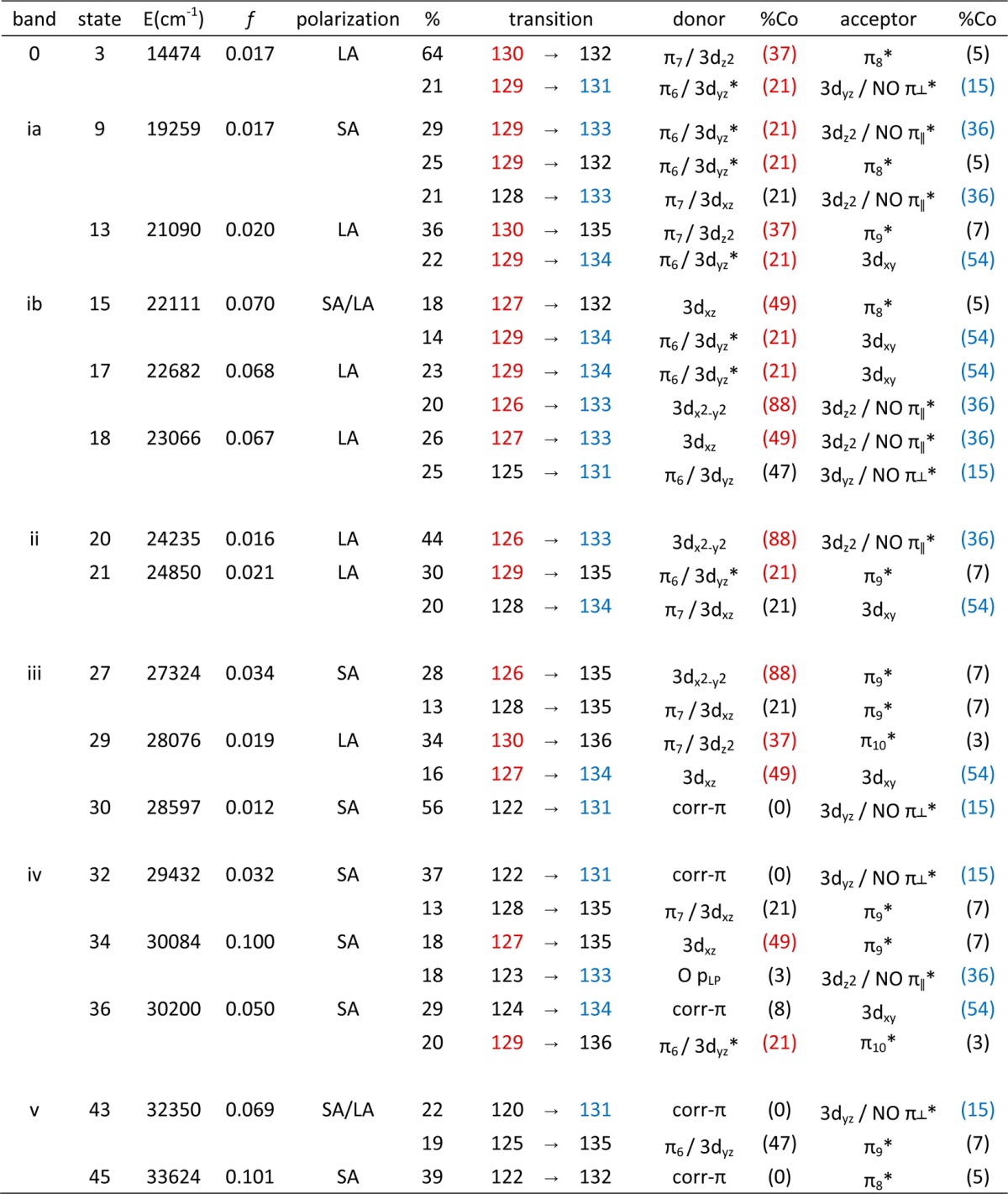
Table 4. TDDFT-Calculated Energies (in cm–1), Polarization, Oscillator Strengths, and Percent Contributions from One-Electron Excitations for the Major Electronic Transitions of NOCbi+ (Band Assignments Are Shown in Figure 6). Donor and Acceptor MOs Are Labeled According to Their Predominant Co/Corrin-π Character, with Percent Co Contributions Shown in Parentheses.
The EDDMs for the dominant transitions in regions ii (excited states 21, 23, and 26) and iii (excited state 37) display large changes in electron density around the DMB and NO ligands, due to one-electron excitations from MOs with significant orbital contributions from the DMB group. Lastly, regions iv and v are dominated by several intense corrin π → π* transitions that also cause a moderate electron density redistribution at the Co center as a result of the sizable Co 3d orbital character in the donor and acceptor MOs. The prediction of numerous intense features in the γ region of the computed Abs spectrum, as opposed to a single, relatively sharp band as is usually observed for Co(III)Cbl species,4 is consistent with the unique Abs spectrum obtained experimentally for NOCbl.
