

Conspectus
Because living organisms are in constant motion, the word “dynamics” can hold many meanings to biologists. Here we focus specifically on the conformational changes that occur in proteins and how studying these protein dynamics may provide insights into enzymatic catalysis.
Advances in integrating techniques such as X-ray crystallography, nuclear magnetic resonance, and electron cryomicroscopy (cryo EM) allow us to model the dominant structures and exchange rates for many proteins and protein complexes. For proteins amenable to atomic resolution techniques, the major questions shift from simply describing the motions to discovering their role in function. Concurrently, there is an increasing need for using perturbations to test predictive models of dynamics–function relationships. Examples are the catalytic cycles of dihydrofolate reductase (DHFR) and cyclophilin A (CypA).
In DHFR, mutations that alter the ability of the active site to sample productive higher energy states on the millisecond time scale reduce the rate of hydride transfer significantly. Recently identified rescue mutations restore function, but the mechanism by which they do so remains unclear. The exact role of any changes in the dynamics remains an open question. For CypA, a network of side chains that exchange between two conformations is critical for catalysis. Mutations that lock the network in one state also reduce catalytic activity.
A further understanding of enzyme dynamics of well-studied enzymes such as dihydrofolate reductase and cyclophilin A will lead to improvement in ability to modulate the functions of computationally designed enzymes and large macromolecular machines. In designed enzymes, directed evolution experiments increase catalytic rates. Detailed X-ray studies suggest that these mutations likely limit the conformational space explored by residues in the active site. For proteins where atomic resolution information is currently inaccessible, other techniques such as cryo-EM and high-resolution single molecule microscopy continue to advance. Understanding the conformational dynamics of larger systems such as protein machines will likely become more accessible and provide new opportunities to rationally modulate protein function.
Introduction
Even simple enzymes must bind a substrate, catalyze a reaction, and release a product. It is difficult to imagine an enzyme performing all of these actions from a single conformational state. The chemical changes after the substrate has converted to product elicit changes in the conformational ensemble populated by the enzyme (Figure 1). While advances in computational simulations are expanding our ability to see these changes at longer time scales,1 here we focus on determining the conformational ensembles that enzymes populate and how these ensembles change during functional catalytic cycles.
Figure 1.

Protein dynamics occur at different time scales. (a) Motions on the picosecond to nanosecond scale involve small changes in backbone or side chain torsion angles.60 Calcium bound calmodulin (1exr, upper) exhibits conformational heterogeneity on the interface of the peptide binding site. The residual conformational entropy of binding61,62 depends on side chains sampling alternative conformations as exemplified by Met36 and Leu39 (lower). Electron density contoured to 2.5 e–/Å3 in a dark blue mesh and 0.8 e–/Å3 in cyan volume representation. The lessons from calmodulin likely apply to enzymes where the loss of conformational entropy associated with the rigidification of active-site loops or side chains can specifically weaken binding to substrate or product complexes63 and promote flux through the catalytic cycle. (b) A model of ubiquitin (2k39) derived from RDC data reporting on motions up to microseconds is shown as cartoon, with the other models in the ensemble shown as transparent ribbons (upper). The dynamic β1β2 loop moves between alternative loop conformations, represented as sticks (upper and lower). The population of the up (cyan), mid (blue), and down (purple) β1β2 conformations can be a critical determinant of binding preferences for protein–protein interactions.64 The rates of transition between these states discriminate between induced fit and conformational selection mechanisms,65,66 which can influence catalytic mechanisms and inhibitor discovery.67 (c) For enzymes, loop motions on the millisecond time scale are often rate limiting for catalytic cycles, with essential roles for governing ligand flux68 and repositioning key catalytic residues for catalysis.69 The WPD loop of protein tyrosine phosphatase 1B (PTP1B) moves between the “closed” (1sug, orange) and “open” (1t49, cyan) form on the millisecond time scale, forming the catalytically competent closed active site conformation.69 Further molecular detail of the two conformations are shown in the lower panel with electron density contoured to 0.3 e–/Å3. (d) The archaeal proteasome, a ∼700-kDa complex, controls active site access through the dynamic exchange of the N-terminus to block or reveal the central pore on the time scale of seconds.70 The structure of the proteasome is shown as a homoheptamer with each subunit in a different color (upper). In the lower panel, the ensemble of structures of the N-terminus of one of the seven subunits is shown in blue (2ku1). (e) Many enzymes enter long-lived states, with distinct catalytic activities, through stochastic fluctuations.71 Quaternary structure reconstruction of two RAS molecules (yellow) and son of sevenless (SOS, gray surface) complex. The Cdc25, REM, DH, histone, and PH domains of SOS are colored blue, green, orange, brown, and red, respectively (1xd2 (RAS) and 3ksy (SOS)). This complex exchanges between long-lived states with distinct catalytic rates. The structural basis of this exchange is currently unknown but likely involves rearrangements of protein–protein interfaces shown schematically in equilibrium.72 (f) The folded crystal structure (1ssx) of α-lytic protease (upper) is a kinetically trapped structure. After folding catalyzed by a proline domain, the kinetic barrier to unfolding makes this protein stable on the scale of years. The benzoyl moiety of Phe228 deviates by 6° from planarity. Removing this distortion can change the unfolding barrier from over a year to less than 2 weeks.73 Electron density contoured to 4.75 e–/Å3 (dark blue mesh, lower) and 0.5 e–/Å3 (cyan volume representation).
