

Abstract
The emergence of multidrug-resistant Staphylococcus aureus (S. aureus) makes the treatment of infectious diseases in hospitals more difficult and increases the mortality of the patients. In this study, we attempted to identify novel potent antibiotic candidate compounds against S. aureus dihydrofolate reductase (saDHFR). We performed three-step in silico structure-based drug screening (SBDS) based on the crystal structure of saDHFR using a 154,118 chemical compound library. We subsequently evaluated whether candidate chemical compounds exhibited inhibitory effects on the growth of the model bacterium: Staphylococcus epidermidis (S. epidermidis). The compound KB1 showed a strong inhibitory effect on the growth of S. epidermidis. Moreover, we rescreened chemical structures similar to KB1 from a 461,397 chemical compound library. Three of the four KB1 analogs (KBS1, KBS3, and KBS4) showed inhibitory effects on the growth of S. epidermidis and enzyme inhibitory effects on saDHFR. We performed structure–activity relationship (SAR) analysis of active chemical compounds and observed a correlative relationship among the IC50 values, interaction residues, and structure scaffolds. In addition, the active chemical compounds (KB1, KBS3, and KBS4) had no inhibitory effects on the growth of model enterobacteria (E. coli BL21 and JM109 strains) and no toxic effects on cultured mammalian cells (MDCK cells). Results obtained from Protein Ligand Interaction Fingerprint (PLIF) and Ligand Interaction (LI) analyses suggested that all of the active compounds exhibited potential inhibitory effects on mutated saDHFR of the drug-resistant strains. The structural and experimental information concerning these novel chemical compounds will likely contribute to the development of new antibiotics for both wild-type and drug-resistant S. aureus.
1. Introduction
S. aureus is a causative microorganism for nosocomial infection, and high rates of morbidity and mortality associated with S. aureus infectious diseases have been observed in many areas around the world.1,2 Moreover, the emergence of multidrug-resistant S. aureus, such as methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA), makes the treatment of nosocomial infections more difficult, thereby increasing the mortality of the patients.3,4S. aureus infections occur not only in hospitals but also in various social communities; furthermore, social community-associated (CA) MRSA and VRSA have been frequently observed in recent years.1,5 CA-MRSA is an epidemic, particularly observed in the USA, that is characterized by rapid spreading and by the production of Panton-Valentine leukocidin (PVL), which causes several deadly illnesses and more strongly virulent diseases than hospital-associated MRSA.6−8 Although new types of resistant S. aureus have been anticipated, the number of new drugs developed against S. aureus has gradually decreased.9 Therefore, the lack of effective antibacterial drugs against the resistant S. aureus strains might become a large threat in the near future. Thus, it is important to develop new antibacterial drugs targeting MRSA, VRSA, and multidrug-resistant S. aureus.
saDHFR is an enzyme that catalyzes the chemical reaction for the reduction of tetrahydrofolate (THF) from dihydrofolate (DHF) through NADPH. DHFR is essential in the pathways for the intracellular production of purines, such as adenine and guanine.10 Therefore, specific inhibitors of DHFR block DNA replication in S. aureus, eventually leading to bacterial death.11 Drugs targeting DHFR include methotrexate (MTX), originally developed as an anticancer drug,12 and trimethoprim (TMP), developed and still used as an antibacterial drug.10,11 TMP is prescribed as cotrimoxazole (Bactrim), comprising TMP and sulfamethoxazole, which inhibits the bacterial-specific enzyme dihydropteroate synthase (DHPS).10,12S. aureus that are resistant to TMP with a diaminopyrimidine (DAP) ring have recently emerged, and the DHFR of the resistant strains contains mutated amino acid residues, including Phe 98 to Tyr (F98Y).10 The Phe to Tyr change at position 98 is the most important mutation residue to cause TMP resistance.13 In addition, it is known that approximately 28% of MRSA show TMP resistance.14 Therefore, the identification of chemical compounds with chemical scaffolds unlike TMP is strongly suggested for the treatment of patients infected with TMP-resistant S. aureus strains.
SBDS is an effective technique for novel drug discovery. In silico SBDS through docking simulations between target proteins and chemical compounds is an efficient screening method to identify candidate compounds from a large chemical database because of the reduced time and cost for hit chemical identification.15 Successful identification of antibacterial chemical compounds through in silico SBDS has been reported.16−19In silico SBDS has been performed using docking simulation tools, such as GOLD,20 DOCK,21 GLIDE,22 FRED,23 and AutoDock.24 Multistep in silico SBDS using combinations of the docking simulation tools have been used to more effectively identify active chemical compounds.15 In a previous study, we identified potent growth inhibitors targeting Mycobacterium through multistep in silico SBDS,17−19 and the strategy of using multiple chemical conformers could improve the accuracy of docking simulations.18,19 In the present study, we performed a three-step in silico SBDS to target the crystal structure of saDHFR from 154,118 chemical compounds library. Subsequently, we rescreened chemical compounds similar to the active hits obtained from the SBDS using 461,397 chemical compounds library. We identified four chemical compounds showing antibacterial effects against a Staphylococcal strain and inhibitory effects on the enzymatic activity of the targeted protein. In addition, we confirmed that three of the four identified chemical compounds did not show inhibitory effects on the growth of model enterobacteria or toxic effects on cultured mammalian cells. These results will contribute to the development of novel antibacterial therapies against drug-resistant S. aureus.
2. Results
2.1. Three-Step in Silico SBDS
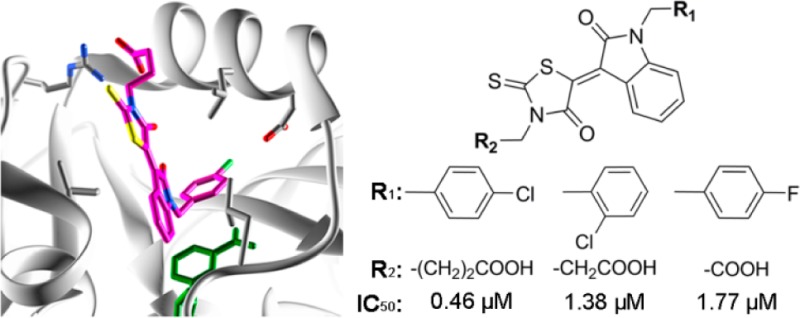
We performed three-step in silico SBDS targeting saDHFR with a virtual chemical compound library (154,118 chemical compounds). The three-step SBDS involved initial screening using DOCK, followed by screening using GOLD with a single chemical conformer and a third screening using GOLD with multiple chemical conformers (Figure 1A). The active site of saDHFR, comprises amino acid residues: Val 6, Ala 7, Leu 20, Pro 25, Asp 27, Leu 28, Val 31, Ser 49, Ile 50, Arg 57, Phe 92, and Thr 111 (Figure 2).10 We screened candidate chemical compounds with high potential of binding affinity for the active site of saDHFR. In the first screen, the docking simulations with DOCK predicted 500 top-ranked chemical compounds (0.3% of the primary chemical compound library) with DOCK scores of less than −48.5 kcal/mol. The calculation speed of DOCK-based screening is fast, reflecting grid-based calculations without hydrogen bond (H-bond) energy through PC clustering. However, the accuracy of the calculations is relatively low [the area under the curve (AUC) values of receiver-operating characteristic (ROC) = 0.56; Figure S1]. In the second screen, we used the top-ranked 500 chemical compounds with conformations outputted after the first DOCK screen. GOLD is a flexible docking simulation tool using genetic algorithm (the AUC values of ROC = 0.89; Figure S1). After the docking simulations with GOLD, we selected 139 top-ranked chemical compounds (GOLD scores >70) from the 500 chemical compounds. In the third screen, we used the multiconformational chemical structures with at most eleven conformations per chemical compound (the AUC values of ROC = 0.95; Figure S1). The 10 top-ranked chemical compounds generated GOLD scores greater than 80. We removed similar chemical compounds with common structures and selected the candidate chemical compounds that had not previously been evaluated in high throughput screening (HTS) using as deposited in the Pubchem Web-based chemical compounds database.25 Finally, we identified five chemical compounds (KB1-KB5) with an average of GOLD scores >82 (Table 1). The GOLD score of KB1 from saDHFR simulation is higher than that of the natural endogenous substrate, DHF (Table 1).
Figure 1.

Flowchart of the in silico SBDS and chemical analog search strategy applied in this study. (A) Flowchart of the identification of candidate chemical compounds using three-step in silico SBDS. (B) Flowchart of the identification of KB1 analogs.
Figure 2.
