

Abstract
Culture extracts from the cave-derived actinomycete Nonomuraea specus were investigated, resulting in the discovery of a new S-bridged pyronaphthoquinone dimer and its monomeric progenitors designated hypogeamicins A–D (1–4). The structures were elucidated using NMR spectroscopy, and the relative stereochemistries of the pyrans were inferred using NOE and comparison to previously reported compounds. Absolute stereochemistry was determined using quantum chemical calculations of specific rotation and vibrational and electronic circular dichroism spectra, after an extensive conformational search and including solute–solvent polarization effects, and comparing with the corresponding experimental data for the monomeric congeners. Interestingly, the dimeric hypogeamicin A (1) was found to be cytotoxic to the colon cancer derived cell line TCT-1 at low micromolar ranges, but not bacteria, whereas the monomeric precursors possessed antibiotic activity but no significant TCT-1 cytotoxicity.
Caves in central south North America are ancient subterranean structures, thought to have been formed as early as the Mesozoic era (145–65 million years ago) and as recently as the Pleistocene era (11.5–1.8 million years ago). So-called hypogean ecosystems are distinct from their surface counterparts in many ways including the complete absence of sunlight, high relative humidity (95–100%), a constant temperature of 13–15 °C, and very low dissolved carbon.1,2 Despite these constraints, members of all microbial kingdoms have been identified (bacteria, archaea, and eukarya).3 The unique environmental properties of these ecosystems have prompted us to initiate a program to investigate the secondary metabolic capacity of cultivatable hypogean actinomycetes.4 This class of microorganisms is a proven prolific source of secondary metabolites in surface and, more recently, oceanic sediments.5−7 Indeed, epigean actinomycetes have been extensively mined8 and, for this reason, form a basis for determining if the unique hypogean microenvironment will foster microorganism diversity possessing a complementary secondary metabolic arsenal.
Herein we describe the isolation of abundant metabolites from a cave-derived actinomycete, Nonomuraea specus (GenBank KJ209502), including a new S-bridged dimeric pyronaphthoquinone, which we designated as hypogeamicin A (1), as well as apparent metabolic precursors, hypogeamicins B–D (2–4), new members of the nanaomycin family. Relative stereochemical assignments were proposed based on NOESY data and comparison to a previously isolated compound, (+)-BE-52440A,9 the only other compound described in this class. Absolute stereochemistry was assigned by comparison of multiple chiroptical data to their corresponding spectra calculated using quantum chemical methods. Hypogeamicin was subsequently demonstrated to be cytotoxic against the colon cancer cell line TCT-1.
Results and Discussion
To investigate the taxonomic and chemical diversity of natural product producing organisms from cave ecosystems, we endeavored to first replicate methods used for isolation of actinomycetes from surface environments.10 We collected sediment samples aseptically from nine locations throughout the Hardin’s cave system located close to Ashland City, Tennessee. It is a horizontal cave that contains a large amount of organic matter and a population of bats. N. specus was obtained from a highly decomposed bark sample collected far from the cave entrance as one of 20 actinomycetes derived via dilution plating from desiccated cave sediments. It was cultured in a variety of complex liquid media as part of a screening for new secondary metabolites. An ethyl acetate extract of a whole fermentation broth of N. specus was fractionated by Sephadex LH-20 chromatography in 100% methanol. Abundant metabolites in Sephadex LH-20 fractions were identified by analytical HPLC and subsequently isolated by preparative HPLC. The most abundant UV-absorbing compound isolated was hypogeamicin A (1). A quantity of 150 μg of compound 1 was isolated as a pale yellow glass (λmax = 356 nm, CH3CN/H2O) of molecular formula C38H44N2O14S, determined from high-resolution electrospray mass spectrometry (m/z 785.2605 [M + H], <3 ppm).
On the basis of the UV spectrum, unsaturation number, and inspection of the 1H NMR, it was apparent that compound 1 was likely a meta-substituted aromatic quinone metabolite (δ 6.7, 7.5 d, J = 2.5 Hz) with aryl O-methyl (δ 4.02, s, 3H) and alkyl group (δ 0.55 t, 3H, J = 7.1 Hz) substitution. Formula constraints were provided by the 13C NMR, which provided evidence for only 19 carbons, and high-resolution mass spectrum (HRMS), suggesting a symmetric dimer bridged by sulfur.