X-ray crystallography and NMR spectroscopy can often be used in concert to answer a question that could not be answered by any single technique2,3 High-resolution (<1.4 Å) X-ray crystallography, ideally at room temperature, and NMR experiments, including measuring J-coupling constants,4 can be combined to determine the structural basis of excited states critical for transit through catalytic cycles (as in the case of dihydrofolate reductase (DHFR) and cyclophilin A (CypA) discussed below).5,6 However, not all enzymes are amenable to these techniques, and many of the central functional questions of protein dynamics at the domain and complex level can be answered using relatively low-resolution structural data paired with kinetic analyses.
For enzymes amenable to high-resolution techniques, such as DHFR and CypA, these technical advances are catalyzing a shift from asking “can we describe protein dynamics” to “what is the role of protein dynamics in function”. Gaining a deeper understanding of these protein dynamics can be extremely challenging; especially important is resisting the urge to ascribe a function to all motions observed by a specific experimental or computational approach. Structural models of enzyme motions are key to understanding whether sampling of specific conformations is essential for properly orienting the substrate during catalysis,7 directly addressing the controversy of whether protein motions are coupled to catalytic function.8
Making the link between dynamics and function requires making some perturbation to the system and assessing the effect of this perturbation. These perturbations could include mutations, changes in the environment of the enzyme, or in more complicated systems, changes in other components interacting with the enzyme. It is critical when making mutations to consider all possible effects of the mutation in addition to the effect on protein dynamics, in order to truly understand the role of the mutated residue.9 This includes understanding the role the mutated residue plays in the structural integrity of the protein and the function of the protein. It is important to note that rigidity and flexibility are simply two extremes of the “protein dynamics” spectrum. Functionally important changes to protein dynamics can act by enhancing rigidity, enhancing flexibility, or altering the correlated motions of residues. For larger and more complicated systems, including protein machines, our level of understanding is often not as detailed compared with that for simple, single-domain, small enzymes. Therefore, the major questions for these machines may still be focused on defining the conformational changes that occur and the relative repositioning of subunits within the folded complex.
The Role of Dynamics in Dihydrofolate Reductase
Dihydrofolate reductase (DHFR) catalyzes the stereospecific reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) (Figure 2a). Five stable intermediates are observed in the catalytic cycle of Escherichia coli DHFR (ecDHFR): the holoenzyme, ecE:NADPH; the Michaelis complex, ecE:NADPH:DHF; and the three product ternary complexes ecE:NADP+:THF, ecE:THF, and ecE:NADPH:THF.10−13 Crystal structures of several complexes were solved by Kraut and co-workers, yielding insights into the structural mechanism of ecDHFR.14−17 For crystallographic work, E:NADP+:FOL was used as a model for the Michaelis complex (E:NADPH:DHF), and 5,10-dideazatetrahydrofolic acid (ddTHF) was used as a stable analog of THF, due to the instability of THF. The crystal structures revealed that ecDHFR undergoes a conformational change in the active site loop (Met20 loop, residues 9–24) that depends on the ligands bound. The conformational change in ecDHFR is therefore coincident with the different stages of the catalytic cycle. The Met20 loop was observed in three dominant conformations: “closed”, “occluded”, and “open”. In the “closed” conformation, the Met20 loop packs tightly against the nicotinamide ring of the cofactor, while in the “occluded” conformation, it projects into the active site and sterically blocks (“occludes”) the nicotinamide-binding pocket, which is compatible with the diffusion of the nicotinamide ring out of the active site (Figure 2a).
Figure 2.
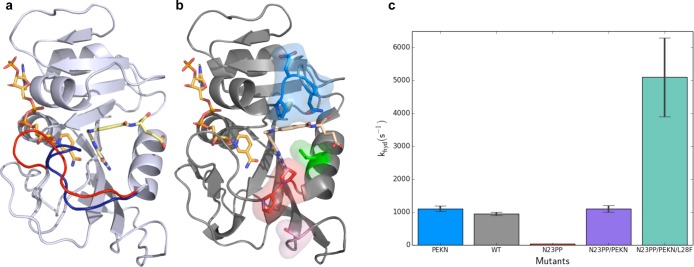
Dynamics in DHFR. (a) Crystal structures of E. coli DHFR show the Met20 loop in the occluded (1rx4, blue) and closed (1rx2, red) conformations. During the catalytic cycle of DHFR, this loop fluctuates between these conformations on the millisecond time scale. The ligands NADPH (left ligand) and folate (right ligand) are shown in orange and yellow, respectively. (b) Mutation of Asn23 to two proline residues (N23PP) shown as sticks in red and Ser148 to alanine (S148A) shown in pink reduce activity of ecDHFR. Mutation of Gly51 to the sequence PEKN (shown in blue) partially recovers the catalytic activity. The activity is increased further by the Leu28Phe (L28F, green) mutation. The structure of N23PP/PEKN (4gh8) is shown with NADPH shown in orange, with substrate mimic methotrexate in tan. (c) pH independent hydride transfer rates of different mutants show the quantitative effects of mutations that alter the dynamics of the Met20 loop and packing around the substrate.