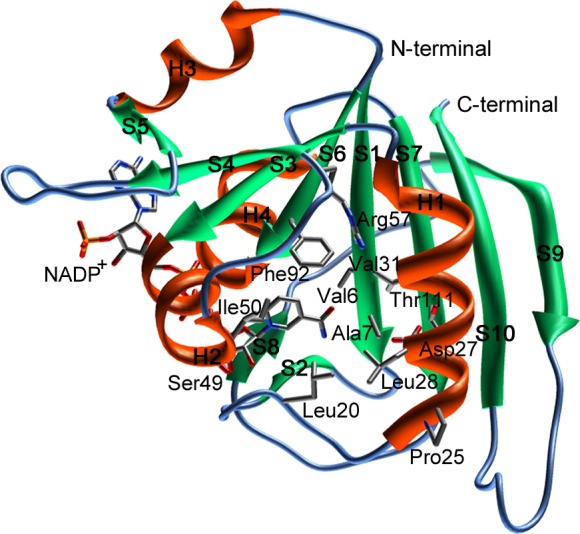
The three-dimensional structure of saDHFR with the NADP+ cofactor. The secondary structures are indicated using ribbon representations [α-helix (H1–H4): orange, β-sheet (S1–S10): green]. The amino acid residues of the active site are shown as a stick model.
Table 1. Five Chemical Compounds Identified through SBDS Analysis Using a Natural Substratea.
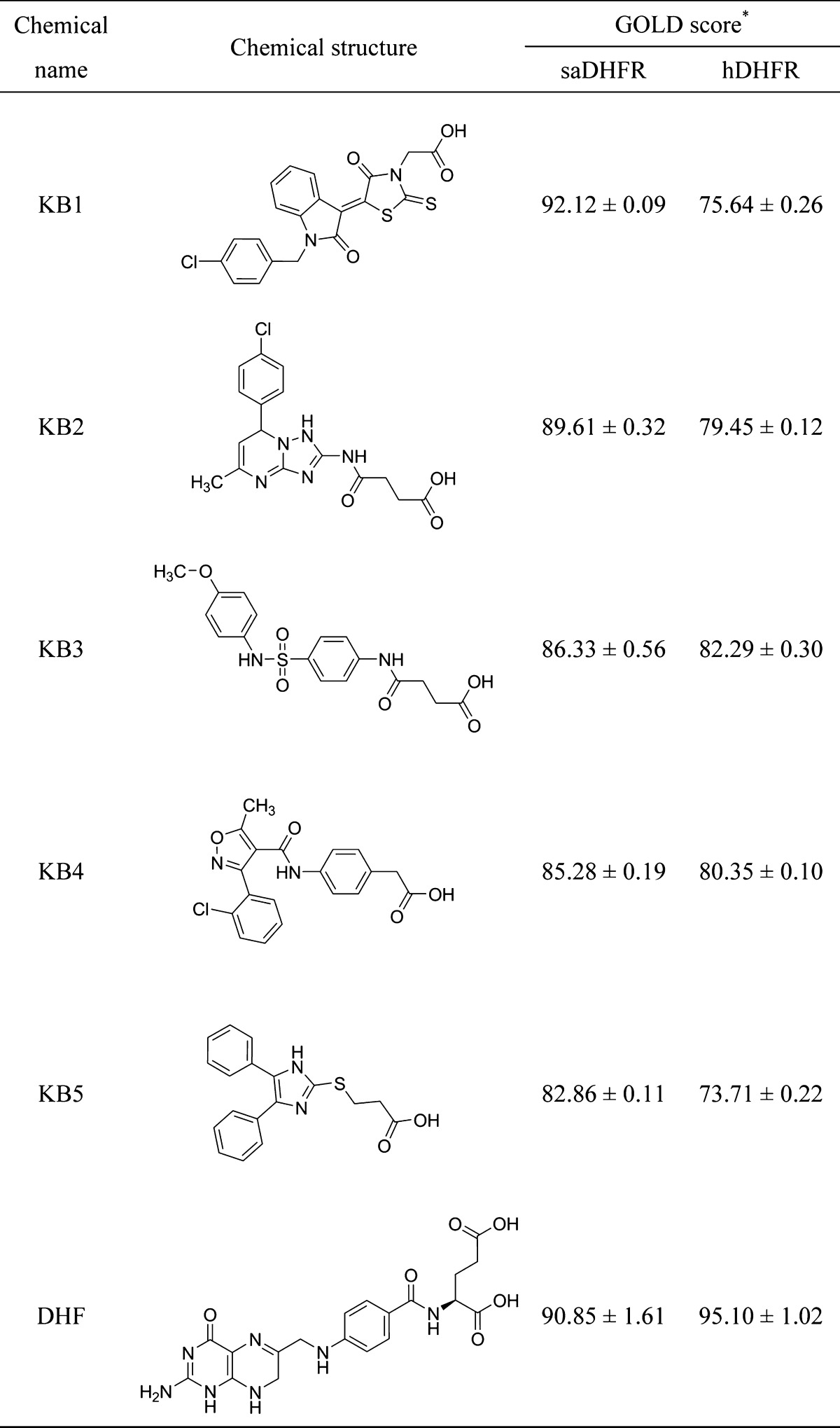
The asterisk denotes the following: each value represents the mean ± SEM.
We compared the amino acid sequence of human DHFR (hDHFR) with that of saDHFR using the Basic Local Alignment Search Tool (BLAST)26 and the Universal Protein Resource database (Uniprot)27 and observed that the amino acid sequence homology between both proteins was low (similarity rate = 48%, identity rate = 26%). The GOLD scores of KB1-KB5 when hDHFR is the target are less than those scores arising when targeting saDHFR (Table 1), indicating that KB1-KB5 were predicted to specifically bind to the active site of saDHFR.
2.2. Bacterial Growth Assay of the Candidate Chemical Compounds
We examined the inhibitory effects of the five candidate chemical compounds (KB1-KB5) on bacterial growth, as predicted using three-step in silico SBDSs. Because S. aureus (Biosafety Level 2) carries infectious risks for humans, we could not perform complex bacterial experiments (time course growth and IC50 determination assays with 96-well assay plates) using S. aureus. For the bacterial growth assays, we used wild-type S. epidermidis ATCC 12228, which does not pose a risk of infection to humans (Biosafety Level 1), as a model bacterial strain. The BLAST26 and the Uniprot27 analyses showed that the amino acid sequence of S. epidermidis DHFR (seDHFR) is similar to that of saDHFR (similarly rate = 94%, identity rate = 82%). All of the amino acid residues in the active site of saDHFR are completely conserved in seDHFR (Figure S2A). In addition, we generated the three-dimensional structure of seDHFR by homology modeling. Figure S2B shows the three-dimensional structures of saDHFR and seDHFR. The mean values of the RMSD for the structure of the active site pocket (including substrate recognition sites) between saDHFR and seDHFR are extremely low; 0.47 Å (Figure S2B, C). Thus, it is expected that the active site structure of seDHFR is extremely similar to that of saDHFR. The five candidate chemical compounds (100 μM) showed inhibitory effects on the growth of S. epidermidis. KB1 showed strong inhibitory effects on the growth of S. epidermidis (Figure 3). The other four candidate chemical compounds (KB2-KB5) had no significant or only weak inhibitory effects on bacterial growth compared with that of KB1. KB1 also showed inhibitory effects on the growth of S. epidermidis similar to those of ampicillin.
Figure 3.

Inhibitory effects of the candidate chemical compounds (KB1-KB5) on the growth of S. epidermidis after 6 h. The concentrations of the chemical compounds were 100 μM. DMSO (0.3%) and ampicillin (100 μg/mL) were used as the negative and positive controls, respectively. Each value represents the mean ± SEM of four independent experiments. Bonferroni’s all-pair comparison test was performed (n.s. not significant, *** p < 0.001).
2.3. Effects of Compound KB1 on E. coli and Mammalian Cells
We examined whether KB1 exhibited inhibitory effects on the growth of the model enterobacterium E. coli BL21 and JM109 strains and toxic effects on mammalian cells (Madine-Darby Canine Kidney: MDCK and human neuroblastoma: SH-SY5Y cells). KB1 did not show inhibitory effects on the growth of E. coli BL21 (Figure 4A) and JM109 (data not shown) strains after 4 and 8 h. Additionally, KB1 did not show any toxic effects on MDCK cells (Figure 4B); however, toxic effects on SH-SY5Y cells were observed (data not shown; 43.3% of cell death is induced).
Figure 4.