The symmetric unit was assembled by analysis of COSY, HSQC, and HMBC data (Figure 1A). Structure determination began with three fragments, a benzoquinone and two distinct aliphatic spin systems. On the basis of 13C chemical shifts and 1H coupling constants, the aromatic ring was assumed to be a meta-substituted aromatic (δ 6.7, 7.5 d, J = 2.5 Hz), and this regiochemistry was confirmed by the HMBC correlation of C-7, C-8, and the C-7 OCH3 to C-7. HMBC data from the aromatic region suggested two aromatic O-substitutions (δ 166.9, 163.8) and two carbonyls (δ 196.4, 189.2). The substitution pattern on the tetrasubstituted aromatic ring was determined as follows: C-6 was linked to quinone carbonyl C-5 by a strong HMBC correlation and a weaker correlation to the carbonyl at C-10. Together with the UV spectrum, these features implied the 1,4-naphthoquinone substitution. The H-1 to H-18 spin system was separated from the H-4 to H-14 spin system by careful inspection of COSY and TOCSY data, which revealed that the methine at δ 3.79 ppm coupled with a propyl side chain and the methine at δ 3.76 coupled to two diastereotopic methylenes, one of which was adjacent to a carbonyl at C-15. On the basis of the HMBC correlation of C-1 and C-3, we assigned these spin systems as part of a pyran ring system with quaternary carbons C-10a and C-4a, also assigned by HMBC from H-1 and H-4 respectively. The symmetric unit was completed by linking the pyran to the quinone by HMBC correlation of the C-4 methylene to quinone C-5 and weak correlation of methine C-1 to the quinone C-10, thereby accounting for all carbons in the symmetric unit.
Figure 1.

(A) The most significant 2D COSY, NOESY, and HMBC correlations for hypogeamicin A (1). (B) Hypogeamicins A–D (1–4).
The assignment of the sulfide bridge, previously inferred from HRMS data, was based on analysis of chemical shift values of C-4a and C-10a, which are consistent with C-4a being S-substituted, and comparison to BE-52440A. Additionally, an NOE signal was observed between the H-3 of the pyranose and H-6 of the naphthoquinone, suggesting a 180 deg orientation between monomeric units in solution. The relative stereochemistry was established based on NOE correlations observed between H-3 and H-16, indicating that protons H-1 and H-3 are likely in trans arrangement in relative configuration (Figure S5). The pyronaphthoquinone framework of the symmetric unit is found in several biologically active metabolites related to the nanaomycins.11−13 More recently two structurally novel S-bridged dimeric pyronaphthoquinones have been described,9 BE-52440A and B, and the absolute stereochemistry of BE-52440A, its pyran ring and mercaptan linkage, was recently established by total synthesis and X-ray analysis.14 Inspection of the diagnostic 13C chemical shift, particularly the quaternary carbons C-4a and C-10a, along with the nominal mass, suggested that compound 1 was similarly bridged by a sulfur.