X-ray structures within the same space group of the holoenzyme (E:NADPH) and the model Michaelis complex (E:NADP+:FOL) show that these complexes adopt the closed conformation, whereas the three product complexes (E:NADP+:THF, E:THF, and E:NADPH:THF) adopt the occluded conformation. The occluded conformation is stabilized by hydrogen bonds between Asn23 and the backbone and side chain of Ser148. For binary substrate complexes, the M20 loop conformation was dependent on space-group, being “occluded”, “open”, or even disordered, with no clear electron density for the majority of the loop. While crystallographic analysis in a variety of space groups showed the Met20 loop to be in three dominant conformations, NMR data including chemical shift analyses and NOEs showed that in solution the enzyme is predominantly either in the “closed” conformation or in the “occluded” conformation and never stably in the “open” conformation.18,19
NMR relaxation dispersion experiments on the five stable intermediates of the catalytic cycle showed that each intermediate samples higher energy “excited states”, whose structural features resemble the preceding or following intermediates.20 Substrate and cofactor exchange likely depends on these excited states, suggesting that the ligand-dependent modulation of these protein conformational dynamics is important as the enzyme proceeds through its catalytic cycle. A mutation in the Met20 loop of ecDHFR (N23PP or N23PP/S148A) inhibits the closed-to-occluded transition and also inhibits the sampling of productive higher-energy states on the millisecond time scale, as assayed by NMR relaxation dispersion21 (Figure 2b). Remarkably, enzyme kinetic experiments revealed that hydride transfer, the chemical step of the enzyme reaction, was severely impaired in this mutant. Further analysis using high-resolution room temperature crystallography coupled with multiconformer model building using qFit22 and automated “pathway analysis” using CONTACT6 suggested that the mutation results in an increase of nonproductive, frustrated motions, while the concerted dynamics in the Met20 loop are inhibited. These results led to our current view: the mutation alters millisecond time scale conformational fluctuations, affecting the probability of populating conformations that are conducive to the optimal transition state configuration. As expected, the N23PP and N23PP/S148A mutations also affect ligand flux.
Guided by comparisons to the homologous human DHFR,23 “rescue” mutations in the N23PP background have been identified24,25 (Figure 2b,c). These mutations increase the hydrophobic packing around the substrate molecule. It is currently an open question how these mutations affect conformational dynamics of the protein, if at all, and the mechanism by which they rescue hydride transfer rates remains to be determined. Many computational and experimental studies have also examined flexibility changes introduced by the G121V mutation.26,27 While simulations have provided initial explanations of these effects, a current challenge for computational approaches is to rationalize both the local and long-range effects across the large activity ranges spanned by these mutations and to make predictions of how other mutations that tune these parameters will manifest experimentally.28−31
The Role of Dynamics in Cyclophilin A
Human cyclophilin A (CypA) belongs to the proline isomerase family of enzymes, which play critical roles in protein folding, signaling, and the immune response.32,33 The reaction catalyzed by CypA, cis–trans proline isomerization, does not make or break new chemical bonds, making it possible to detect conformational exchange of both the substrate and the saturated enzyme during catalysis by NMR. Both backbone and side chain NMR dynamics experiments have been performed at various time scales revealing a network of residues that undergo two-state conformational exchange, which are thought to represent the enzyme’s conformation when bound to cis- or trans-proline substrates (Figure 3a).34,35 Interestingly, NMR relaxation dispersion experiments show that similar conformational switching is observed in the free enzyme. These rates correspond closely to the sum of the rate constants of cis-to-trans and trans-to-cis isomerization (kex = 2500 s–1) suggesting that the rate of the catalytic cycle is governed by the intrinsic protein dynamics.
Figure 3.