From NOCbl to NOCbi+, TD-DFT predicts the most intense feature in the visible region of the Abs spectrum to blue shift by 3500 cm–1, in qualitative agreement with the ∼1000 cm–1 shift of the α/β bands observed experimentally (Figure 6). However, additional features are present in the near-IR region of the computed Abs spectrum that have no discernible counterparts in the experimental spectrum. As revealed by their EDDMs (Supporting Information, Figure S15), the transitions associated with the first two features in regions 0 and ia cause a large electron redistribution within the Co–NO moiety (see Table 4). The fact that analogous features are not observed experimentally indicates that TDDFT incorrectly predicts the intensities and/or energies of these charge transfer (CT)-type transitions of NOCbi+; similar cases where TDDFT fails to properly describe CT excited states are well-documented in the literature.57,58 The remaining changes in the computed Abs spectra from NOCbl to NOCbi+ pertain to a significant intensity redistribution among the dominant features in the γ region, which is again in good qualitative agreement with our experimental results.
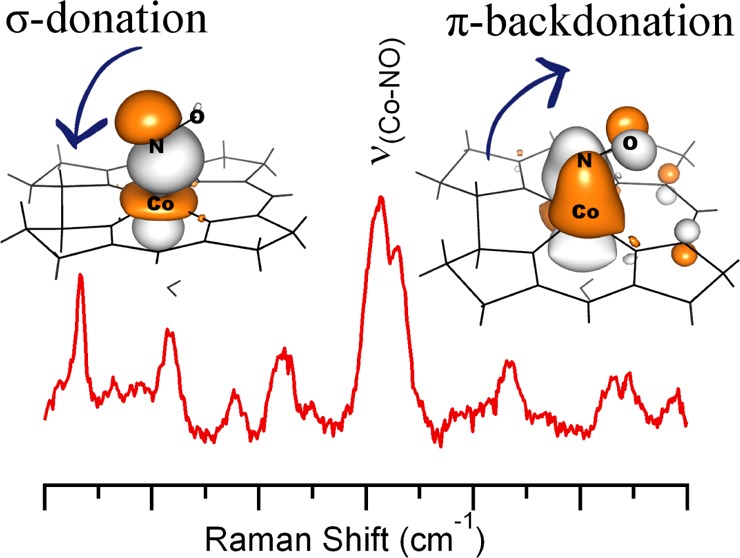
Because our TDDFT computations reproduce the key differences between the Abs spectra of NOCbl and NOCbi+ observed experimentally quite well, the computed MO diagrams can be used as the basis for exploring the electronic structural origin of these differences. As described above, the DMB → H2O ligand substitution causes a stabilization of MOs with significant Co 3dz2 orbital contributions relative to the other MOs. Of particular importance with respect to the α/β band transition is the stabilization of the corrin π7-based donor MO relative to the corrin π8-based acceptor MO from NOCbl (MOs #163 and #166) to NOCbi+ (MOs #128 and #132), which readily explains the blue shift of the α/β bands observed experimentally (Figures 2, 3, and 6). Interestingly, in the case of alkylcobalamins that also possess a strong σ-donor ligand in the upper axial position, the blue shift of the α/β bands in response to DMB → H2O ligand substitution is considerably larger than it is in the case of NOCbl; for example, ∼2500 cm–1 for MeCbl4 versus ∼1000 cm–1 for NOCbl. This difference can be understood in terms of the larger σ-donation from the upper ligand in NOCbl than in alkylcobalamins, which leads to a weaker Co–N(DMB) bond and, thus, to less pronounced changes in the electronic structure upon lower-ligand substitution in the former species. In fact, our computed MO description for NOCbl bears some intriguing similarities to that developed previously for Co(II)Cbl, especially with regard to the energies of the Co 3d-based MOs relative to the corrin π/π*-based MOs.23 This observation indicates that the high degree of σ-donation from the occupied NO π∥* orbital into the formally unoccupied Co 3dz2 orbital leads to a significant decrease in the effective nuclear charge experienced by the Co 3d orbitals in NOCbl. However, the NO → Co charge donation is partially compensated for by the strong π-backbonding interaction involving the doubly occupied Co 3dyz orbital and formally empty NO π⊥* orbital. A more detailed analysis of the salient Co–NO bonding interactions within the NBO formalism is presented next.