In subsequent scale-up fermentations of the producing organism we isolated the likely monomeric progenitors of the dimeric pyronaphthoquinone. Again, an ethyl acetate extract of whole fermentation broth was first fractionated on a Sephadex LH-20 column with 100% methanol, and abundant metabolites were then purified on an RP-HPLC preparative column with a water/acetonitrile gradient. We isolated and characterized the three most abundant metabolites. Analysis of NMR and MS data (see Supporting Information) indicated these to be monomeric analogues of compound 1. The corresponding hypogeamicins B–D (2–4) are structural analogues of previously reported pyronaphthoquinones of the actinorhodin class. Hypogeamicin B (2) was isolated as a pale yellow glass with λmax of 360 nm (CH3CN/H2O) and of molecular formula C19H21NO7 determined from high-resolution mass spectrometry (m/z 376.1388 [M + H] <3 ppm). On the basis of the molecular formula, inspection of 1H NMR, COSY, HSQC, and HMBC spectral data, and the comparison to previously characterized hypogeamicin A (1), we were able to establish the structure of compound 2, which turned out to be a monomeric unit of compound 1 with the epoxide moiety in positions C-4a and C-10a, which was determined from 13C chemical shift values for carbons C-4a and C-10a (δ 61.3, 61.8). Hypogeamicin C (3) was also isolated as pale yellow glass with a λmax of 360 nm (CH3CN/H2O) and a molecular formula of C19H23NO8 derived from HRMS (m/z 394.1513 [M + H] <3 ppm). The structure of 3 was determined via comparison of the 1H NMR, COSY, HSQC, and HMBC spectral data to previously characterized hypogeamicin B (2). The presence of an additional oxygen and two hydrogens in the molecular formula and the upfield shift of carbons C-4a and C-10a (δ 75.8, 75.4) suggested substitution of the epoxide moiety with two hydroxyls in positions C-4a and C-10a for compound 3. Hypogeamicin D (4) was isolated as a pale yellow glass with a λmax of 360 nm, CH3CN/H2O, and a molecular formula of C19H21NO8 determined from HRMS (m/z 392.1345 [M + H] <3 ppm). Inspection of the 1H NMR, COSY, HSQC, and HMBC spectral data suggested that the epoxide moiety in positions C-4a and C-10a was still present (δ 62.9, 63.2), but the methylene group at position C-14 was substituted with a hydroxyl. Hoping to establish the absolute stereochemistry of the series, we compared the optical rotation of hypogeamicin B (2) ([α]24D −80 (c 0.2, CH3CN)) to nanaomycin E ([α]24D +127 (c 1.00, CHCl3)) and its enantiomer frenolicin ([α]24D −37.7 (c 1.5, MeOH)).
Determination of the absolute stereochemistry of hypogeamicin B (2) would allow inference of A (1) and C (3) via antiperiplanar epoxide ring opening via sulfide or water, respectively. Unfortunately, the assignment of the absolute stereochemistry from optical rotation data alone was not possible15 due to the structural differences between these compounds and previously reported analogues, and therefore we were unable to determine the absolute stereochemistry using these data. Extensive crystallization attempts of this series were also unsuccessful, possibly due to low levels of isolated hypogeamicins. An emerging alternative approach for determination of the absolute stereochemistry of sample-limited materials is comparison of experimental chiroptical spectra to corresponding spectra predicted by modern quantum chemical theories.16 This method has proven to be a robust method for absolute stereochemistry determination, given the following precaution that at least two chiroptical experimental data sets are found to be in agreement with quantum chemical calculations for only one stereoisomer among all those possible.17,18 Thus, we endeavored to assign the stereochemistry of hypogeamicin B (2) via correlation of experimental electronic and vibrational circular dichroism (ECD and VCD), optical rotatory dispersion (ORD) spectra, and ab initio quantum chemical calculations.19−25 Given that the epoxide linkage must be syn, and the NOE signal between H-3 and H-16 was the only one observed across the pyranose ether linkage (Figure S10), four different stereoisomers of epoxide 2 were investigated computationally: (a) 1R,3R,4aS,10aR (2A); (b) 1R,3S,4aS,10aR (2B); (c) 1S,3R,4aS,10aR (2C); and (d) 1S,3S,4aS,10aR (2D) (see Figure 2). Since the relative configurations at positions 4a and 10a are fixed, the remaining possible stereoisomers are mirror images of the four considered. As described in detail in the Supporting Information (section 1), we aquired optical rotary disperion, electronic circular dichroism, and vibrational circular dichrosim spectra for hypogeamicin B (2) and calculated the corresponding spectra using multiple optimized geometries with and without added solvent molecules. The similarity between experimental and calculated spectra was determined via similarity overlap, using CDspecTech, which provides the numerical measures of similarity between experimental and calculated spectra.26,27 The consensus of quantum mechanical calculations, along with the NOE-derived relative stereochemistry, suggested the absolute configuration of (−)-2 to be (1R,3S,4aS,10aR). This is also the configuration of the frenolicin pyronaphthoquinone.28
Figure 2.
