

Abstract
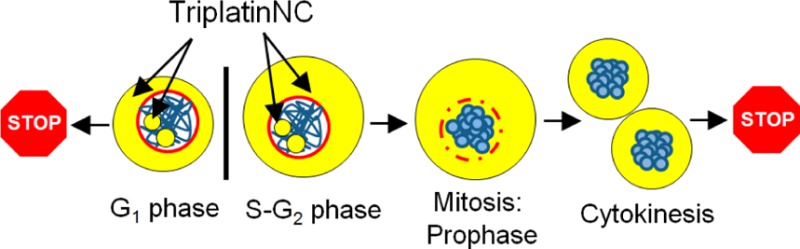
TriplatinNC is a highly positively charged, substitution-inert derivative of the phase II clinical anticancer drug, BBR3464. Such substitution-inert complexes form a distinct subset of polynuclear platinum complexes (PPCs) interacting with DNA and other biomolecules through noncovalent interactions. Rapid cellular entry is facilitated via interaction with cell surface glycosoaminoglycans and is a mechanism unique to PPCs. Nanoscale secondary ion mass spectrometry (nanoSIMS) showed rapid distribution within cytoplasmic and nucleolar compartments, but not the nucleus. In this article, the downstream effects of nucleolar localization are described. In human colon carcinoma cells, HCT116, the production rate of 47S rRNA precursor transcripts was dramatically reduced as an early event after drug treatment. Transcriptional inhibition of rRNA was followed by a robust G1 arrest, and activation of apoptotic proteins caspase-8, -9, and -3 and PARP-1 in a p53-independent manner. Using cell synchronization and flow cytometry, it was determined that cells treated while in G1 arrest immediately, but cells treated in S or G2 successfully complete mitosis. Twenty-four hours after treatment, the majority of cells finally arrest in G1, but nearly one-third contained highly compacted DNA; a distinct biological feature that cannot be associated with mitosis, senescence, or apoptosis. This unique effect mirrored the efficient condensation of tRNA and DNA in cell-free systems. The combination of DNA compaction and apoptosis by TriplatinNC treatment conferred striking activity in platinum-resistant and/or p53 mutant or null cell lines. Taken together, our results support that the biological activity of TriplatinNC reflects reduced metabolic deactivation (substitution-inert compound not reactive to sulfur nucleophiles), high cellular accumulation, and novel consequences of high-affinity noncovalent DNA binding, producing a new profile and a further shift in the structure–activity paradigms for antitumor complexes.
Keywords: BBR3464, TriplatinNC, platinum, nucleolus, rRNA, DNA compaction
Introduction
Cancer is the result of tumor suppressor mutation (e.g., p53, AMPK, ARF), activation of oncogenes (e.g., EGFR, Ras), and modulation of growth factor signaling pathways (e.g., mTOR, Akt). Alterations affecting these pathways result in high levels of rRNA biogenesis, a coordinated process that largely takes place in the nucleolar compartment of the cell.1−4 The nucleolus is formed around five acrocentric chromosome pairs containing head-to-tail arrays of more than 400 DNA copies of rRNA genes as they are transcribed, processed, and assembled into ribosomal subunits.1−4 The nucleolus as drug intervention is increasing in importance, and rRNA synthesis has emerged as a shared target of many clinically important anticancer agents.5 Examples include the platinum-based drugs, oxaliplatin and cisplatin, which inhibit the transcriptional rate of the long 47S rRNA precursor transcript, and the antimetabolite, 5-fluorouracil (5-FU), which disrupts processing of the precursor into shorter, mature 28S, 18S, and 5.8S rRNA transcripts.6 Surprisingly, these effects are observed immediately after cell treatment, implying that inhibition of rRNA processes is an early determinant of efficacy. Oxaliplatin, cisplatin, and 5-FU interactions are not limited to the nucleolus. Such drugs are nonselective, genotoxic agents that incorporate into the total pool of nucleic acid.7−9 In this context, an important challenge for small molecule therapeutics is to specifically target rRNA synthesis, thereby limiting genotoxic events.