vi. NBO-Based Co–NO Bonding Description
While a relatively straightforward correlation can be established between the DFT/TDDFT-predicted and experimentally observed properties of NOCbl and NOCbi+, a direct evaluation of the key Co–NO bonding interactions is complicated by the partial delocalization of the relevant MOs over the corrin ring. To overcome this challenge, we resorted to the NBO formalism within which the calculated electron density is partitioned into chemically intuitive bonding MOs and electron pairs so as to generate a bonding description closely adhering to the Lewis structure formalism.59 Because the Co–N(O) bonding interactions in NOCbl and NOCbi+ are essentially identical as judged on the basis of our experimental rR data and DFT results (vide supra), the following analysis will focus on the electronic structure of NOCbi+. The relevant NBOs, labeled by their primary orbital contributors and classified either as bonding (BD) or antibonding (BD*) with respect to the Co–NO bond, are shown in Figure 10 (left). As expected on the basis of the DFT results described above, the Co 3dz2 BD-NBO is characterized by a highly covalent Co–NO σ-bonding interaction. However, shortcomings of the Lewis structure-like description for NOCbi+ are evident from the relatively large occupancies of several formally empty orbitals, most importantly the antibonding Co 3dxy BD*-NBO and Co 4s BD*-NBO, as well as the occupancies of considerably less than 2 of some of the formally doubly occupied orbitals. These observations indicate that deviations from a single Lewis structure, treated within the NBO framework as donor–acceptor hyperconjugation interactions,60 are important for properly describing the bonding interactions in NOCbi+. These interactions can be fully accounted for by a transformation of the canonical MOs to a set of natural localized MOs (NLMOs), which are constructed to retain a large amount of NBO character. These NLMOs possess integer occupancies, like the canonical MOs, and thus provide a useful connection between the canonical MOs and the NBOs.59,61,62
Figure 10.

Isosurface plots of the metal-based NBOs (in blue/gray) and corresponding NLMOs (in orange/gray) of NOCbi+. Occupancies are shown below each orbital. The composition of the NBOs and their percent contributions to the NLMOs are provided in Table 4
The relevant NLMOs for NOCbi+ are shown in Figure 10 (right), and their compositions are provided in Table 5. Inspection of the Co 3dz2 BD-NLMO discloses a very covalent σ-bonding interaction between the Co ion and NO (58% Co 3dz2 and 42% N(O) 2pz orbital contributions, see Table 5), as anticipated on the basis of the canonical MO and NBO compositions. Intriguingly, while the Co 3dz2 BD-NLMO as well as the nonbonding Co 3dx2–y2 and 3dxz NLMOs are very similar to their corresponding NBOs, the 3dyz BD-NLMO shows distinct differences. The composition of this NLMO reveals that it only retains 83.1% of “pure” Co 3dyz NBO character (in comparison to >95% for the other Co-based NLMOs) due to a sizable π-backbonding interaction between the Co 3dyz orbital and the NO π⊥* orbital (Figure 10, center columns). Since the Co 3dyz orbital only mixes with the NO π⊥* orbital (Figure 10), the amount of charge backdonation from Co to NO can be estimated to be ∼0.32 electrons. Because of the presence of this π-backbonding interaction, the Co ion does not retain a Co(II)-like electronic structure as might be anticipated on the basis of the large σ donation from the NO π∥* orbital to the Co 3dz2 orbital. Lastly, the unoccupied Co 3dz2 BD*-NLMO and Co 3dxy BD*-NLMO are σ-antibonding with respect to the corrin ring and the axial ligand orbitals, as expected, and retain >95% of NBO character (see Table 5 and Figure 10, outside columns).
Table 5. Summary of NBO and NLMO Results for NOCbi+. Energies, Occupancies, and Percent Co/NNO Compositions of the Co-Based NBOs Are Shown, in Addition to the Percent Contributions of These NBOs to the Corresponding NLMOs. Low-Occupancy NBOs, which Correspond to Formally Empty Orbitals, Are Highlighted in Reda.
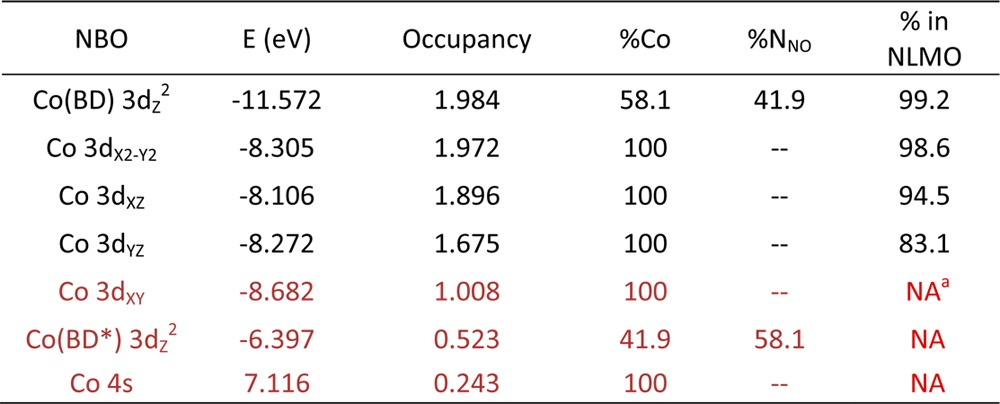
NA indicates values not availalable from NLMO composition analysis, but estimated to be close to 100%.