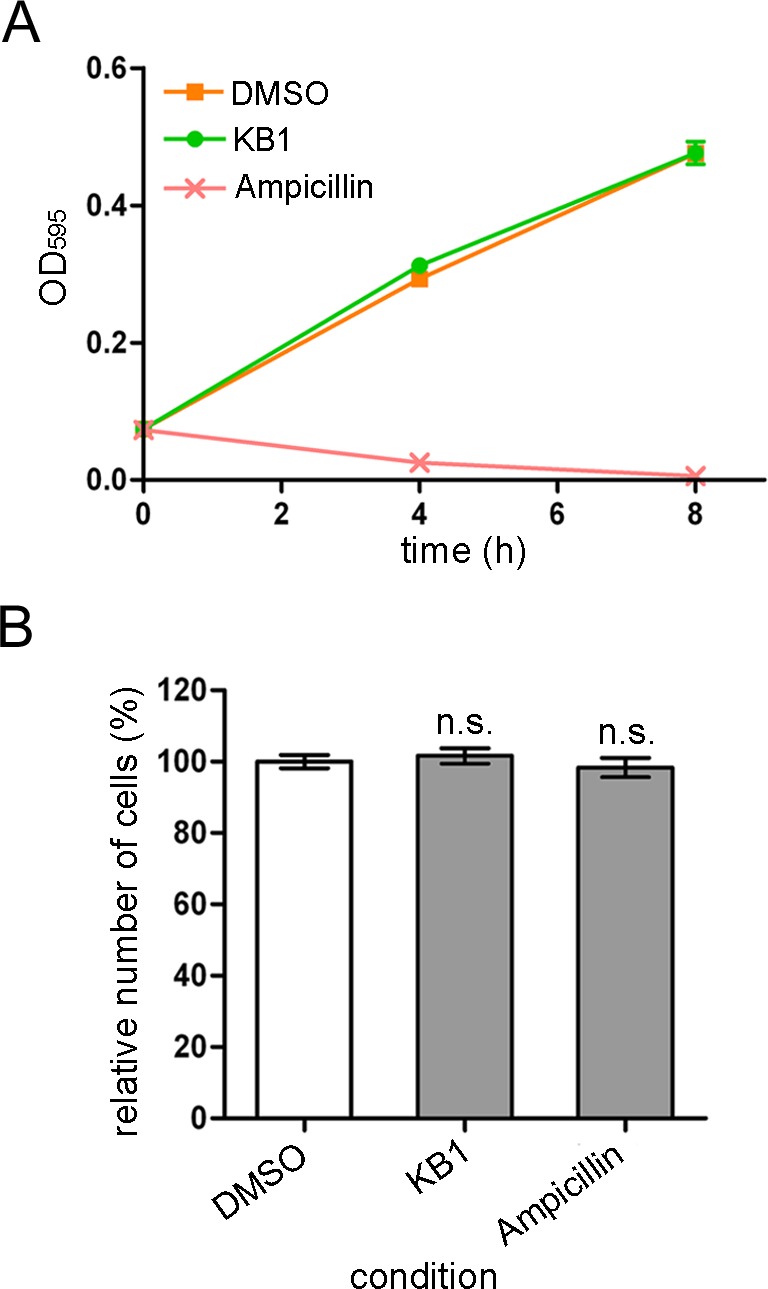
Effects of the chemical compound (KB1) on E. coli and MDCK cells. (A) Effect of KB1 on E. coli BL21 strains. The concentrations of the chemical compounds was 100 μM. DMSO (0.3%) and ampicillin (100 μM) were used as the negative and positive controls, respectively. (B) Effects of KB1 on MDCK cells. The chemical compounds (30 μM; IC99 value for S. epidermidis bacterial growth) were added to the cultures. Each value represents the mean ± SEM of four independent experiments. Dunnett’s multiple comparison tests were performed (n.s. not significant, ** p < 0.01).
2.4. Screening KB1 Analogs Based on Chemical Structure Similarities
We screened KB1 analogs expected to have inhibitory effects similar to those of KB1 (Figure 1B). We selected 23 KB1 analogs from a Web-based database (Hit2LEAD.com).28 The 23 KB1 analogs had chemical structures with a similarity rate (Tanimoto coefficient) greater than 85%. Subsequently, we performed docking simulations targeting saDHFR with multiconformation of the 23 KB1 analogs using GOLD. The simulations predicted five top-ranked chemical compounds (KBS1-KBS5, Table 2), which were not previously identified using HTS (as available in the PubChem database).25 The GOLD scores of the five KB1 analogs (KBS1-KBS5) were calculated through docking simulations with saDHFR, showing higher score values than those calculated through docking simulations with hDHFR (Table 2). The analogs KBS1-KBS5 have substituted carboxyl and chlorobenzyl groups and similar structures compared with KB1. KBS1 was the only analog with a GOLD score higher than that of KB1 (Table 2).
Table 2. KB1 Analogs with Different R1 and R2 Substituentsa.
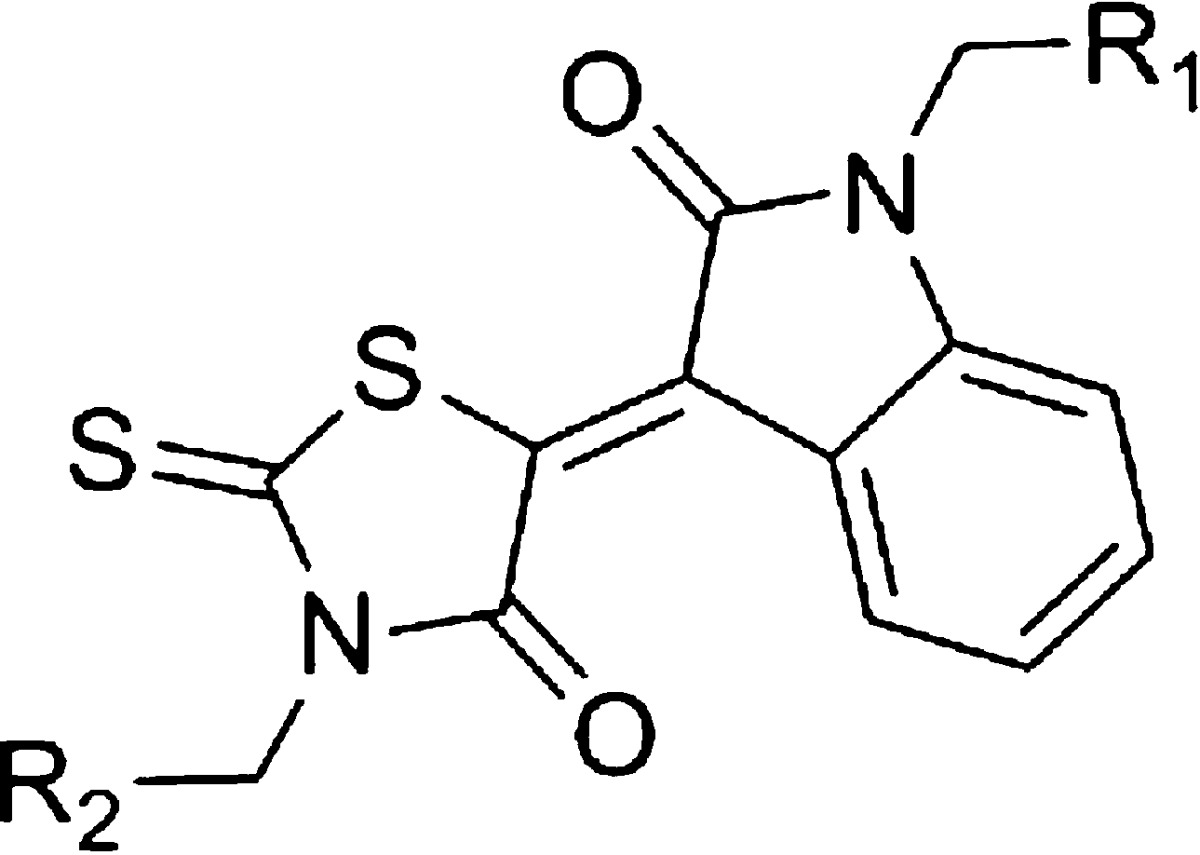
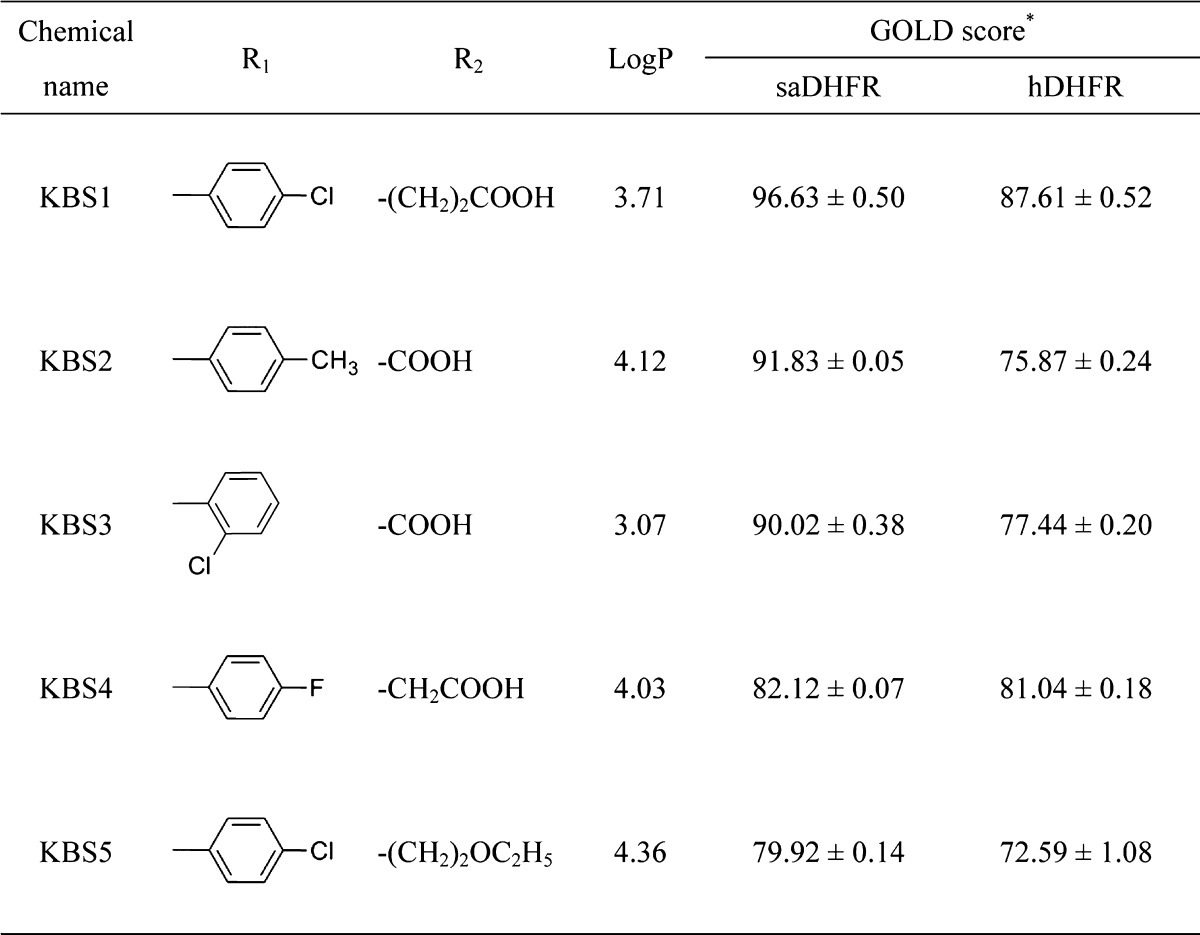
The asterisk denotes the following: each value represents the mean ± SEM.
2.5. Effects of KB1 Analogs on S. epidermidis, E. coli, and Mammalian Cells