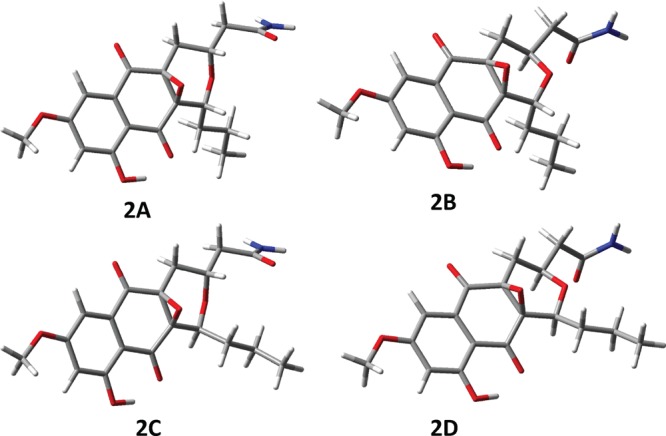
Four stereoisomers of 2 investigated in the present work: (a) 1R,3R,4aS,10aR (2A); (b) 1R,3S,4aS,10aR (2B); (c) 1S,3R,4aS,10aR (2C); and (d) 1S,3S,4aS,10aR (2D). The conformation shown for each case is the lowest energy optimized conformer at the B3LYP/6-31G(d)/PCM(DMSO) level with close intramolecular O–H···O=C bond.
Given that the regiochemisty of the epoxide opening was previously established by Tatsuta et al.14 in the BE-52440 series by chemical synthesis and X-ray structure analysis, we propose that the conversion of hypogeamicin B (2) to A (1) likely occurs by ring opening of the epoxide by a sulfur nucleophile (formally H2S) to the less sterically congested epoxide position C-4a followed by antiperiplanar addition of this intermediate species again to the C-4a epoxide of hypogeamicin B (2) to generate the dimeric natural product (Figure 3). To test this hypothesis, we incubated hypogeamicin B (2) with one molar equivalent of sodium sulfide in dioxane in accordance with literature precedent in the BE-52440 series. Notably, the regiochemisty of the epoxide opening was established in the BE-52440 series by chemical synthesis and X-ray structure analysis.14 We observed conversion of hypogeamicin B (2) to a new dimeric species, compound 1 (m/z = 785.2605), and subsequent to isolation by C18-HPLC, determined it to be spectroscopically identical to hypogeamicin A (1) by 1H and 13C NMR analysis (Table S5). On the basis of the likely conservation of epoxide ring opening stereo- and regiochemistry and our stereochemical assignment of hypogeamicin B (2), we propose that hypogeamycin A (1) as shown in Figure 1 possessed the (1R,3R,4aR,10aS,10R,3′R,4a′R,10a′S)-configuration. Notably, this is the same configuration as observed in the BE-52440 series.
Figure 3.

Chemical thiolysis of hypogeamycin B (2) generating hypogeamycin A (1).
Hypogeamicin A (1) demonstrated no detectable activity against B. subtillis, suggesting that dimerization of this class of molecules obviates antibacterial activity. However, in assays against human colon cancer cell line TCT-1, compound 1 displayed activity comparable to the BE-52440 series (IC50 = 6.4–12.8 μM) but still significantly lower than the cytotoxicity of paclitaxel against human tumor cell lines (IC50 = 2.5–7.5 nM).29 Notably, hypogeamicins B–D (2–4) possess no observable cytotoxicity but detectable antimicrobial activity against B. subtilis (an MIC of ∼7 μg/mL for hypogeamicins B (2) and D (4) and an MIC of ∼28 μg/mL for hypogeamicin C (3)) comparable to the activity of nanaomycins (MICs of 6–8 ug/mL). However, when compared to other antibiotics, hypogeamicins B–D (2–4) are not as potent in antimicriobial activity against B. subtilis as for example erythromycin or gentamicin (MIC of 0.125 μg/mL for both).30 In conclusion, we have isolated four pyronaphthoquinone natural products from an actinomycete derived from a hypogean ecosystem that are structurally related to BE-52440 A and B, the only other reported S-bridged pyronaphthoquinone natural products. The absolute stereochemistry of hypogeamicin B (2) was determined from quantum calculations and comparison to experimental chiroptical spectra (see the Supporting Information), and the absolute stereochemistry of hypogeamicin A (1) was proposed by chemical transformation. Future studies of the biodiversity of hypogean actinomycetes will determine if the hypogean ecosystem fosters a complementary chemical diversity.