The NBO/NLMO results can also be used to estimate the number of electrons in the Co 3d atomic orbitals (AOs) and thus an effective oxidation state of the Co ion. As the number of d electrons is sensitive to the covalency of the metal–ligand bonding interactions as well as the delocalization of the 3d orbitals, the following approach was used to estimate this value (see Supporting Information for additional details). First, the %NBO composition of each NLMO (see Table 5) was used to calculate the fractional occupancy of the Co 3d-based NBO(s) contributing to this particular NLMO. This electron count was then partitioned into Co 3d-based and ligand-based contributions, estimated based on the %Co 3d contributions to each NBO. Finally, the fractional Co 3d electron counts for all NLMOs obtained using this procedure were added up to estimate the total number of Co 3d electrons. Using this approach, an effective number of Co 3d electrons of 6.27 was obtained for NOCbi+, larger than a value of 6 expected for a Co(III) species. This result corroborates the conclusion drawn from the energies and compositions of the canonical MOs regarding the relatively high Co(II) character in the electronic structures of NOCbl and NOCbi+. Thus, while the large amount of electron donation from NO to Co in the Co 3dz2 BD-NLMO is partially offset by the strong π-backbonding interaction in the Co 3dyz BD-NLMO, our NBO/NLMO analysis clearly shows that a Co(III)–NO– description for NOCbi+, and by analogy NOCbl, is inaccurate. Rather, these species are better described as resonance hybrids with both Co(III)–NO– and Co(II)–NO• limiting structures being major contributors.
Discussion
Given the roles of NO as a blood-pressure regulator, neurotransmitter, and second messenger,63 as well as a cytotoxic agent in immunological response,63 the ability of Co(II)Cbl to scavenge this molecule in vivo to form NOCbl is of considerable interest.9,10 Additionally, NOCbl has been shown to be potentially useful for targeted NO delivery as a chemotherapeutic or vasodilating agent,64,12 since its Co–NO bond can be broken under physiological conditions for controlled NO release. Thus, elucidating the nature of the bonding interaction between the NO ligand and the Co center in NOCbl represents an important step toward the development of an improved understanding of the chemical and biological properties of this molecule. Previous reports have highlighted the unique structural properties of NOCbl relative to other biologically relevant cobalamins, most notably the presence of an unprecedentedly long Co–N(DMB) bond in this species. However, because NOCbl exhibits similar spectroscopic signatures as the well-characterized alkyl-Co(III)Cbls, such as MeCbl and AdoCbl,4 it has generally been described as a Co(III)–NO– species.
To test this assumption, we developed experimentally validated electronic structure descriptions for NOCbl in its base-on and base-off states. While our Abs spectrum of NOCbl in fluid solution at 300 K is similar to those reported in the literature,25,2 we discovered a previously unobserved thermochromism for this species (Supporting Information, Figures S3). From a comparison to the Abs data obtained for NOCbi+, a model of base-off NOCbl, we conclude that an increase in temperature from 4.5 to 300 K causes a shift in the base-on to base-off equilibrium for NOCbl, favoring dissociation of the N(DMB) ligand at high temperatures. This change in Co coordination environment has, however, negligible effects on the Co–NO bonding interaction, as evidenced by our rR data, TDDFT results, and NBO analysis. The origin and implications of these findings are discussed below.
Co–NO Bonding
Collectively, our spectroscopic and computational data reveal that NOCbl is inadequately described as a Co(III)–NO– species, because significant charge donation from the NO ligand notably alters the effective nuclear charge of the metal center. Compared to the methyl group of MeCbl, the NO ligand of NOCbl is an even stronger σ-donor, therefore inducing additional Co 3dz2 orbital character into the HOMO and further enhancing the Co–N(DMB) σ-antibonding interaction in this orbital. Our DFT-computed MO description indicates that the formally unoccupied Co 3dz2 orbital contributes as much as 29% to the HOMO of NOCbl, as compared to 7% in MeCbl.4 This prediction is consistent with the observed lengthening of the Co–N(DMB) bond from 2.16 Å in MeCbl to 2.35 Å in NOCbl as determined by X-ray crystallography.65,16 The high degree of electron donation from NO is particularly evident from an analysis of the NLMOs and NBOs (Figure 10) derived from the canonical MOs obtained with DFT. On the basis of this analysis, the doubly occupied 3dz2 BD-NLMO contains 58% Co 3d orbital character (Table 5), consistent with a very covalent Co–NO bond. Another unique feature of the Co–NO bond in NOCbl and NOCbi+ compared to the axial bonds in other Co(III) corrinoid species is the presence of a sizable π-backbonding interaction involving the Co 3dyz and NO π⊥* orbitals. Inspection of the Co 3dyz BD-NLMO reveals that the NO ligand contribution to this orbital is 16%, leading to an estimate for the extent of π donation of ∼0.32 electrons. This backbonding partially compensates for the large amount of σ donation from the NO ligand into the Co 3dz2 orbital, and represents an unprecedented mechanism by which the electronic properties of the Co(III) ion can be modulated by the axial ligands.
The high degree of NO → Co σ donation in NOCbl weakens the Co–N(DMB) bond to the point that at high temperature, the base-on to base-off equilibrium favors a unique pentacoordinate species in aqueous solution. In the case of NOCbi+, an effectively five-coordinate species is present at all temperatures, suggesting that the strong trans influence exerted by the NO ligand precludes the binding of a weakly donating water molecule even at 4.5 K. Nonetheless, during the geometry optimization of a complete NOCbi+ model, the lower water ligand did not fully dissociate. A comparison of this structure and that obtained for the truncated model reveals major differences in terms of the orientation of the water molecule, indicating that the propionamide side chains of the corrin ligand, which were absent in the truncated model, may play an important role in modulating the lower axial bonding interaction in cobalamins.