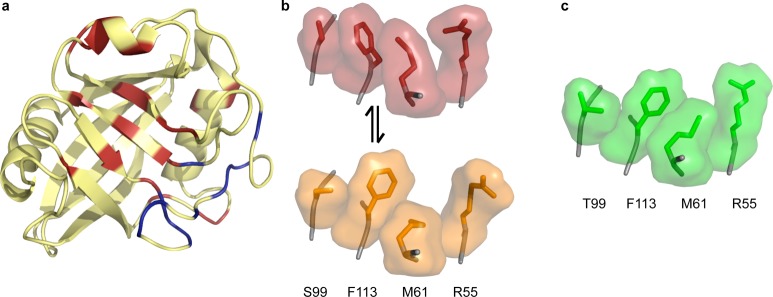
Dynamics in CypA. (a) Exchanging residues detected by CPMG experiments show two groups with exchange rate kex = 1140 ± 200 s–1 (red) and kex = 2260 ± 200 s–1 (blue) in the absence of substrate. All residues with detectable dynamic exchange can be fit to one rate (∼2400 s–1) when the protein is saturated with substrate, which interconverts from cis to trans on the enzyme (1rmh). (b) Wild-type CypA (3k0n) shows two sets of conformations at room temperature. The network of side chains of residues S99, F113, M61, and R55 are shown with surface representations around sticks, with the major conformation in red and the minor conformation in orange. These residues lie across the central β strands shown in panel a. (c) The network of these four residues for the S99T mutant at room temperature only occupies the minor-like conformation, shown in green (3k0o).
While NMR studies provided numerous insights and hinted that the minor conformation sampled in the free enzyme was relevant to catalysis, a detailed structural picture of the minor conformation was not decipherable from NMR experiments alone. The chemical shift differences between the excited and ground state provided few hints about the structural nature of the “excited” state. Examination of low levels of electron density, derived from new high resolution X-ray data collected at room temperature, uncovered a network of minor side-chain conformations for the residues exhibiting NMR dispersion (Figure 3b). It is worth noting that both visual and automated examination, using Ringer,36 of the electron density on data collected under cryogenic conditions did not uncover the full subset of minor conformations. These results point to several complications for deriving a structural basis for conformational dynamics. First, even though the CPMG experiments conducted here probed changes in the backbone environment, the conformational changes are mostly at the side chain level. Changes in side chain dihedral angles and the proximity of aromatic groups, in particular Phe113, create these changes with little change in backbone torsions. Second, the small energy differences (∼1–2 kcal/mol) separating these states render them very susceptible to perturbations like cryocooling or lattice contacts and can have unforeseen consequences, including eliminate evidence for the “excited state” even in high resolution X-ray data.
To further link X-ray and NMR measurements, a mutation (Ser99Thr) was designed to stabilize the network of side-chain rotamers associated with the minor conformation in solution as well as in the crystal (Figure 3c). Like the DHFR N23PP mutant, this mutation ablated detectable exchange on the microsecond to millisecond time scale. Furthermore, enzyme assays showed that shifting the equilibrium toward the side-chain rotamers populated by the minor conformation results in slower enzyme catalysis. Simulations have provided further evidence of a coupling between enzyme and substrate conformations during the catalytic cycle.37 Recent studies incorporating new NMR measurements and simulations point to the importance of considering a conformational ensemble in creating the environment compatible with catalysis and binding both the cis and trans forms of the substrate.38
Dynamics in Designed Enzymes
One of the most exciting recent developments in enzymology is the application of protein design methodologies to create enzymes that catalyze reactions not found in nature.39 Several simple reactions have now been created based on the designs that place catalytic groups in specific orientations relative to a computational model of the transition state.40−42 These initial designs are generally weak catalysts, and directed evolution strategies have been implemented to improve them by several orders of magnitude.43−45 While these evolved enzymes have greatly increased catalytic rates, structural and enzymological studies have revealed that some of the principles implemented in the designs are subverted over the course of multiple rounds of selection. For example, a detailed study of a designed retroaldolase revealed only a modest shift in the reactivity of the lysine and no role for a designed water-mediated interaction in catalysis.46 The design was successful in creating strong hydrophobic binding interactions with the substrate,46 which parallels lessons from simple micelle systems.47 A separate study on a distinct designed retroaldolase revealed that a second lysine residue acquired during directed evolution was responsible for greater rate enhancement than the original “catalytic” lysine and that the substrate occupied an entirely new position in the catalytic cavity.48 Similarly, positioning of the substrate changed dramatically during the course of directed evolution studies of a designed Kemp eliminase (Figure 4a,b).49,50 Many rounds of selection resulted in catalysis with efficiency approaching that of natural enzymes and an intricate series of interactions in the active site (Figure 4c).50 However, examination of low levels of electron density in this high-resolution data set suggests there are still interactions available for optimization (Figure 4d,e). Therefore, even in the most proficient synthetic catalysts, an abundance of highly flexible residues in the active site may be limiting the formation of precise and stable interactions required for catalysis.
Figure 4.