Experimental Section
General Experimental Procedures
Mass spectrometry was performed on a ThermoFinnigan LTQ ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) (ESI, positive and negative mode). HRESIMS was obtained on a Synapt hybrid quadrupole/oa-TOF mass spectrometer (Waters Corp., Milford, MA, USA) equipped with a dual chemical ionization/electrospray (ESCI) source. NMR spectra were recorded on a 600 MHz Bruker AV-II FT NMR spectrometer with cryoprobe & LC-SPE (Bruker BioSpin Corp., Billerica, MA, USA). Methanol-d4 (Sigma-Aldrich) and dimethyl sulfoxide-d6 (Sigma-Aldrich) were used as a solvents, and the chemical shifts were referenced to the solvent signal of methanol-d4 (3.30 ppm).
Chiroptical Spectra
The optical rotations at discrete wavelengths, 633, 589, and 546 nm, were measured at a concentration of 2 mg/mL in CH3CN, with a 0.5 dm cell using an Autopol IV polarimeter. The ORD spectra are presented as specific rotations (in units of deg cm3 g–1 dm–1) as a function of wavelength (in units of nm). The electronic absorption (EA) and ECD spectra were recorded on a Jasco J720 spectrometer, at a concentration of 3 mg/mL in CH3CN using a 0.01 cm path length quartz cell. The solvent spectrum has been subtracted from the experimental ECD spectrum of the sample solution. The EA and ECD spectra are presented respectively as molar extinction and differential molar extinction, in units of L mol–1 cm–1, as a function of wavelength (in units of nm). The vibrational absorption (VA) and VCD spectra were recorded in the 2000–900 cm–1 region using a ChiralIR Fourier transform VCD spectrometer (BioTools, USA). The VCD spectra were recorded with a 1 h data collection time at 8 cm–1 resolution. VA and VCD spectra were measured in deuterated dimethysulfoxide (DMSO-d6) at a concentration of 3 mg/100 μL. The samples were held in a 100 μm fixed path length cell with BaF2 windows. In the VA spectrum, the solvent absorption was subtracted. Similarly, the solvent VCD spectrum was subtracted from that of the sample solution. The region below 1050 cm–1 has been excluded from presentation due to interference from solvent absorption. The VA and VCD spectra are presented respectively as molar extinction and differential molar extinction, in units of L mol–1 cm–1, as a function of wavenumber (in units of cm–1).
The computational details for EA, ECD, ORD, VA, and VCD spectra and the comparison of computed spectra to experimental spectra are presented in the Supporting Information
Isolation of Nonomuraea specus from Cave Sediments
Soil samples were collected from nine locations throughout the Hardin’s cave system located close to Ashland City, Tennessee, using sterile Falcon tubes and a spatula. A 100 mg portion of desiccated sediment sample was vortexed with 1 mL of sterile water, and the supernatant was subjected to serial dillution and plated on the agars selective for actinobacteria. Agar plates were then incubated at 30 °C for about 3 weeks and screened for the presence of actinobacteria single colonies. N. specus was obtained from a highly decomposed bark sample collected far from the cave entrance as one of 20 other actinomycetes derived from cave sediments.
Taxonomy of Producing Organism
The 16S rRNA was amplified with 27F (AGA GTT TGA TCC TGG CTC AG) and 1525R (AAG GAG GTG ATC CAG CCG CA) universal primers, transformed into the TOP10 cells, and sequenced with M13F (GTT TTC CCA GTC ACG AC) and M13R (CAG GAA ACA GCT ATG AC), resulting 98.8% identity with Nonomuraea salmonea (DSM 43678). A phylogenetic tree was constructed by using the neighbor-joining method in MEGA5 (see Figure S45).