Further experimental support for our bonding description for NOCbl is provided by published NMR data, which indicate that the 15N-resonance of the NO ligand undergoes a 40 ppm upfield shift upon protonation and dissociation of the DMB ligand in the lower axial position.11 Although the observed change in electron shielding experienced by the 15N nucleus was originally attributed to an increase in the Co–N–O bond angle from base-on to base-off NOCbl, our rR and DFT results provide compelling evidence that the Co–NO core structures of these two species are in fact virtually identical. Instead, our computational data suggest that the enhanced shielding of the 15N nucleus in base-off NOCbl reflects the weaker σ-antibonding interaction between Co and the lower axial ligand in that species (cf. MOs #164 of NOCbl and #130 of NOCbi+ in Figures 8 and 9, respectively), with the consequent increase in N(O) natural charge by 4% (see Table 2).
Extension to O2Cbl
Previous crystallographic26 and EPR studies66 have indicated that Co(II)Cbl binds molecular oxygen reversibly to yield superoxocobalamin (O2Cbl) as the first step in the autoxidation process that eventually yields H2OCbl+. O2Cbl may also be of physiological importance as free cobalamin is present predominantly in the Co(II)Cbl state in vivo.67 Given the similar frontier orbitals of NO and O2, which has been widely exploited to mimic the binding of dioxygen to metal centers in proteins by using nitric oxide, it is interesting to compare the electronic structures of NOCbl and O2Cbl. While crystallographic data for these species have revealed that both the NO and O2 molecules ligate to the Co center in a bent end-on fashion, the axial Co–N(DMB) bond lengths differ by 0.29 Å between NOCbl (2.35 Å)16 and O2Cbl (2.06 Å).26 Hence, the Co–N(DMB) bond length in O2Cbl is also considerably shorter than it is in MeCbl (2.16 Å)52 but very similar to that reported for CNCbl (2.041 Å),52 which is considered to possess a moderately strong σ donor in the upper axial position based on computational analyses and the fact that this species exhibits a “typical” Co(III)Cbl Abs spectrum.4
The NLMOs for O2Cbl are shown in Figure 11, and the compositions of the relevant NBOs and NLMOs are listed in Table 6 (note that our electronic structure description for O2Cbl is supported by a previous single-crystal EPR characterization of this species,66 given the good agreement between the computed and experimental g values and hyperfine coupling constants, see Table 7). Because of the spin-unrestricted formalism used in these calculations, the NLMOs have occupancies of 1.00 and are divided into a set of spin-up (α, majority spin. Figure 11, left) and spin-down orbitals (β, minority spin. Figure 11, right). Inspection of the NLMOs of O2Cbl reveals that these orbitals are very similar in composition to the NLMOs of NOCbl, although with some important differences. For both sets of spin orbitals, the Co 3dxz and Co 3dx2–y2 NLMOs of O2Cbl are nonbonding with respect to the ligand framework, in an analogous manner as the NLMOs of NOCbl. Additionally, the compositions of the β NLMOs of O2Cbl (Table 6, right) reflect the presence of a relatively covalent Co–O2 bond, with a Co 3d orbital contribution to the Co 3dz2(BD) β NLMO of 41%, as compared to 58% predicted for NOCbl (see Table 5). However, the Co contribution to the spin-up counterpart (i.e., the Co 3dz2(BD) α NLMO) is merely 24%. Thus, the net σ donation to the Co(III) ion from O2 in O2Cbl is smaller than it is from NO in NOCbl, which is consistent with the greater oxidizing power of O2 (E0 = −0.16 V vs NHE) versus NO (E0 = −0.8 V NHE).68 Likewise, the effective number of Co 3d electrons of O2Cbl, computed as described above for NOCbl, is 6.08, consistent with the classification of this species as a typical Co(III)Cbl species based on its short Co–N(DMB) bond. Thus, our computational results indicate that O2Cbl is adequately described as a Co(III)–O– species.
Figure 11.

Isosurface plots of the metal-based NLMOs of O2Cbl. The α orbitals (majority spin, on left) are shown in orange/gray, while β orbitals (minority spin, on right) are shown in yellow/gray. The horizontal dashed line separates singly occupied from empty orbitals. NLMOs are labeled according to the Co-based NBO providing the largest contribution. The compositions of the NLMOs in terms of the corresponding NBOs are provided in Table 5
Table 6. Summary of NBO and NLMO Results for O2Cbl. Energies, Occupancies, and Percent Co/OO2 Compositions of the Co-Based NBOs Are Shown for Alpha (Majority Spin) and Beta (Minority Spin) Orbitals, in Addition to the Percent Contributions of These NBOs to the Corresponding NLMOs. Low-Occupancy NBOs, which Correspond to Formally Empty Orbitals, Are Highlighted in Reda.