We examined whether the five KB1 analogs (KBS1-KBS5) exhibited an inhibitory effect on the growth of S. epidermidis. We did not examine the effects of compound KBS2, as this compound was not soluble at 100 μM in DMSO. The three chemical compounds (KBS1, KBS3, and KBS4) exhibited significant inhibitory effects on the growth of S. epidermidis (Figure 5).
Figure 5.

Inhibitory effects of the candidate chemical compounds (KBS1 and KBS3-KBS5) on the growth of the S. epidermidis after 6 h. The concentrations of the chemical compounds were 100 μM. DMSO (0.3%) and ampicillin (100 μg/mL) was used as the negative and positive controls, respectively. Each value represents the mean ± SEM of four independent experiments. Bonferroni’s all-pair comparison test was performed (n.s. not significant, *** p < 0.001).
Furthermore, we examined whether these hit chemical compounds exhibited inhibitory effects on the growth of E. coli (BL21 and JM109 strains) and toxic effects on cultured mammalian cells. The results showed that these hit chemical compounds (KBS1, KBS3, and KBS4) showed no inhibitory effects on the growth of E. coli BL21 (Figure 6A) and JM109 (data not shown) strains. KBS3 and KBS4 did not exhibit toxic effects on MDCK cells, although a weak toxic effect of KBS1 on MDCK cells was detected (Figure 6B). The hit compounds (KBS1, KBS3, and KBS4) also showed toxic effects on SH-SY5Y cells (data not shown; 35.4–52.1% of cell death is induced).
Figure 6.
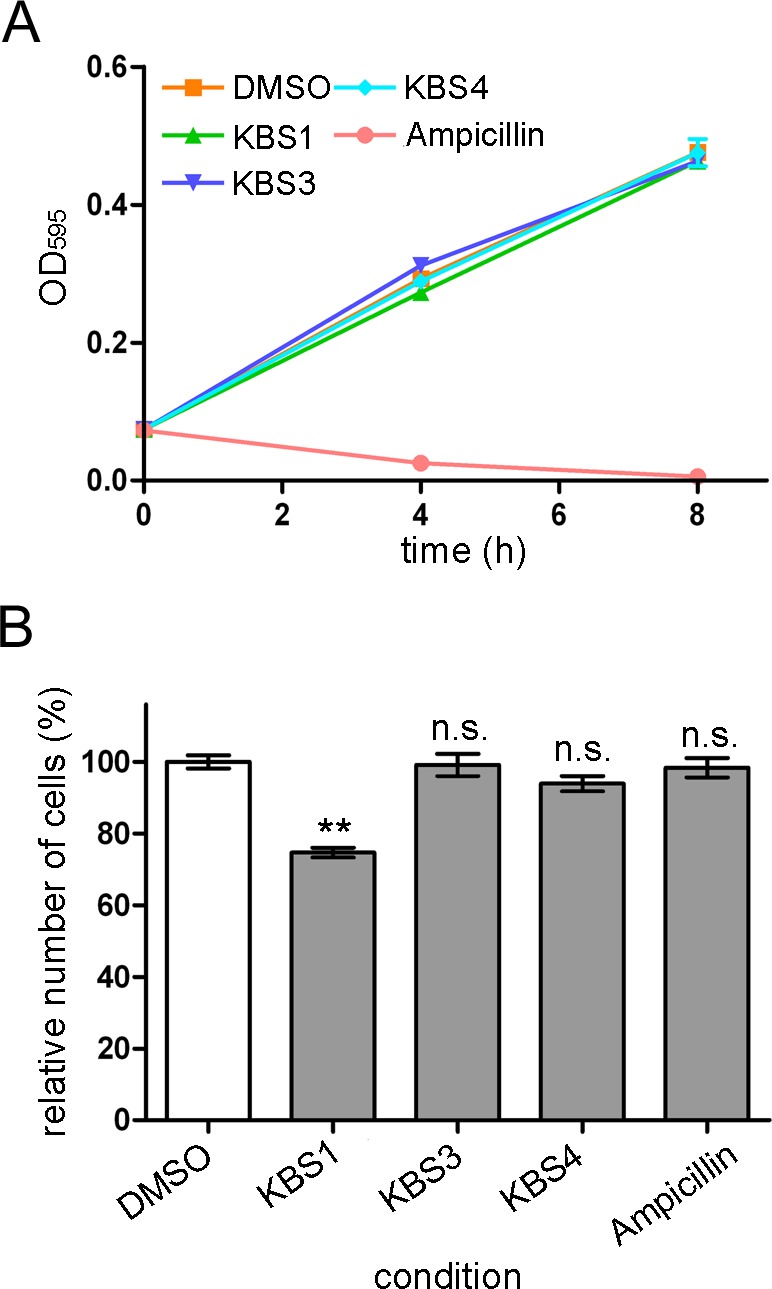
Effects of the hit chemical compounds (KBS1, KBS3, and KBS4) on E. coli and MDCK cells. (A) Effects of KB1 analogs on E. coli BL21 strains (∼8 h). The concentrations of the hit chemical compounds were 100 μM. DMSO (0.3%) and ampicillin (100 μM) were used as the negative and positive controls, respectively. (B) Effects of KB1 analogs on MDCK cells. The chemical compounds (30 μM; IC99 value for S. epidermidis bacterial growth) were added to the cultures. Each value represents the mean ± SEM of four independent experiments. Dunnett’s multiple comparison tests were performed (n.s. not significant, ** p < 0.01).