Designed enzymes. (a) The Kemp eliminase reaction scheme. (b) Structure of designed Kemp eliminase prior to directed evolution (3nyd) with two alternative ligand conformations (cyan and purple). Electron density contoured to 1.5 e–/Å3 as a dark blue mesh, with a lower contour shown as a cyan volume representation at 0.3 e–/Å3. (c) Structure of final designed Kemp eliminase (4BS0) highlighting the hydrogen bond network. Disordered water replaces the acetate of the previous structure. Electron density contoured to 2.65 e–/Å3 as a dark blue mesh, with a lower contour shown as a cyan volume representation at 0.3 e–/Å3. Difference density contoured to 0.3 e–/Å3, colored green for positive and red for negative. Hydrogen bonds in hydrogen bond network drawn as dashed black lines. (d) Disorder of tryptophan residue 44 in final Kemp eliminase. The modeled alternative conformation deposited in the PDB is shown in dark green. Electron density contoured as in panel b. (e) Possible alternative conformation of catalytic Asp127 as detected by two positive difference peaks indicating alternative positions of the carboxylic oxygens that stabilize interactions with alternative water conformations. Electron density contoured as in panel c.
A current focus of computational studies on designed enzymes is to use molecular dynamics simulations to screen for candidates with overly flexible active sites51 or excess solvent penetration.52 However, collectively these studies suggest that increased active-site conformational flexibility, while detrimental to the chemical step, are likely key to the evolvability of these enzymes:53 the imprecision of the design process and of nonspecific hydrophobic interactions allows for the eventual tuning of catalytic residues during the course of directed evolution procedures. As the reactions performed by designed enzymes become more demanding, the role of directed evolution in rigidifying the active site will become more important;54 however, it is important to note that flexibility of second shell residues may still be desirable to ensure efficient transit through the catalytic cycle.55 Characterizing the energy landscapes of designed enzymes and using this information to improve the design process will be key to realizing the dream of designing synthetic catalysts for many important, yet currently difficult, reactions.56
Extending Protein Dynamics to Large “Protein Machines”
For larger enzymes that often undergo large-scale conformational changes, NMR spectroscopy and high-resolution X-ray crystallography may not be feasible (Figure 5). In these cases, electron cryomicroscopy (cryo-EM) and single-molecule Förster resonance energy transfer (smFRET) or single molecule two-color localization experiments can yield important insights into protein dynamics. For example, smFRET experiments have revealed excited states of the ribosome.57 Our understanding of protein dynamics using these methods is less atomically detailed and tends to focus on reorientation of large subdomains. However, these experiments will provide key information in guiding future studies on macromolecular machines and complexes. Advances such as recently developed direct electron detectors58 and sophisticated methods of 3D classification59 have catapulted cryo-EM to the forefront of understanding protein conformational landscapes in larger systems, at resolutions overlapping with the low end of those achieved by X-ray crystallography. While the resolutions achievable are still lower than what is required to understand enzyme catalysis at as detailed a level as available for DHFR or CypA, a fairly thorough picture of larger scale conformational changes can be developed through comparison of density maps and de novo models. Using these tools to probe the conformational landscape of larger protein machines will provide exciting new insights into their function.
Figure 5.
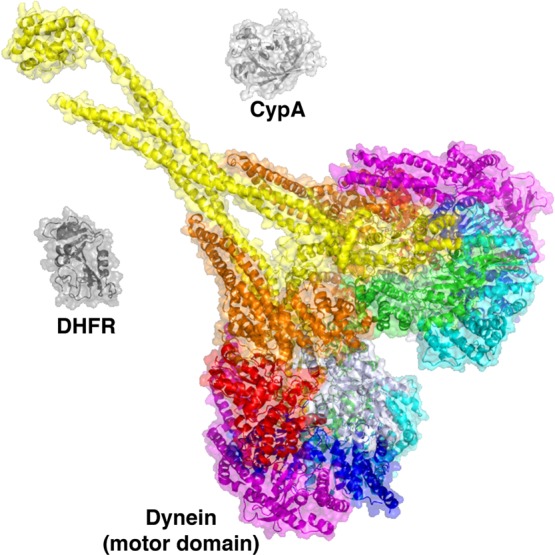
The challenge of larger protein machines. The motor domain of dynein, a microtubule-based motor protein belonging to the AAA family of enzymes, is shown, colored by domain (3VKH). The entire dynein heavy chain is considerably larger and is difficult to produce in quantities required for structural biology studies. For comparison, DHFR (1RX2) and CypA (2CPL) are shown to scale.
Acknowledgments
G.B. is the Merck Fellow of the Damon Runyon Cancer Research Foundation (Grant DRG-2136-12). J.T.B. is an NSF Graduate Research Fellow. J.S.F. is a Searle Scholar and a Pew Scholar. Work in the lab of J.S.F. is supported by NIH Grants OD009180 and GM110580 and NSF Grant STC-1231306. We thank Tony Liu and Steven Benkovic for discussions of mutant DHFR kinetics and Peter Wright and Daniel Keedy for helpful comments.
Biographies
Gira Bhabha was a graduate student with Peter Wright at The Scripps Research Institute, where she studied the role of protein dynamics in enzyme catalysis and the evolution of protein conformational dynamics. Since 2012, Gira has been investigating the mechanism of the motor protein dynein in Ron Vale’s lab at UCSF, as the Merck Fellow of the Damon Runyon Cancer Research Foundation.