Fermentation, Extraction, and Isolation
Seed cultures (20 mL) were transferred to a 2.8 L Fernbach flask containing 500 mL of production media (glucose 1.0%, glycerol 0.5%, corn steep liquor 0.3%, beef extract 0.3%, malt extract 0.3%, CaCO3 0.2%, and thiamin 0.01%, added after autoclave by sterile filtration (pH 7.2)). Fermentation was carried out for 7 days at 30 °C on a rotary shaker at 180 rpm. Ethyl acetate (250 mL) was added to the culture, and the biphasic mixture was agitated to produce an emulsion via rotary shaking at 180 rpm for 30 min. The phases were separated by centrifugation, and the ethyl acetate layer was decanted, dried with anhydrous MgSO4, filtered by vacuum filtration, and evaporated to dryness via rotary evaporation to produce a crude extract. The Crude extract was subjected to LH20 chromatography (Sephadex, GE Healthcare, Buckinghamshire, England) by resuspension in 3 mL of methanol and elution with methanol. Fractions containing abundant metabolites were combined based on thin-layer chromatographic analysis and evaporated to dryness. Hypogeamicins were purified by reverse-phase HPLC (XBridge C18, 10 × 150 mm, 5 μm) with a water/acetonitrile/ammonium acetate gradient (5% acetonitrile in 10 mM ammonium acetate to 95% acetonitrile over 60 min and held at 95% acetonitrile for 10 min). The flow rate was 5 mL/min, and the injection volume was 500 μL.
Chemical Thiolysis of Hypogeamicin B (2)
Hypogeamicin B (2) (5 mg) was incubated with one molar equivalent of sodium sulfide in 1,4-dioxane at 25 °C for 30 min and monitored with TLC. After 30 min of stirring the solvent was evaporated in vacuo and the residue was subjected to RP-HPLC, yielding 4.2 mg of pure hypogeamicin A (1).
Biological Activity
Colon cancer (RKO) and human mucoepidermoid pulmonary carcinoma-derived NCI-H292 cells were obtained from ATCC. Cells were cultured in RPMI-1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (Atlas Biologicals) and were maintained in humidified cell culture incubators under 5% CO2, 95% air. For viability assays, cells were dislodged from culture vessels by trypsinization, counted using a model Z1 cell counter (Coulter), and seeded in 96-well culture plates at a density of 7500 cells per well. After the cells had adhered for 24 h, compound 1 was dissolved in DMSO and serial dilutions were performed to achieve the desired concentrations (200×), such that final DMSO in the cell culture was 0.5%. After 48 h treatment with compound, media was aspirated and culture plates were washed once with Dulbecco’s phosphate-buffered saline (D-PBS), then replaced with a solution of 1 μM calcein-AM (Molecular Probes) in D-PBS. After 30 min, fluorescence was read using a SpectraMax multiwell plate reader (Molecular Devices) with an excitation of 494 nm and emission of 517 nm.
Acknowledgments
This work was supported by the National Institutes for Health (GM092218), the National Science Foundation (CHE-0804301), and Vanderbilt Institute for Chemical Biology. Computation work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University, Nashville, TN.