2.6. Determination IC50 Values of Hit Chemical Compounds
We investigated the dose-dependent effects of the four chemical compounds (KB1, KBS1, KBS3, and KBS4) on the growth of S. epidermidis. The experimentally determined IC50 values of KB1, KBS1, KBS3, and KBS4 were 3.84 ± 0.27, 15.7 ± 5.15, 12.0 ± 0.60, and 9.46 ± 0.48 μM, respectively (Figure 7A-D, Table 3).
Figure 7.

The dose-dependent effects of compounds KB1, KBS1, KBS3, and KBS4 on the growth of S. epidermidis (A) KB1, (B) KBS1, (C) KBS3, and (D) KBS4. The bacterial growth rate (%) during the 6 h cultivation of S. epidermidis. Each plotted value represents the mean ± SD of four independent experiments. Each IC50 value was determined through nonlinear regression analysis.
Table 3. Predicted Interaction Residues in the Active/Inactive Chemical Compoundsa.
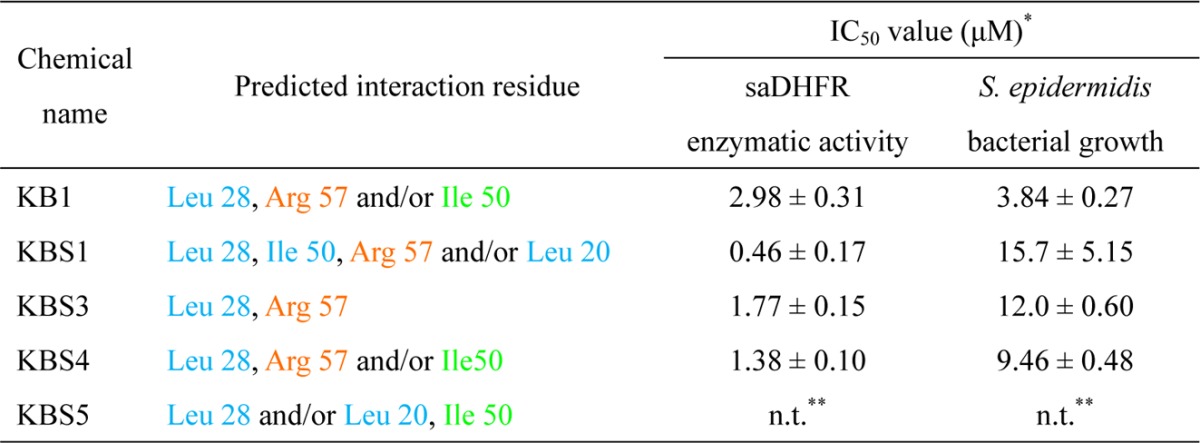
The asterisks denote the following: (*) each value represents the mean ± SEM; (**) not tested.
2.7. S. aureus DHFR Enzymatic Assay
We investigated the enzyme inhibitory activity of the hit compounds (KB1, KBS1, KBS3, and KBS4). In the enzyme inhibition assay, the reaction carried out with wild-type saDHFR was assessed under saturating conditions of substrate and cofactor, and the level of inhibition was determined relative to an uninhibited negative control reaction. All hit compounds inhibited saDHFR activity in a manner similar to that of the competitive inhibitor TMP (IC50 value = 0.008 ± 0.003 μM). The experimentally determined IC50 values of KB1, KBS1, KBS3, and KBS4 were 2.98 ± 0.31, 0.46 ± 0.17, 1.77 ± 0.15, and 1.38 ± 0.10 μM, respectively (Figure 8A-D, Table 3).
Figure 8.

The dose-dependent effects (percent inhibition) of compounds KB1, KBS1, KBS3, and KBS4 on SaDHFR enzymatic activity (A) KB1, (B) KBS1, (C) KBS3, and (D) KBS4. The level of inhibition is expressed as a percentage of the remaining saDHFR activity compared to a reaction with no chemical compound (i.e., 0% inhibition). The result of each hit compound (KB1, KBS1, KBS3, and KBS4) is shown as a representative of four independent experiments. Each IC50 value represents the average of four independent experiments and was determined by performing nonlinear regression analysis.
2.8. Prediction of the Binding Modes of the Hit Chemical Compounds to saDHFR
We evaluated the detailed interactions between the target protein (saDHFR) and the hit chemical compounds (KB1, KBS1, KBS3, and KBS4) using PLIF and LI analyses.29 Figure 9 shows the predicted binding modes of the hit chemical compounds with the highest GOLD scores from multiconformation simulations. All of the hit chemical compounds were predicted to be located near the active site of saDHFR. The carboxyl group in the hit compounds forms H-bond with Arg 57 (Table 3). In addition, the 4-oxo-2-thioxo-1,3-thiazolidin group in the hit compounds forms van der Waals (vdW) contacts with Leu 28. The 2-pyrrolidone group in KB1 and KBS4 forms arene-H interactions with Ile 50 and the 2-oxo-1,2-dihydro-3H-indol-3-ylidene group in KBS1, and the 2-pyrrolidone group in KBS1 forms vdW contacts with Leu 20 and Ile 50, respectively. Moreover, all hit compounds were not predicted to interact with Phe 98 (mutation of the residue in TMP-resistant saDHFR). In contrast, the inactive compound KBS5, without a carboxyl group, did not form an H-bond with Arg 57. Moreover, the carboxyl groups of all hit chemical compounds were predicted to be located near one of two carboxyl groups similar to the natural endogenous substrate DHF. The carboxyl group of DHF forms an H-bond with Arg 57 (Figure 10).
Figure 9.
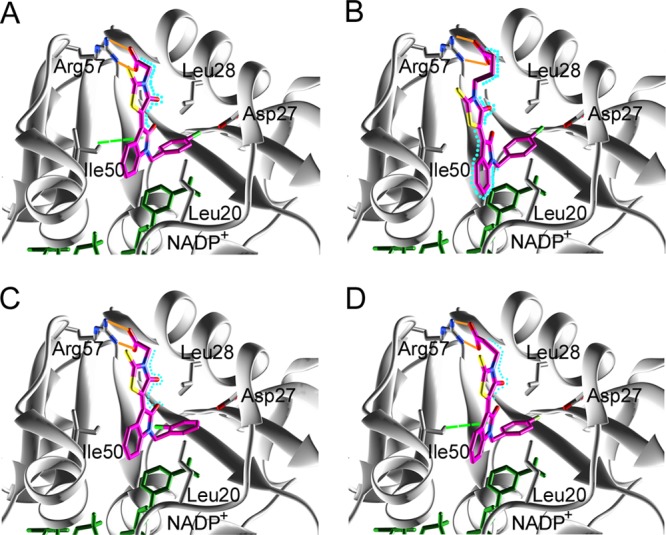
The predicted binding modes of hit chemical compounds with the saDHFR based on PLIF and LI analyses: (A) KB1, (B) KBS1, (C) KBS3, and (D) KBS4. The H-bond, arene-H, and vdW interactions are indicated with orange, light green dashed, and light blue dashed lines, respectively. The color of the amino acid interaction is identical to the colors of the amino acids indicated in Table 3.
Figure 10.
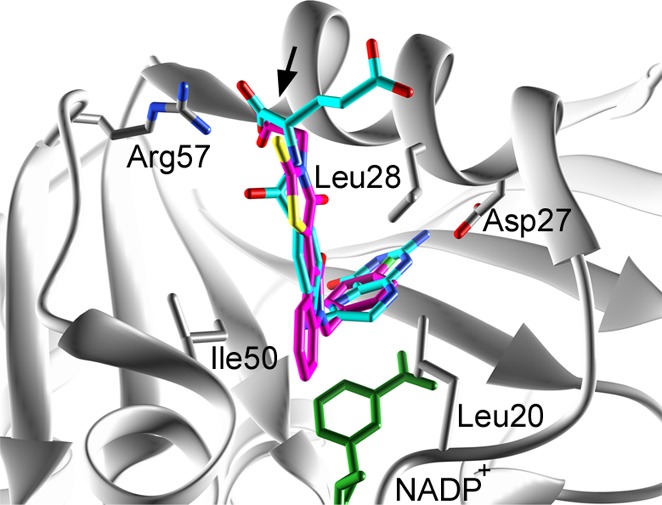
Predicted binding modes of KB1 of the hit chemical compound and the DHF substrate to the saDHFR. KB1 and DHF are indicated in magenta and light blue, respectively. The black arrow indicates the overlapping carboxyl group between DHF and KB1. KB1 is shown as a representative of active compounds.