Justin Biel received his bachelors in biochemistry and biophysics at Oregon State University, working on ultra high resolution X-ray crystallography analysis in the lab of P. Andrew Karplus. In 2013, Justin began graduate school at University of California, San Francisco, where he is an NSF Graduate Research Fellow.
James Fraser was a graduate student at UC Berkeley with Tom Alber, where he developed new experimental and computational methods to investigate protein conformational dynamics. In 2011, James moved to UCSF, where his lab focuses on the role of protein motions in catalysis, ligand binding, and allostery.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Protein Motion in Catalysis”.
Funding Statement
National Institutes of Health, United States
References
- Dror R. O.; Dirks R. M.; Grossman J. P.; Xu H.; Shaw D. E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [DOI] [PubMed] [Google Scholar]
- Ward A. B.; Sali A.; Wilson I. A. Biochemistry. Integrative structural biology. Science 2013, 339, 913–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle L. M.; Dyson H. J.; Wright P. E. Side-chain conformational heterogeneity of intermediates in the Escherichia coli dihydrofolate reductase catalytic cycle. Biochemistry 2013, 52, 3464–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick R. B.; van den Bedem H.; Fraser J. S.; Wright P. E. Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bedem H.; Bhabha G.; Yang K.; Wright P. E.; Fraser J. S. Automated identification of functional dynamic contact networks from X-ray crystallography. Nat. Methods 2013, 10, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. A 21st century revisionist’s view at a turning point in enzymology. Nat. Chem. Biol. 2009, 5, 543–550. [DOI] [PubMed] [Google Scholar]
- Kamerlin S. C.; Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis?. Proteins 2010, 78, 1339–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D.; Natarajan A. Fundamental challenges in mechanistic enzymology: Progress toward understanding the rate enhancements of enzymes. Biochemistry 2013, 52, 2050–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R.; Dyson H. J.; Wright P. E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [DOI] [PubMed] [Google Scholar]
- Benkovic S. J.; Fierke C. A.; Naylor A. M. Insights into enzyme function from studies on mutants of dihydrofolate reductase. Science 1988, 239, 1105–1110. [DOI] [PubMed] [Google Scholar]
- Fierke C. A.; Benkovic S. J. Probing the functional role of threonine-113 of Escherichia coli dihydrofolate reductase for its effect on turnover efficiency, catalysis, and binding. Biochemistry 1989, 28, 478–486. [DOI] [PubMed] [Google Scholar]
- Fierke C. A.; Johnson K. A.; Benkovic S. J. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 1987, 26, 4085–4092. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [DOI] [PubMed] [Google Scholar]
- Bystroff C.; Kraut J. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry 1991, 30, 2227–2239. [DOI] [PubMed] [Google Scholar]
- Bystroff C.; Oatley S. J.; Kraut J. Crystal structures of Escherichia coli dihydrofolate reductase: The NADP+ holoenzyme and the folate.NADP+ ternary complex. Substrate binding and a model for the transition state. Biochemistry 1990, 29, 3263–3277. [DOI] [PubMed] [Google Scholar]
- Reyes V. M.; Sawaya M. R.; Brown K. A.; Kraut J. Isomorphous crystal structures of Escherichia coli dihydrofolate reductase complexed with folate, 5-deazafolate, and 5,10-dideazatetrahydrofolate: Mechanistic implications. Biochemistry 1995, 34, 2710–2723. [DOI] [PubMed] [Google Scholar]
- Osborne M. J.; Venkitakrishnan R. P.; Dyson H. J.; Wright P. E. Diagnostic chemical shift markers for loop conformation and substrate and cofactor binding in dihydrofolate reductase complexes. Protein Sci. 2003, 12, 2230–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitakrishnan R. P.; Zaborowski E.; McElheny D.; Benkovic S. J.; Dyson H. J.; Wright P. E. Conformational changes in the active site loops of dihydrofolate reductase during the catalytic cycle. Biochemistry 2004, 43, 16046–16055. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Lee J.; Ekiert D. C.; Gam J.; Wilson I. A.; Dyson H. J.; Benkovic S. J.; Wright P. E. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 2011, 332, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bedem H.; Dhanik A.; Latombe J. C.; Deacon A. M. Modeling discrete heterogeneity in X-ray diffraction data by fitting multi-conformers. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Ekiert D. C.; Jennewein M.; Zmasek C. M.; Tuttle L. M.; Kroon G.; Dyson H. J.; Godzik A.; Wilson I. A.; Wright P. E. Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis K.; Stojkovic V.; Kohen A. Preservation of protein dynamics in dihydrofolate reductase evolution. J. Biol. Chem. 2013, 288, 35961–35968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. T.; Hanoian P.; French J. B.; Pringle T. H.; Hammes-Schiffer S.; Benkovic S. J. Functional significance of evolving protein sequence in dihydrofolate reductase from bacteria to humans. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10159–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekko K.; Yamagami K.; Kunori Y.; Ichihara S.; Kodama M.; Iwakura M. Effects of point mutation in a flexible loop on the stability and enzymatic function of Escherichia coli dihydrofolate reductase. J. Biochem. 1993, 113, 74–80. [DOI] [PubMed] [Google Scholar]