Positive charge is a major factor in localization and retention of molecules to the nucleolus. Mutagenesis studies of nucleolar proteins, such as nucleolin, fibrillarin, and the viral HIV TAT, show that clusters of positively charged amino acids, arginine and lysine, serve as nucleolar localization signals.10,11 Polyarginine peptides efficiently penetrate cellular membranes and localize to the nucleolar region of cells.12−14 Recently, using nanoscale secondary ion mass spectrometry (nanoSIMS), we have shown that TriplatinNC (Figure 1B), a highly positively charged (+8) substitution-inert derivative of the phase II clinical platinum drug, BBR3464 (Figure 1B), localizes to the nucleolus and cytoplasm, but not the nucleus.15
Figure 1.

Chemical structures of (A) cisplatin, (B) BBR3464, and (C) TriplatinNC, the arginine fork (top) and phosphate clamp (bottom).
In addition to nucleolar localization, TriplatinNC has three unique biological properties. First, the substitution inertness of TriplatinNC means that binding to thiol-containing serum proteins is reversible and the interactions do not impede cell entry nor reduce cytotoxicity as is the case of cisplatin and BBR3464 containing substitution-labile Pt–Cl bonds.16 Second, rapid cellular uptake of TriplatinNC is mediated by cell surface glycosaminoglycans (GAGs) and inhibits uptake of the fluorescently labeled nonaarginine, TAMRA-R9.17 This mode of accumulation may be extremely beneficial for selective uptake in tumors that exhibit high levels of GAGs and is an accumulation mechanism not shared with cisplatin or oxaliplatin. The high affinity of TriplatinNC for oligosaccharides has been used as proof of principle for “metalloshielding”, whereby enzyme cleavage of the oligosaccharide is inhibited in the presence of TriplatinNC.18 Third, the crystal and molecular structure of TriplatinNC associated with the double-stranded B-DNA dodecamer 5′-[d(CGCGAATTCGCG)]2 at 1.2 Å resolution (PDB: 2DYW) shows formation of phosphate clamps.19,20 These interactions are mediated through hydrogen bonding and have structural similarities with the arginine fork, an important motif for protein–DNA interactions, where positively charged guanidino groups of arginine interact with negatively charged oxygen of DNA phosphate backbone21 (Figure 1C). Indeed, the analogy of TriplatinNC to a polyarginine peptide has proven useful in exploring its properties. The phosphate clamp is a third mode of ligand–DNA binding, distinct from the canonical intercalation and minor-groove binding. Through the phosphate clamp, TriplatinNC proves to be a highly efficient condensing agent for both DNA and tRNA, at concentrations significantly lower than spermine.22−24
It is of interest therefore to see how these biophysical properties, along with that of discrete accumulation mechanisms, manifest in cells. Initial studies confirm that overall cell growth inhibitory activity is similar to that of cisplatin in a panel of tumor cell lines, in the micromolar range for IC50, a unique result in itself for a compound, which in principle does not form covalent Pt-DNA bonds, Table S1 (Supporting Information).25,26 In this article, we show the immediate downstream effects of nucleolar localization, including decrease in rRNA transcription, G1 arrest, and eventually apoptosis. Additionally, we find the fate of cells to be different depending on whether they are treated with TriplatinNC in G1 or later in the cell cycle. Cells treated while in G1 arrest immediately, whereas the majority of cells in S or G2 successfully complete one round of mitosis, and then arrest in G1 as well. Unexpectedly, at this point, nearly one-third of the cells contain highly compacted DNA that cannot be associated with mitosis, senescence, or apoptosis. This cellular effect is unique to TriplatinNC treatment and has not, to our knowledge, been previously described for other platinum compounds or in general. Interestingly, the mechanistic combination of apoptosis and DNA compaction confers equal activity in cell lines that differ in p53-status or that are platinum-resistant. The ability to achieve such cellular effects is unique for a noncovalent compound representing a further paradigm change in platinum therapeutics.
Materials and Methods
Compound Synthesis
Cisplatin,27 TriplatinNC,20,26 and BBR346428 were synthesized as described previously. The stock solutions of platinum compounds were prepared in water at 1 mM concentration and stored at −20 °C.