3. Discussion
The in silico SBDS targeting of saDHFR revealed four hit compounds (KB1, KBS1, KBS3, and KBS4) exhibiting inhibitory effects on the growth of S. epidermidis and enzymatic activity of saDHFR. The PLIF and LI analyses predicted that the hit chemical compounds made vdW contacts with Leu 20 and Leu 28 similar to TMP, an inhibitor of DHFR.10 Particularly, vdW contacts with Leu 20 were only predicted in KBS1, suggesting that this interaction is considerably associated with the highest enzyme inhibitory activity of KBS1. We performed an SAR analysis of KB1, KBS1, and KBS3-KBS5 to identify the correlative relationships between the chemical structure scaffolds, biological activities, and interaction residues. The carboxyl groups in R2 were essential for the inhibitory effects on bacterial growth (Table 2 and Figure 5). In addition, the carboxyl groups in R2 were predicted to H-bond with Arg 57 at an interaction residue similar to that observed in the natural substrate DHF. Therefore, the interaction with Arg 57 likely plays an important role in the inhibitory effects of this compound. Furthermore, the activities of the compounds were altered through differences in the substituent groups in R1 (Tables 2 and 3).
Although the hit compounds showed inhibitory effects on both saDHFR enzymatic activity and S. epidermidis bacterial growth, different IC50 values were observed. It is possibly suggested that membrane penetration contributes to the inhibitory effects of these compounds on bacterial growth. The results of the bacterial growth assay showed that the activities of those compounds are enhanced by the smaller structure of R2 (Tables 2 and 3). In particular, the R2 of KB1, with a relatively small carbon chain, exhibits an inhibitory effect on S. epidermidis growth that is five times higher than that of KBS1. However, the results of enzyme activity assay showed that the longer carbon chain of R2 enhanced the activities of the compounds. Drug absorption and membrane permeability are influenced by molecular weight and LogP value.30 Although KBS1 and KBS4 with longer R2 carbon chains exhibit higher inhibitory activity against saDHFR, KBS1 and KBS4 have relatively lower inhibitory effects on bacterial growth. An SAR analysis of the hit compound will provide important information for development of chemical derivatives targeting S. aureus.
We used docking simulations with multiple chemical conformers and PLIF and LI analyses to estimate whether the chemical compounds identified in this study have inhibitory effects on TMP-resistant S. aureus strains. Most interaction residues predicted in wild-type saDHFR through PLIF and LI analyses (Table 3) were also identified in PLIF and LI analyses of TMP-resistant saDHFR (data not shown). These candidate chemical compounds were predicted not to interact with Tyr 98, which is mutated residues in TMP-resistant saDHFR.10 Therefore, the hit compounds identified in this study are expected to exhibit inhibitory effects on the growth of TMP-resistant S. aureus.
In general, HTS methods are used for hit identification and in the lead discovery phase of drug development.31 Although HTS have the advantage of hit compound identification from chemical libraries containing large numbers of compounds, this technique requires high-cost and long-term experiments, and moreover, the hit rate of HTS targeting for S. aureus was approximately 1%.32,33 In contrast, in silico strategies such as molecular docking, quantitative structure–activity relationship (QSAR),34 molecular dynamics (MD),35 and pharmacophore modeling36 have been developed to identify novel drugs,37 and these methods can be performed with lower costs and shorter times than HTS. The three-step in silico SBDS utilized in the present study initially obtained a 20% hit rate, including the identification of a high-efficacy growth inhibitor from five candidate chemical compounds. Furthermore, the screening of the active compound analogs considerably improved the hit rate of the drug screening; three growth inhibitors were identified from four candidate compounds (75% hit rate, IC50 < 15.7 μM). Therefore, this in silico drug screening strategy is expected to be effective for the identification of novel active compounds against other disease-causing agents.
4. Conclusion
In the present study, we identified five chemical compounds using three-step in silico SBDSs. These compounds were predicted to have a high binding affinity to saDHFR. The in vitro biological assay revealed that one of these compounds, KB1, exhibited antibiotic effects against S. epidermidis, as a model for the S. aureus strain, and enzyme inhibitory effects against saDHFR. KB1 showed no toxic effects on MDCK cells, and no inhibition of E. coli growth was observed. Moreover, five KB1 analogs (KBS1-KBS5) were identified using docking simulations. Among these, three KB1 analogs (KBS1, KBS3, and KBS4) exhibited inhibitory effects on the growth of S. epidermidis and enzyme inhibitory effects against saDHFR. KBS3 and KBS4 did not have any toxic effects on MDCK cell growth, and no inhibitory effects on E. coli growth were observed. We performed an SAR analysis of KB1, KBS1, and KBS3-KBS5, and the results of SAR analysis will likely be useful for the identification and development of more potent chemical derivatives. In addition, the candidate chemical compounds identified in this study are predicted to have potential antibiotic effects against resistant S. aureus stains based on structural and protein–ligand interaction. Therefore, we expect that these compounds will serve as the lead chemical for the development of novel antimicrobial agents targeting S. aureus.
5. Materials and Methods
5.1. Chemical Compounds Library
All chemical compounds libraries were obtained from the Web-based database, Ressource Parisienne en Bioinformatique Structurale (RPBS).38 For the in silico SBDS, we used a single conformation chemical compounds library (ChemBridge, 154,118 chemical compounds)28 and a multiconformation chemical compounds library including a maximum of eleven conformations per chemical compound. These compound libraries were ADME/Tox (Absorption, Distribution, Metabolism, Excretion and Toxicity) filtered, using the most lax filtering method.39
For structure-based screening with hit chemical similarity, we used a multiconformation chemical compounds library (ChemBridge, 461,397 chemical compounds)28 generated using a LowMode MD module in the Molecular Operating Environment (MOE) version 2010. 10., with default parameters.29,40
5.2. Protein Structure Preparation
The crystal structure data for the saDHFR (PDB ID: 2W9G),10 hDHFR (PDB ID: 3NTZ),41 and TMP-resistant saDHFR (PDB ID: 3M09)10 were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank.42 Before using the saDHFR crystal structure in the in silico SBDS, we removed all atoms of the water molecule and TMP atoms of the inhibitor from the original crystal structure data. In addition, hydrogens were added to the saDHFR crystal structure using the MMFF94x force field, and the energy was minimized using the Protonate 3D and Energy Minimize modules, with all heavy atoms tethered in MOE version 2010. 10.29,40 TMP-resistant saDHFR and hDHFR were treated using the same methods.
5.3. Three-Step in Silico SBDS
We performed in silico SBDS using UCSF DOCK version 6.421 and CCDC GOLD suite version 5.0.1.20 In the first screening using DOCK, we searched and extracted the molecular surface of DHFR using the DMS program.43 In addition, we explored clefts potentially interacting with chemical compounds and determined the potential interaction space using the SPHGEN program.44 The protein–ligand interaction space was restricted to the active site of saDHFR. A scoring function of DOCK is calculated using vdW and electrostatic interaction energies to estimate the potential binding affinity.21 We performed the docking simulations using rigid ligand conditions from the virtual chemical compounds library (154,118 chemical compounds).
In the second screening, we estimated the binding affinity between saDHFR and the chemical compounds selected from the first screening using GOLD, which is a flexible docking simulation tool using genetic algorithm (GA).20 The docking simulations are performed with default settings in GOLD. In the final screening using GOLD, we performed docking simulations with the multiconformation virtual chemical compounds library (the selected chemical compounds from the second screening).
5.4. Sequence Alignment and Homology Modeling
We compared the amino acid sequences of saDHFR, seDHFR, and hDHFR. We obtained the sequences of wild-type saDHFR (Entry name: DYR_STAAU), wild-type seDHFR (Entry name: DYR_STAES), and wild-type hDHFR (Entry name: DYR_HUMAN) from the Uniprot Web site,27 and these amino acid sequences were compared using the blastp Web tool on the BLAST Web site.26
The three-dimensional structure of seDHFR is not available on the in PDB Web site.42 Therefore, we generated the three-dimensional structure of seDHFR using the homology modeling module in MOE version 2010. 10., with default parameters.29,40
5.5. Screening of Analog Compounds
For screening based on chemical structure similarities, we searched chemical compounds similar to the initial hit chemical compound KB1 from the data for all chemical compounds using ChemBridge by 2D and 3D similarity search methods.28 The searching methods are based on the Tanimoto coefficient. We selected 23 chemical compounds (similarity rate against KB1 > 85%). We performed docking simulations between the target protein and the multiconformation of the 23 virtual chemical compounds using GOLD.