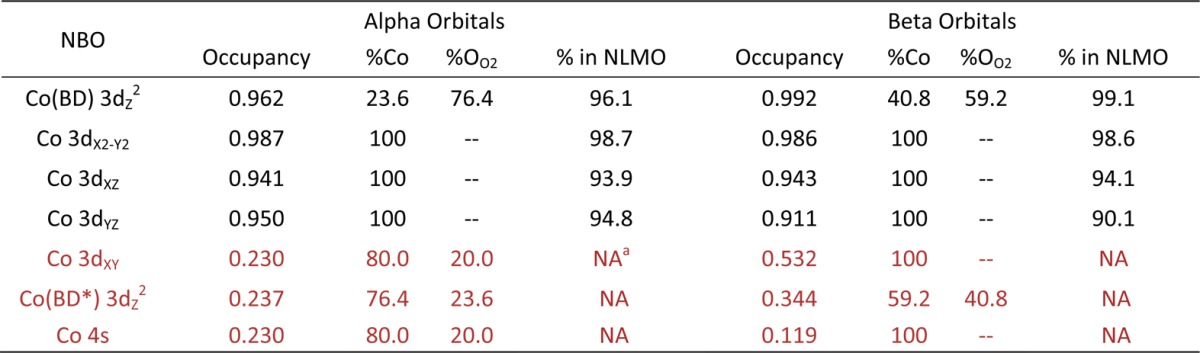
NA indicates values not availalable from NLMO composition analysis, but estimated to be close to 100%.
Table 7. Experimental and DFT-Computed Spin Hamiltonian Parameters for O2Cbl. Hyperfine Coupling Constants Are Given in Megahertz (MHz).
|
g values |
A(Co) |
A(17Oα) |
A(17Oβ) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g1 | g2 | g3 | A1 | A2 | A3 | A1 | A2 | A3 | A1 | A2 | A3 | |
| experimenta | 1.993 | 2.013 | 2.089 | –30 | –64.8 | –18.7 | –167 | 59 | *b | –201 | 70 | *b |
| DFT | 1.992 | 2.008 | 2.041 | –28 | –50.8 | –1.1 | –146 | 77 | 44 | –191 | 81 | 46 |
From ref (65).
Values estimated to be between 0 and 20 MHz.
Implications for Biological Systems
Biochemical studies have shown that NO can inhibit the two B12-dependent enzymes found in humans, namely, MetH and MMCM,7,8 which could provide an additional pathway for the regulation of these enzymes. Specifically, it was shown that inhibition of MMCM in the presence of NO donors resulted in the formation of NOCbl in the active site of the enzyme.8 For this and related enzymes, the formation of NOCbl in the active site and consequent inhibition of catalytic activity would require a repair mechanism involving NO dissociation or replacement of NOCbl by AdoCbl. Given the strong Co–NO bond in NOCbl as revealed by the present investigation, NOCbl removal from the active site of these enzymes is expected to be the main pathway for reactivation. This process could be facilitated by the strong trans influence exerted by the NO ligand. MMCM binds AdoCbl in the “base-off/His-on” conformation,6 and on the basis of the results obtained in this study, it can be speculated that NO coordination to the transiently formed Co(II)Cbl species in the enzyme active site will considerably weaken the Co–N(His) bond. Similarly, for B12-dependent enzymes that bind AdoCbl in the base-on conformation,69 the Co–N(DMB) bond elongation and additional geometric changes in response to NO coordination to the Co(II)Cbl intermediate will likely facilitate removal of the NOCbl product from the active site. Although more experimental evidence is needed to support these hypotheses, the results obtained from this study offer unprecedented insights into the spectral and electronic properties of NOCbl and provide useful spectroscopic markers for probing the interaction of this species with B12-dependent enzymes.
Acknowledgments
This work was supported in part by the National Science Foundation Grant Nos. MCB-0238530 (to T.C.B.) and CHE-0840494. I.G.P. was supported in part by Grant 5T32GM08349 via the Biotechnology Training Grant at University of WI-Madison. The authors thank Prof. Clark Landis for his assistance with the NBO analysis.