Cell Culture and Drug Treatments
The human colorectal cancer cell line HCT116 and its knockout derivatives, HCT116 p53–/– and HCT116 p21–/– (kind gift of Dr. Bert Vogelstein), were cultured in RPMI 1640 (Invitrogen), supplemented with 10% calf serum (Atlanta Biologicals) and 1% penicillin/streptomycin (Invitrogen). Cells were maintained in logarithmic growth as a monolayer in T75 culture flasks at 37 °C in a humidified atmosphere containing 5% CO2. For drug treatment studies, unless otherwise noted, the molar drug-to-cell ratio was kept constant by seeding 5 × 104 cells/mL media for all experiments. The human ovarian carcinoma IGROV-1 cell line and the platinum-resistant sublines, IGROV-1/Pt1, IGROV-1/OHP, and IGROV/CP, were maintained in RPMI-1640 medium (BioWhittaker Lonza, Lonza Milano S.r.l., Italy) supplemented with 10% fetal bovine serum (Life Technologies, Monza, Italy). The platinum-resistant variants were generated as described.29−31 The human NSCLC H460 and A549 cell lines and the cisplatin-resistant sublines H460/Pt and A549/Pt were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum. The A549/Pt and H460/Pt were generated as described.32 The human ovarian carcinoma A2780 cell line and the platinum-resistant sublines A2780/CP and A2780/BBR were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum. The platinum-resistant variants were generated as described.33 The human osteosarcoma U2-OS cell line and the cisplatin resistant variant U2-OS/Pt were grown in McCoy’s 5A medium (BioWhittaker Lonza), supplemented with 10% fetal bovine serum.34 Resistance of sublines was stable for at least 6 months when cells were grown in the absence of selecting agent. Cell cultures were routinely checked for being mycoplasma-free, and experiments were carried out using cell lines at similar passages following thawing from a frozen stock.
Polyarginine Competition Assay
HCT116 cells (1 × 104) were seeded in 8-well chamber slides (Lab-Tek II) in 500 μL of media and allowed 24 h to attach. Cells were treated with 10 μM of each drug for 10 min followed by the addition of 1 μM Tamra-R9 (nona-arginine peptide labeled with 5-(and 6-)-carboxytetramethylrhodamine) (Anaspec, cat# 61208) for 15 min. Slides were washed 3× with ice-cold PBS, fixed with 3% paraformaldehyde, washed with 3× PBS again, and mounted with Vectashield mounting medium (Vector Laboratories). Tamra-R9 fluorescence within cells was analyzed quantitatively using a Zeiss LSM 510 confocal microscope and Zen imaging software (settings: 561 nm laser, 578–696 filter, and 63× objective).
Metabolic Labeling and rRNA Analysis
HCT116 cells (2 × 105) were seeded in 6-well plates in 4 mL of RPMI/10% FBS media and allowed to attached for 24 h. Cells were drug treated for 5 h. For phosphate depletion, complete media was replaced with phosphate-free DMEM containing 10% dialyzed FBS and drug. The cells were incubated 30 min before the addition of 15 μCi/mL of 32P-orthophosphate (PerkinElmer). Cells were incubated another 30 min. Medium was changed to RPMI containing 10% FBS and drug for 3 h. Cells were harvested, and total RNA was isolated using RNeasy (Qiagen). RNA concentration was determined using a Nanodrop (Thermo Scientific). Then 1.5 μg of total RNA was separated on a 1% agarose-formaldehyde gel. After electrophoresis, 28S rRNA quantities were visualized with ethidium bromide as a loading control. Gels were placed on Whatman paper and dried for 2 h at 80 °C under vacuum suction. Dried agarose gels were exposed to X-ray film.
Cell Cycle Analysis
Cells were seeded in 100 mm dishes and allowed to attach for 24 h. Cells were treated with 20 μM TriplatinNC for 6, 24, and 48 h time points. Both attached and floating cells were harvested at each time point and counted. Cells (1 × 106 ) were suspended in 1 mL of propidium iodide solution (3.8 mM sodium citrate; 0.05 mg/mL propidium iodide; 0.1% Triton X-100) with added RNase B (7 Kunitz units/ml) and kept in the dark at 4 °C. Cells were analyzed by flow cytometry on a CoulterElite XL-MCL (Beckman Coulter) using a 488 nm argon laser. Twenty thousand events were acquired and analyzed using Modfit software.
Quantitative PCR
Total RNA was isolated using TRIzol (Invitrogen) and reverse-transcribed with SuperScript III (Invitrogen) and random hexamers. Gene expression was determined using qPCR with Quantitect SYBR Green PCR Mastermix (Qiagen). Values were normalized to β-Actin. p53 primers, forward 5′-ATGTGTAACAGTTCCTGCATGGGC-3′, reverse 5′-TTGCGGAGATTCTCTTCCTCTGTG-3′; p21 primers, forward 5′-TGTCACTGTCTTGTACCCTTGTGC-3′, reverse, 5′-GGCGTTTGGAGTGGTAGAAATCTG-3′; β-Actin primers, forward 5′-TTCTACAATGAGCTGCGTGTGGCT-3′, reverse 5′-TAGCACAGCCTGGATAGCAACGTA-3′.