Supporting Information Available
1D and 2D NMR spectra for compounds 1–4, quantum chemical calculations method, ECD and VCD plots for compound 2, and 16S rRNA phylogenetic tree are presented in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Gaussian, Inc. 340 Quinnipiac Street, Building 40 Wallingford, CT 06492, USA.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Barton H. A. J. Cave Karst. Stud. 2006, 68, 43. [Google Scholar]
- Barton H. A.; Taylor N. M.; Kreate M. P.; Springer A. C.; Oehrle S. A.; Bertog J. L. Int. J. Speleol. 2007, 36, 93. [Google Scholar]
- Barton H. A.; Taylor M. R.; Pace N. R. Geomicrobiol. J. 2004, 21, 11. [Google Scholar]
- Bhullar K.; Waglechner N.; Pawlowski A.; Koteva K.; Banks E. D.; Johnston M. D.; Barton H. A.; Wright G. D. PLoS One 2012, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdy J. J. Antibiot. 2005, 58, 1. [DOI] [PubMed] [Google Scholar]
- Jensen P. R.; Mincer T. J.; Williams P. G.; Fenical W. Anton. Leeuw. Int. J. G 2005, 87, 43. [DOI] [PubMed] [Google Scholar]
- Magarvey N. A.; Keller J. M.; Bernan V.; Dworkin M.; Sherman D. H. Appl. Environ. Microb. 2004, 70, 7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nett M.; Ikeda H.; Moore B. S. Nat. Prod. Rep. 2009, 26, 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto M.; Nakajima S.; Murooka K.; Naito M.; Suzuki H.; Hirayama M.; Hirano K.; Kojiri K.; Suda H. J. Antibiot. 2000, 53, 687. [DOI] [PubMed] [Google Scholar]
- Porter J. N.; Wilhelm J. J.; Tresner N. D. Appl. Microbiol. 1960, 8, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Koyama Y.; Awaya J.; Marumo H.; Oiwa R.; Katagiri M.; Nagai T.; Omura S. J. Antibiot. 1975, 28, 860. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Koyama Y.; Nagai T.; Marumo H.; Omura S. J. Antibiot. 1975, 28, 868. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Marumo H.; Nagai T.; Okada M.; Taniguchi K.; Omura S. J. Antibiot. 1975, 28, 925. [DOI] [PubMed] [Google Scholar]
- Tatsuta K.; Suzuki Y.; Toriumi T.; Furuya Y.; Hosokawa S. Tetrahedron Lett. 2007, 48, 8018. [Google Scholar]
- Stephens P. J.; Pan J. J.; Devlin F. J.; Cheeseman J. R. J. Nat. Prod. 2008, 71, 285. [DOI] [PubMed] [Google Scholar]
- Berova N.; Polavarapu P. L.; Nakanishi K. Woody R. W.. Comprehensive Chiroptical Spectroscopy, 2012 ed.; Wiley & Sons, 2012; Vols. 1–2. [Google Scholar]
- Polavarapu P. L. Chirality 2008, 20, 664. [DOI] [PubMed] [Google Scholar]
- Polavarapu P. L. Chirality 2012, 24, 909. [DOI] [PubMed] [Google Scholar]
- Autschbach J.; Ziegler T.; van Gisbergen S. J. A.; Baerends E. J. J. Chem. Phys. 2002, 116, 6930. [Google Scholar]
- Autschbach J. Chirality 2009, 21, E116. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. J. Phys. Chem. A 2009, 113, 767. [DOI] [PubMed] [Google Scholar]
- Cheeseman J. R.; Frisch M. J.; Devlin F. J.; Stephens P. J. Chem. Phys. Lett. 1996, 252, 211. [Google Scholar]
- Stephens P. J.; Devlin F. J.; Cheeseman J. R.; Frisch M. J. J. Phys. Chem. A 2001, 105, 5356. [Google Scholar]
- Crawford T. D.; Tam M. C.; Abrams M. L. J. Phys. Chem. A 2007, 111, 12057. [DOI] [PubMed] [Google Scholar]
- Pecul M.; Ruud K. Adv. Quantum Chem. 2005, 50, 185. [Google Scholar]
- Convington C. L.; Polavarapu P. L.. CDSpecTech: A program for calculating similarity overlap between experimental and calculated chiroptical spectra http://sites.google.com/site/cdspectech1/.
- Covington C. L.; Polavarapu P. L. J. Phys. Chem. A 2013, 117, 3377. [DOI] [PubMed] [Google Scholar]
- Ellestad G. A.; Kunstman M. P.; Whaley H. A.; Patterson E. L. J. Am. Chem. Soc. 1968, 90, 1325. [DOI] [PubMed] [Google Scholar]
- Liebmann J. E.; Cook J. A.; Lipschultz C.; Teague D.; Fisher J.; Mitchell J. B. Br. J. Cancer 1993, 68, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. T.; Connelly M. B.; Amolo C.; Otani S.; Yaver D. S. Antimicrob. Agents Chemother. 2005, 49, 1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.