Supporting Information Available
Additional details regarding the synthesis of NOCbl and NOCbi+, rR spectra at room temperature as well as calculated rR/IR spectra, Gaussian deconvolution of rR spectra, tabulated values of orbital compositions, and further information regarding NBO calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hodgkin D. C.; Kamper J.; Mackay M.; Pickworth J.; Trueblood K. N.; White J. G. Nature 1956, 178, 64. [DOI] [PubMed] [Google Scholar]
- Hassanin H. A.; Hannibal L.; Jacobsen D. W.; Brown K. L.; Marques H. M.; Brasch N. E. Dalton Trans. 2009, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson J. D.; Zayas C. L.; Escalante-Semerena J. C. J. Bacteriol. 2003, 185, 7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich T. A.; Brooks A. J.; Buan N. R.; Brunold T. C. J. Am. Chem. Soc. 2003, 125, 5897. [DOI] [PubMed] [Google Scholar]
- Banerjee R. V.; Matthews R. G. FASEB J. 1990, 4, 1450. [DOI] [PubMed] [Google Scholar]
- Ludwig M. L.; Matthews R. G. Annu. Rev. Biochem. 1997, 66, 269. [DOI] [PubMed] [Google Scholar]
- Danishpajooh I. O.; Gudi T.; Chen Y. C.; Kharitonov V. G.; Sharma V. S.; Boss G. R. J. Biol. Chem. 2001, 276, 27296. [DOI] [PubMed] [Google Scholar]
- Kambo A.; Sharma V. S.; Casteel D. E.; Woods V. L.; Pilz R. B.; Boss G. R. J. Biol. Chem. 2005, 280, 10073. [DOI] [PubMed] [Google Scholar]
- Brouwer M.; Chamulitrat W.; Ferruzzi G.; Sauls D. L.; Weinberg J. B. Blood 1996, 88, 1857. [PubMed] [Google Scholar]
- Zheng D. H.; Birke R. L. J. Am. Chem. Soc. 2001, 123, 4637. [DOI] [PubMed] [Google Scholar]
- Wolak M.; Zahl A.; Schneppensieper T.; Stochel G.; van Eldik R. J. Am. Chem. Soc. 2001, 123, 9780. [DOI] [PubMed] [Google Scholar]
- Broderick K. E.; Alvarez L.; Balasubramanian M.; Belke D. D.; Makino A.; Chan A.; Woods V. L.; Dillmann W. H.; Sharma V. S.; Pilz R. B.; Bigby T. D.; Boss G. R. Exp. Biol. Med. 2007, 232, 1432. [DOI] [PubMed] [Google Scholar]
- Goodrich L. E.; Paulat F.; Praneeth V. K. K.; Lehnert N. Inorg. Chem. 2010, 49, 6293. [DOI] [PubMed] [Google Scholar]
- Enemark J. H.; Feltham R. D. Curr. Contents/Eng. Tech. Appl. Sci. 1988, 16. [Google Scholar]
- Wolak M.; van Eldik R. Coord. Chem. Rev. 2002, 230, 263. [Google Scholar]
- Hassanin H. A.; El-Shahat M. F.; DeBeer S.; Smith C. A.; Brasch N. E. Dalton Trans. 2010, 39, 10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemistry and Biochemistry of B12; Banerjee R., Ed.; John Wiley & Sons, Inc.: New York, 1999. [Google Scholar]
- Hamza M. S. A.; Zou X.; Brown K. L.; van Eldik R. Eur. J. Inorg. Chem. 2003, 268. [DOI] [PubMed] [Google Scholar]
- Brunold T. C.; Conrad K. S.; Liptak M. D.; Park K. Coord. Chem. Rev. 2009, 253, 779. [Google Scholar]
- Reig A. J.; Conrad K. S.; Brunold T. C. Inorg. Chem. 2012, 51, 2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liptak M. D.; Fleischhacker A. S.; Matthews R. G.; Telser J.; Brunold T. C. J. Phys. Chem. B 2009, 113, 5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. T.; Majtan T.; Freeman K. M.; Su Y.; Kraus J. P.; Burstyn J. N. Inorg. Chem. 2011, 50, 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich T. A.; Buan N. R.; Brunold T. C. J. Am. Chem. Soc. 2004, 126, 9735. [DOI] [PubMed] [Google Scholar]
- Park K.; Mera P. E.; Escalante-Semerena J. C.; Brunold T. C. Biochemistry 2008, 47, 9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V. S.; Pilz R. B.; Boss G. R.; Magde D. Biochemistry 2003, 42, 8900. [DOI] [PubMed] [Google Scholar]
- Hohenester E.; Kratky C.; Krautler B. J. Am. Chem. Soc. 1991, 113, 4523. [Google Scholar]
- Liptak M. D.; Brunold T. C. J. Am. Chem. Soc. 2006, 128, 9144. [DOI] [PubMed] [Google Scholar]
- ADF2012, S. In ADF2012, SCM; Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2012. http://www.scm.com. [Google Scholar]
- Velde G. T.; Bickelhaupt F. M.; Baerends E. J.; Guerra C. F.; Van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. J. Comput. Chem. 2001, 22, 931. [Google Scholar]
- Guerra C. F.; Snijders J. G.; te Velde G.; Baerends E. J. Theor. Chem. Acc. 1998, 99, 391. [Google Scholar]
- Vosko S. H.; Wilk L.; Nusair M. Can. J. Phys. 1980, 58, 1200. [Google Scholar]