5.6. Evaluation of Docking Accuracy
ROC analysis was performed to evaluate docking performance using the Lactobacillus casei DHFR (PDB ID: 3DFR(45)) and the directory of useful decoy (DUD) ligands and decoys set.46 The chemical structure data for all ligands and decoys were generated using the Energy Minimize and LowMode MD modules in the MOE version 2010. 10., with default parameters.29,40 The ROC curve and AUC values were calculated using the R source of Dr. S. Aoki (Gunma University).47 The docking studies were performed using the same methods and parameters as the first step of DOCK and the second and third steps of the GOLD screening process.
5.7. Chemical Compounds
All of the chemical compounds (KB1-KB5 and KBS1-KBS5) were purchased from the ChemBridge Corporation28 and dissolved in dimethyl sulfoxide (DMSO, Sigma). The following chemical compounds were obtained from ChemBridge (supplier IDs given in parentheses): KB1: {5-[1-(4-chlorobenzyl)-2-oxo-1,2-dihydro-3H-indol-3-ylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}acetic acid (7469513); KB2: 4-{[7-(4-chlorophenyl)-5-methyl-4,7-dihydro[1,2,4]triazolo[1,5-a]pyrimidin-2-yl]amino}-4-oxobutanoic acid (6584644); KB3: 4-[(4-{[(4-methoxyphenyl)amino]sulfonyl}phenyl)amino]-4-oxobutanoic acid (6952265); KB4: [4-({[3-(2-chlorophenyl)-5-methyl-4-isoxazolyl]carbonyl}amino)phenyl]acetic acid (9009041); KB5: 3-[(4,5-diphenyl-1H-imidazol-2-yl)thio]propanoic acid (6528030); KBS1: 4-{5-[1-(4-chlorobenzyl)-2-oxo-1,2-dihydro-3H-indol-3-ylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}butanoic acid (7424742); KBS2: {5-[1-(4-methylbenzyl)-2-oxo-1,2-dihydro-3H-indol-3-ylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}acetic acid (6660051); KBS3: {5-[1-(2-chlorobenzyl)-2-oxo-1,2-dihydro-3H-indol-3-ylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}acetic acid (7365728); KBS4: 3-{5-[1-(4-fluorobenzyl)-2-oxo-1,2-dihydro-3H-indol-3-ylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}propanoic acid (6666591); and KBS5: 1-(4-chlorobenzyl)-3-[3-(3-ethoxypropyl)-4-oxo-2-thioxo-1,3-thiazolidin-5-ylidene]-1,3-dihydro-2H-indol-2-one (7397168).
5.8. Bacterial Strains
The model bacterial strain S. epidermidis ATCC 12228 was obtained from the RIKEN BioResource Center (Saitama, Japan).48 The E. coli BL21 and JM109 strains were kindly gifted from Dr. S. Sueda (Kyushu Institute of Technology).
5.9. Growth Conditions and the Bacterial Growth Assay
S. epidermidis was incubated overnight in 3 mL of culture medium [1% peptone (BD), 1% beef extract (BD) and 0.5% NaCl (Wako), pH 7.1] on a rotary shaker (37 °C, 300 rpm). E. coli was incubated overnight in 3 mL of culture medium [0.5% yeast extract (BD), 1% tryptone (BD) and 0.5% NaCl (Wako), pH 7.0] on a rotary shaker (37 °C, 300 rpm). We used 15 × dilutions of the S. epidermidis culture and 10 × dilutions of the E. coli culture for the growth assays. The bacterial strains were seeded in 96-well assay plates (Nunc) with a 100 μL total reaction volume and incubated in a culture medium containing each chemical compound, 0.3% DMSO as a negative control or ampicillin (Sigma) as a positive control in the S. epidermidis and E. coli growth assays. S. epidermidis and E. coli were cultured at 300 rpm at 37 °C. After 4, 6, and 8 h, we measured the absorbance (595 nm) of the culture media using a micro plate reader (BioRad).
5.10. Mammalian Cells Growth Assay
The mammalian cells toxicity assay was performed as previously described.18 The effect of the chemical compounds on mammalian cells was assayed using the Cell Counting Kit-8 (DOJIN), to measure the number of living cells. The MDCK and SH-SY5Y cells were seeded in 100 mm dishes and maintained in DMEM medium (Wako) supplemented with 10% FBS (GIBCO), 100 units/mL penicillin, 100 mg/mL streptomycin (GIBCO), and 2 mM l-glutamine (GIBCO). The MDCK and SH-SY5Y cells were seeded in 96-well assay plates (CORNING) with 100 μL of total reaction volume at 5.0 × 103 and 1.5 × 104 cells/well, respectively, and subsequently incubated for 6 h and overnight, respectively, at 37 °C in 5% CO2. For cell starvation, the culture medium of the MDCK and SH-SY5Y cells was replaced with fresh DMEM medium containing 0.25% FBS, and the cells were then incubated overnight and for 2 days, respectively. After cell starvation, the culture medium of the MDCK and SH-SY5Y cells was replaced with fresh medium (0.25% FBS) containing one of the chemical compounds or ampicillin (30 μM) as a negative control. The MDCK and SH-SY5Y cells subsequently were incubated for 1 and 2 days, respectively. We added 10 μL/well of Cell Counting Kit-8 and after 3 h measured the absorbance (450 nm) of WST-8 formazan using a micro plate reader (BioRad).
5.11. Enzymatic Assay
Purified recombinant wild-type saDHFR protein49 was used in this enzymatic assay. The assay was performed using previously described methods with slight modifications.49,50 The assay was carried out in a 96-well assay plate (BD) with a 200 μL total reaction volume using a Biomek 2000 liquid handling robot. The enzyme and dilutions of each chemical compound were preincubated at room temperature for 3 min. Subsequently, 120 nM NADPH (Sigma) was added and incubated at 30 °C; the reaction was initiated by the addition of 226 μM DHF (Sigma) and was monitored for 3 min; the reaction rate remained linear. The enzyme concentration (5.0 nM) yielded an activity of 1.4 nmol DHF reduced per minute for wild-type DHFR. The redox-sensitive tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS, Promega), was utilized as a reagent for the detection of DHFR activity. MTS is reduced by the product THF to yield an increased absorbance at 450 nm. The absorbance (450 nm) of MTS was measured using a DTX880 plate reader (Beckman Coulter). The change in reaction rate signal was calculated as the percentage of a reaction with no chemical compounds over 2.8 min of reaction.
5.12. Statistical Analysis
All statistical analyses were performed using R version 2.15.1 (The R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism version 4 (GraphPad Prism software, Inc., San Diego, CA).
Acknowledgments
The authors are very thankful to Dr. S. Fujii (Kyushu Institute of Technology) for valuable comments and Mr. K. Tsuruta (Kyushu Institute of Technology) for excellent technical support. The enzymatic assay experiments were funded by the National Institutes of Allergy and Infectious Diseases, award R01 AI090685 to W.W.B.