- Mauldin R. V.; Sapienza P. J.; Petit C. M.; Lee A. L. Structure and dynamics of the G121V dihydrofolate reductase mutant: lessons from a transition-state inhibitor complex. PloS One 2012, 7, e33252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczyk A. J.; Cao J.; Kamerlin S. C.; Warshel A. Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14115–14120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Rajagopalan P. T.; Benkovic S. J.; Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dametto M.; Antoniou D.; Schwartz S. D. Barrier Crossing in Dihydrofolate Reductase does not involve a rate-promoting vibration. Mol. Phys. 2012, 110, 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan A.; Savol A.; Burger V.; Chennubhotla C. S.; Agarwal P. K. Protein conformational populations and functionally relevant substates. Acc. Chem. Res. 2014, 47, 149–156. [DOI] [PubMed] [Google Scholar]
- Lu K. P.; Finn G.; Lee T. H.; Nicholson L. K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [DOI] [PubMed] [Google Scholar]
- Sokolskaja E.; Luban J. Cyclophilin, TRIM5, and innate immunity to HIV-1. Curr. Opin. Microbiol. 2006, 9, 404–408. [DOI] [PubMed] [Google Scholar]
- Eisenmesser E. Z.; Millet O.; Labeikovsky W.; Korzhnev D. M.; Wolf-Watz M.; Bosco D. A.; Skalicky J. J.; Kay L. E.; Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [DOI] [PubMed] [Google Scholar]
- Eisenmesser E. Z.; Bosco D. A.; Akke M.; Kern D. Enzyme dynamics during catalysis. Science 2002, 295, 1520–1523. [DOI] [PubMed] [Google Scholar]
- Lang P. T.; Ng H. L.; Fraser J. S.; Corn J. E.; Echols N.; Sales M.; Holton J. M.; Alber T. Automated electron-density sampling reveals widespread conformational polymorphism in proteins. Protein Sci. 2010, 19, 1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan L. C.; Hamelberg D. Conformational plasticity of an enzyme during catalysis: Intricate coupling between cyclophilin A dynamics and substrate turnover. Biophys. J. 2013, 104, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C.; Sahakyan A. B.; Holliday M. J.; Isern N. G.; Zhang F.; Eisenmesser E. Z.; Vendruscolo M. Cyclophilin A catalyzes proline isomerization by an electrostatic handle mechanism. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 10203–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss G.; Celebi-Olcum N.; Moretti R.; Baker D.; Houk K. N. Computational enzyme design. Angew. Chem. 2013, 52, 5700–5725. [DOI] [PubMed] [Google Scholar]
- Bolon D. N.; Mayo S. L. Enzyme-like proteins by computational design. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14274–14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlisberger D.; Khersonsky O.; Wollacott A. M.; Jiang L.; DeChancie J.; Betker J.; Gallaher J. L.; Althoff E. A.; Zanghellini A.; Dym O.; Albeck S.; Houk K. N.; Tawfik D. S.; Baker D. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Althoff E. A.; Clemente F. R.; Doyle L.; Rothlisberger D.; Zanghellini A.; Gallaher J. L.; Betker J. L.; Tanaka F.; Barbas C. F. 3rd; Hilvert D.; Houk K. N.; Stoddard B. L.; Baker D. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Rothlisberger D.; Dym O.; Albeck S.; Jackson C. J.; Baker D.; Tawfik D. S. Evolutionary optimization of computationally designed enzymes: Kemp eliminases of the KE07 series. J. Mol. Biol. 2010, 396, 1025–1042. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Rothlisberger D.; Wollacott A. M.; Murphy P.; Dym O.; Albeck S.; Kiss G.; Houk K. N.; Baker D.; Tawfik D. S. Optimization of the in-silico-designed kemp eliminase KE70 by computational design and directed evolution. J. Mol. Biol. 2011, 407, 391–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Kiss G.; Rothlisberger D.; Dym O.; Albeck S.; Houk K. N.; Baker D.; Tawfik D. S. Bridging the gaps in design methodologies by evolutionary optimization of the stability and proficiency of designed Kemp eliminase KE59. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 10358–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassila J. K.; Baker D.; Herschlag D. Origins of catalysis by computationally designed retroaldolase enzymes. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J.; Ehasz C.; Epperson M.; Klas K.; Wyatt J.; Hennig M.; Forconi M. The effect of the hydrophobic environment on the retro-aldol reaction: comparison to a computationally-designed enzyme. Org. Biomol. Chem. 2013, 11, 8419–8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger L.; Caner S.; Obexer R.; Kast P.; Baker D.; Ban N.; Hilvert D. Evolution of a designed retro-aldolase leads to complete active site remodeling. Nat. Chem. Biol. 2013, 9, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privett H. K.; Kiss G.; Lee T. M.; Blomberg R.; Chica R. A.; Thomas L. M.; Hilvert D.; Houk K. N.; Mayo S. L. Iterative approach to computational enzyme design. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 3790–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg R.; Kries H.; Pinkas D. M.; Mittl P. R.; Grutter M. G.; Privett H. K.; Mayo S. L.; Hilvert D. Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature 2013, 503, 418–421. [DOI] [PubMed] [Google Scholar]