- Becke A. D. Phys. Rev. A 1988, 38, 3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Phys. Rev. B 1986, 33, 8822. [DOI] [PubMed] [Google Scholar]
- Neese F.ORCA—An Ab initio, Density Functional, and Semiempirical Program Package, 2.9.1 ed.; Universität Bonn: Bonn, Germany, 2008. [Google Scholar]
- Bauernschmitt R.; Ahlrichs R. Chem. Phys. Lett. 1996, 256, 454. [Google Scholar]
- Casida M. E.; Jamorski C.; Casida K. C.; Salahub D. R. J. Chem. Phys. 1998, 108, 4439. [Google Scholar]
- Stratmann R. E.; Scuseria G. E.; Frisch M. J. J. Chem. Phys. 1998, 109, 8218. [Google Scholar]
- Hirata S.; Head-Gordon M. Chem. Phys. Lett. 1999, 302, 375. [Google Scholar]
- Hirata S.; Head-Gordon M. Chem. Phys. Lett. 1999, 314, 291. [Google Scholar]
- Eichkorn K.; Treutler O.; Ohm H.; Haser M.; Ahlrichs R. Chem. Phys. Lett. 1995, 240, 283. [Google Scholar]
- Schafer A.; Horn H.; Ahlrichs R. J. Chem. Phys. 1992, 97, 2571. [Google Scholar]
- Neese F.; Olbrich G. Chem. Phys. Lett. 2002, 362, 170. [Google Scholar]
- Glendening E. D.; Badenhoop J. K.; Reed A. E.; Carpenter J. E.; Bohmann J. A.; Morales C. M. Weinhold F.. NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, 2001. [Google Scholar]
- Park K.; Brunold T. C. J. Phys. Chem. B 2013, 117, 5397. [DOI] [PubMed] [Google Scholar]
- Andruniow T.; Kozlowski P. M.; Zgierski M. Z. J. Chem. Phys. 2001, 115, 7522. [DOI] [PubMed] [Google Scholar]
- Conrad K. S.; Brunold T. C. Inorg. Chem. 2011, 50, 8755. [DOI] [PubMed] [Google Scholar]
- Kornobis K.; Kumar N.; Lodowski P.; Jaworska M.; Piecuch P.; Lutz J. J.; Wong B. M.; Kozlowski P. M. J. Comput. Chem. 2013, 34, 987. [DOI] [PubMed] [Google Scholar]
- Rovira C.; Kozlowski P. M. J. Phys. Chem. B 2007, 111, 3251. [DOI] [PubMed] [Google Scholar]
- Pratt D. A.; van der Donk W. A. J. Am. Chem. Soc. 2005, 127, 384. [DOI] [PubMed] [Google Scholar]
- Brooks A. J.; Vlasie M.; Banerjee R.; Brunold T. C. J. Am. Chem. Soc. 2004, 126, 8167. [DOI] [PubMed] [Google Scholar]
- Randaccio L.; Geremia S.; Nardin G.; Wuerges J. Coord. Chem. Rev. 2006, 250, 1332. [Google Scholar]
- Randaccio L.; Geremia S.; Demitri N.; Wuerges J. Molecules 2010, 15, 3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton L. E.; Bowen H. J. M.. Tables of Interatomic Distances and Configuration in Molecules and Ions; The Chemical Society: London, U.K., 1958. [Google Scholar]
- Becke A. D.J. Chem. Phys. 2014, 140, 18A301, p 18. [DOI] [PubMed] [Google Scholar]
- Lee D.; Albrecht A. C.. A Unified View of Raman, Resonance Raman, and Fluorescence Spectroscopy; Wiley: Great Britain, 1985. [Google Scholar]
- Lever A. B. P.Inorganic Electronic Spectroscopy, 1st ed.; Elsevier Publishing Company: New York, NY, 1968. [Google Scholar]
- Vlcek A.; Zalis S. Coord. Chem. Rev. 2007, 251, 258. [Google Scholar]
- Dreuw A.; Head-Gordon M. J. Am. Chem. Soc. 2004, 126, 4007. [DOI] [PubMed] [Google Scholar]
- Weinhold F.; Landis C. L.. Valency and Bonding; Cambridge University Press: Cambridge, U.K., 2005. [Google Scholar]
- Glendening E. D.; Landis C. R.; Weinhold F. J. Comput. Chem. 2013, 34, 2134. [DOI] [PubMed] [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Chem. Rev. 1988, 88, 899. [Google Scholar]
- Reed A. E.; Weinhold F. J. Chem. Phys. 1985, 83, 1736. [Google Scholar]
- Koshland D. E. Science 1992, 258, 1861. [DOI] [PubMed] [Google Scholar]
- Bauer J. A. Anti-Cancer Drugs 1998, 9, 239. [DOI] [PubMed] [Google Scholar]
- Perry C. B.; Marques H. M. S. Afr. J. Sci. 2004, 100, 368. [Google Scholar]
- Bayston J. H.; King N. K.; Looney F. D.; Winfield M. E. J. Am. Chem. Soc. 1969, 91, 2775. [DOI] [PubMed] [Google Scholar]
- Stich T. A.; Buan N. R.; Escalante-Semerena J. C.; Brunold T. C. J. Am. Chem. Soc. 2005, 127, 8710. [DOI] [PubMed] [Google Scholar]
- Blaesi E. J.; Gardner J. D.; Fox B. G.; Brunold T. C. Biochemistry 2013, 52, 6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh N. G.; Holloway D. E.. Adenosyl-cobalamin dependent enzymes. In Enzyme-Catalyzed Electron and Radical Transfer, Holzenburg, A., Scrutton, N. S., Eds.; Chapter 11; Kluwer Academic Publishers: New York, 2000; Vol. 35. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.