Supporting Information Available
Evaluation of docking accuracy by analyzing the ROC curve (Figure S1). Sequence alignment and superposition of the three-dimensional structure of saDHFR and seDHFR (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rakette S.; Donat S.; Ohlsen K.; Stehle T. Structural analysis of Staphylococcus aureus serine/threonine kinase PknB. PLoS One 2012, 7, e39136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H.; Sang G.; You Y.; Xue X.; Zhou Y.; Hou Z.; Meng J.; Luo X. Targeting RNA polymerase primary sigma(70) as a therapeutic strategy against methicillin-resistant Staphylococcus aureus by antisense peptide nucleic acid. PLoS One 2012, 7, e29886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiran M. D.; Adikesavan N. V.; Cirioni O.; Giacometti A.; Silvestri C.; Scalise G.; Ghiselli R.; Saba V.; Orlando F.; Shoham M.; Balaban N. Discovery of a quorum-sensing inhibitor of drug-resistant staphylococcal infections by structure-based virtual screening. Mol. Pharmacol. 2008, 73, 1578–1586. [DOI] [PubMed] [Google Scholar]
- Iwase T.; Uehara Y.; Shinji H.; Tajima A.; Seo H.; Takada K.; Agata T.; Mizunoe Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 2010, 465, 346–349. [DOI] [PubMed] [Google Scholar]
- Lee D.-S.; Burd H.; Liu J.; Almaas E.; Wiest O.; Barabasi A.-L.; Oltvai Z. N.; Kapatral V. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J. Bacteriol. 2009, 191, 4015–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould I. M.; David M. Z.; Esposito S.; Garau J.; Lina G.; Mazzei T.; Peters G. New insights into meticillin-resistant Staphylococcus aureus (MRSA) pathogenesis, treatment and resistance. Int. J. Antimicrob. Agents 2012, 39, 96–104. [DOI] [PubMed] [Google Scholar]
- DeLeo F. R.; Otto M.; Kreiswirth B. N.; Chambers H. F. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010, 375, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep B. A.; Chan L.; Tattevin P.; Kajikawa O.; Martin T. R.; Basuino L.; Mai T. T.; Marbach H.; Braughton K. R.; Whitney A. R.; Gardner D. J.; Fan X.; Tseng C. W.; Liu G. Y.; Badiou C.; Etienne J.; Lina G.; Matthay M. A.; DeLeo F. R.; Chambers H. F. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5587–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellberg B.; Guidos R.; Gilbert D.; Bradley J.; Boucher H. W.; Scheld W. M.; Bartlett J. G.; Edwards J. Infectious Diseases Society of America, The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 155–164. [DOI] [PubMed] [Google Scholar]
- Heaslet H.; Harris M.; Fahnoe K.; Sarver R.; Putz H.; Chang J.; Subramanyam C.; Barreiro G.; Miller J. R. Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins 2009, 76, 706–717. [DOI] [PubMed] [Google Scholar]
- Duch D. S.; Bigner D. D.; Bowers S. W.; Nichol C. A. Dihydrofolate-reductase in primary brain tumors, cell-cultures of central nervous-system origin, and normal brain during fetal and neonatal growth. Cancer Res. 1979, 39, 487–491. [PubMed] [Google Scholar]
- Coughter J. P.; Johnston J. L.; Archer G. L. Characterization of a staphylococcal trimethoprim resistance gene and its product. Antimicrob. Agents Chemother. 1987, 31, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale D. E.; Broger C.; Hartman P. G.; Langen H.; Page M. G.; Then R. L.; Stüber D. Characterization of the gene for the chromosomal dihydrofolate reductase (DHFR) of Staphylococcus epidermidis ATCC14990: The origin of the trimethoprim-resistant S1 DHFR from Staphylococcus aureus?. J. Bacteriol. 1995, 177, 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huovinen P.; Sundström L.; Swedberg G.; Sköld O. Trimethoprim and Sulfonamide Resistance. Antimicrob. Agents Chemother. 1995, 39, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miteva M. Hierarchical structure-based virtual screening for drug design. Biotechnol. Biotechnol. Equip. 2008, 22, 634–638. [Google Scholar]
- Ballester P. J.; Mangold M.; Howard N. I.; Robinson R. L. M.; Abell C.; Blumberger J.; Mitchell J. B. O. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J. R. Soc. Interface 2012, 9, 3196–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumizono Y.; Arevalo S.; Koseki Y.; Kuroki M.; Aoki S. Identification of novel potential antibiotics for tuberculosis by in silico structure-based drug screening. Eur. J. Med. Chem. 2011, 46, 1849–1856. [DOI] [PubMed] [Google Scholar]
- Koseki Y.; Kinjo T.; Kobayashi M.; Aoki S. Identification of novel antimycobacterial chemical agents through the in silico multi-conformational structure-based drug screening of a large-scale chemical library. Eur. J. Med. Chem. 2013, 60, 333–339. [DOI] [PubMed] [Google Scholar]
- Kinjo T.; Koseki Y.; Kobayashi M.; Yamada A.; Morita Y.; Yamaguchi K.; Tsurusawa R.; Gulten G.; Komatsu H.; Sakamoto H.; Sacchettini J. C.; Kitamura M.; Aoki S. Identification of compounds with potential antibacterial activity against Mycobacterium through structure-based drug screening. J. Chem. Inf. Model. 2013, 53, 1200–1212. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Lang P. T.; Brozell S. R.; Mukherjee S.; Pettersen E. F.; Meng E. C.; Thomas V.; Rizzo R. C.; Case D. A.; James T. L.; Kuntz I. D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Miteva M. A.; Lee W. H.; Montes M. O.; Villoutreix B. O. Fast structure-based virtual ligand screening combining FRED, DOCK, and Surflex. J. Med. Chem. 2005, 48, 6012–6022. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pubchem Web site, http://pubchem.ncbi.nlm.nih.gov/ (accessed July 28, 2011).
- BLAST, http://blast.ncbi.nlm.nih.gov/ (accessed June 8, 2011).
- Uniprot, http://www.uniprot.org/ (accessed April 28, 2011).
- ChemBridge, San Diego, CA, http://www.chembridge.com (accessed May 18, 2011). [Google Scholar]
- Vilar S.; Cozza G.; Moro S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- Keseru G. M.; Makara G. M. Hit discovery and hit-to-lead approaches. Drug Discovery Today 2006, 11, 831–839. [DOI] [PubMed] [Google Scholar]
- Firestine S. M.; Paritala H.; McDonnell J. E.; Thoden J. B.; Holden H. M. Identification of inhibitors of N-5-carboxyaminoimidazole ribonucleotide synthetase by high-throughput screening. Bioorg. Med. Chem. 2009, 17, 3317–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello D.; Barnes M. H.; Biswas E. E.; Biswas S. B.; Gu S.; Williams J. D.; Bowlin T. L.; Moir D. T. Discovery, characterization and comparison of inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicases. Bioorg. Med. Chem. 2009, 17, 4466–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artemenko A. G.; Muratov E. N.; Atamanyuk D. V.; Kuz’min V. E.; Hromov A. I.; Kutsyk R. V.; Lesyk R. B. QSAR analysis of antimicrobial activity of 4-thiazolidone derivatives. QSAR Comb. Sci. 2009, 28, 194–205. [Google Scholar]
- Zacharias M. Rapid protein-ligand docking using soft modes from molecular dynamics simulations to account for protein deformability: Binding of FK506 to FKBP. Proteins 2004, 54, 759–767. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A. K.; Hartell M. G.; Nichols D. A.; Hicks R. P.; Stanton B.; van Hamont J. E.; Milhous W. K. Structure-activity relationship study of antimalarial indolo 2,1-b quinazoline-6,12-diones (tryptanthrins). Three dimensional pharmacophore modeling and identification of new antimalarial candidates. Eur. J. Med. Chem. 2004, 39, 59–67. [DOI] [PubMed] [Google Scholar]
- Cao R.; Liu M.; Yin M.; Liu Q.; Wang Y.; Huang N. Discovery of novel tubulin inhibitors via structure-based hierarchical virtual screening. J. Chem. Inf. Model. 2012, 52, 2730–2740. [DOI] [PubMed] [Google Scholar]
- RPBS, Paris, France, http://bioserv.rpbs.jussieu.fr/RPBS/cgi-bin/Ressource.cgi?chzn_lg=an&chzn_rsrc=Collections (accessed June 10, 2012). [Google Scholar]
- Yu H. S.; Adedoyin A. ADME-Tox in drug discovery: Integration of experimental and computational technologies. Drug Discovery Today 2003, 8, 852–861. [DOI] [PubMed] [Google Scholar]
- Labute P. LowModeMD-implicit low-mode velocity filtering applied to conformational search of macrocycles and protein loops. J. Chem. Inf. Model. 2010, 50, 792–800. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Zhou X.; Kisliuk R. L.; Piraino J.; Cody V.; Gangjee A. Design, synthesis, biological evaluation and X-ray crystal structure of novel classical 6,5,6-tricyclic benzo 4,5 thieno 2,3-d pyrimidines as dual thymidylate synthase and dihydrofolate reductase inhibitors. Bioorg. Med. Chem. 2011, 19, 3585–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissing H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards F. M. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D.; Blaney J. M.; Oatley S. J.; Langridge R.; Ferrin T. E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [DOI] [PubMed] [Google Scholar]
- Bolin J. T.; Filman D. J.; Matthews D. A.; Hamlin R. C.; Kraut J. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. 1. General features and binding of methotrexate. J. Biol. Chem. 1982, 257, 13650–13662. [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K.; Irwin J. J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki S.laboratory homepage, Gunma University, Japan, http://aoki2.si.gunma-u.ac.jp/R/ROC.html (accessed October 10, 2012). [Google Scholar]
- RIKEN, RIKEN BioResource Center, Japan, http://www.jcm.riken.go.jp (accessed June 8, 2011). [Google Scholar]
- Bourne C. R.; Barrow E. W.; Bunce R. A.; Bourne P. C.; Berlin K. D.; Barrow W. W. Inhibition of antibiotic-resistant Staphylococcus aureus by the broad-spectrum dihydrofolate reductase inhibitor RAB1. Antimicrob. Agents Chemother. 2010, 54, 3825–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow E. W.; Bourne P. C.; Barrow W. W. Functional cloning of Bacillus anthracis dihydrofolate reductase and confirmation of natural resistance to trimethoprim. Antimicrob. Agents Chemother. 2004, 48, 4643–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.