- Kiss G.; Pande V. S.; Houk K. N. Molecular dynamics simulations for the ranking, evaluation, and refinement of computationally designed proteins. Methods Enzymol. 2013, 523, 145–170. [DOI] [PubMed] [Google Scholar]
- Kiss G.; Rothlisberger D.; Baker D.; Houk K. N. Evaluation and ranking of enzyme designs. Protein Sci. 2010, 19, 1760–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. Protein dynamism and evolvability. Science 2009, 324, 203–207. [DOI] [PubMed] [Google Scholar]
- Korendovych I. V.; DeGrado W. F. Catalytic efficiency of designed catalytic proteins. Curr. Opin. Struct. Biol. 2014, 27C, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscio J. Z.; Kohn J. E.; Ball K. A.; Head-Gordon T. The influence of protein dynamics on the success of computational enzyme design. J. Am. Chem. Soc. 2009, 131, 14111–14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiswerk N.; Beck T.; Schulz J. D.; Milovnik P.; Mayer C.; Siegel J. B.; Baker D.; Hilvert D. Impact of scaffold rigidity on the design and evolution of an artificial Diels-Alderase. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 8013–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Abeysirigunawarden S. C.; Chen K.; Mayerle M.; Ragunathan K.; Luthey-Schulten Z.; Ha T.; Woodson S. A. Protein-guided RNA dynamics during early ribosome assembly. Nature 2014, 506, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Mooney P.; Zheng S.; Booth C. R.; Braunfeld M. B.; Gubbens S.; Agard D. A.; Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 2013, 10, 584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S. H. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 2012, 415, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittermaier A.; Kay L. E. New tools provide new insights in NMR studies of protein dynamics. Science 2006, 312, 224–228. [DOI] [PubMed] [Google Scholar]
- Marlow M. S.; Dogan J.; Frederick K. K.; Valentine K. G.; Wand A. J. The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol. 2010, 6, 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick K. K.; Marlow M. S.; Valentine K. G.; Wand A. J. Conformational entropy in molecular recognition by proteins. Nature 2007, 448, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alber T.; Gilbert W. A.; Ponzi D. R.; Petsko G. A. The role of mobility in the substrate binding and catalytic machinery of enzymes. Ciba Found. Symp. 1983, 93, 4–24. [DOI] [PubMed] [Google Scholar]
- Lange O. F.; Lakomek N. A.; Fares C.; Schroder G. F.; Walter K. F.; Becker S.; Meiler J.; Grubmuller H.; Griesinger C.; de Groot B. L. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 2008, 320, 1471–1475. [DOI] [PubMed] [Google Scholar]
- Hammes G. G.; Chang Y. C.; Oas T. G. Conformational selection or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13737–13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt A. D.; Di Cera E. Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 2012, 51, 5894–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M.; Coleman R. G.; Fraser J. S.; Shoichet B. K. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014, 6, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; Dyson H. J.; Wright P. E. Conformational relaxation following hydride transfer plays a limiting role in dihydrofolate reductase catalysis. Biochemistry 2008, 47, 9227–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittier S. K.; Hengge A. C.; Loria J. P. Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 2013, 341, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham M. P.; Sekhar A.; Kay L. E. Understanding the mechanism of proteasome 20S core particle gating. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 5532–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X. S.; Choi P. J.; Li G. W.; Lee N. K.; Lia G. Single-molecule approach to molecular biology in living bacterial cells. Annu. Rev. Biophys. 2008, 37, 417–444. [DOI] [PubMed] [Google Scholar]
- Iversen L.; Tu H. L.; Lin W. C.; Christensen S. M.; Abel S. M.; Iwig J.; Wu H. J.; Gureasko J.; Rhodes C.; Petit R. S.; Hansen S. D.; Thill P.; Yu C. H.; Stamou D.; Chakraborty A. K.; Kuriyan J.; Groves J. T. Molecular kinetics. Ras activation by SOS: allosteric regulation by altered fluctuation dynamics. Science 2014, 345, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelch B. A.; Salimi N. L.; Agard D. A. Functional modulation of a protein folding landscape via side-chain distortion. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9414–9419. [DOI] [PMC free article] [PubMed] [Google Scholar]