Cell Synchronization
To obtain G0 cell synchronization, cells were seeded at a density of 5 × 105 cells per 100 mm dish and allowed to attach for 24 h. The cells were serum-starved in media containing 0.5% FBS for 96 h. Flow cytometry analysis of synchronized cells showed approximately 80% cells arrested in G0/G1 phase.35
Immunoblot Analysis
Primary antibodies used were against p53 (Cell Signaling, # 9282), p21 (Santa Cruz, clone F-5), p27 (Cell Signaling, clone SX53G8.5), caspase-3 (Cell-Signaling, 9662), caspase-8 (Cell Signaling, clone IC12), cleaved caspase-9 (Cell Signaling, #9501), cleaved PARP (Cell Signaling, # 9541), and beta-Actin (Abcam, ab8226). After drug treatments, both floating and adherent cells were harvested. Cells were washed with ice-cold PBS, pelleted, and resuspended in SDS lysis buffer (62.5 mM Tris-HCl, pH 7.5, 5% glycerol, 4% SDS, 4% complete protease inhibitor (Roche), 5% BME). After homogenization, proteins were resolved on 7.5–15% polyacrylamide gels, transferred to PVDF membrane, and blocked in 5% nonfat milk at room temperature for 1 h. The membranes were probed with primary antibodies overnight, followed by secondary antibodies conjugated to horseradish peroxidase (Cell Signaling, Thermo Scientific). Chemiluminescent protein bands were visualized on X-ray film.
Immunofluorescence
Cells were seeded in 8-well chamber slides and allowed to attach for 24 h. After drug treatment, cells were washed with PBS, and fixed with 3% paraformaldehyde for 15 min. Cells were permeabilized with 0.5% Triton-X in PBS for 10 min and blocked in PBS/Casein for 1 h at room temperature. Then 1:50 dilution NPM/B23 (Santa Cruz, #sc-5564), 1:100 dilution alpha-Tubulin (Cell Signaling, #3873), or 1:100 dilution of cleaved caspase-3 (Cell signaling, # 9662) was added overnight at 4 °C. A 1:500 dilution of antirabbit IgG Alexa 647 (Cell Signaling, #4414) or antimouse Alexa 555 (Cell Signaling, #4409) secondary antibody conjugates was added for 3 h at room temperature. The antibodies were then fixed with 3% paraformaldehyde for 15 min at room temperature. All slides were mounted in VectaShield with DAPI (Vector Laboratories) and viewed using a Zeiss LSM 510 confocal microscope.
Mitotic Checkpoint Assay
Cells (5 × 105) were seeded in 100 mm dishes and allowed to attach for 24 h. Cells were drug treated for 24 h, harvested, fixed with 1% paraformaldehyde followed by 80% ethanol, and permeabilized with 0.25% Triton X-100 in PBS for 5 min. Cells were stained with antiphospho-histone H3 antibody (Cell Signaling, #3377P), followed by anti-IgG Alexa 647. DNA was stained with propidium iodide in the presence of RNase B. Phospho-histone H3 positive cells were determined using a FACSCanto II flow cytometer (Becton Dickinson, San Jose,CA, USA). Ten thousand events were acquired for each sample.
β-Galactosidase Assay
Cells were plated in 6-well plates, allowed to attach overnight, and drug treated for 5 days. For senescence-activated β-galactosidase (SA-β-Gal) staining, the Senescence-β-Gal Staining Kit (Cell Signaling Technology) was used, following the manufacturer’s instructions.
Analyses of Cell Sensitivity to Drugs
Cell sensitivity to drugs was assessed by growth inhibition assays (MTT, 3,4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; cell counting) and by clonogenic assays allowing measurement of cell survival. For MTT assays, cells were seeded in a 96-well plate at 5 × 103 cells/well in 100 μL of media and allowed 24 h to attach. Cells were then drug treated for a period of 72 h. After drug removal, cells were incubated with 0.5 mg/mL MTT reagent (Sigma) in media for 3 h at 37 °C. After MTT was removed, 100 μL of DMSO was added to each well. The plate was then incubated on a shaker at room temperature in the dark. The spectrophotometric reading was taken at 570 nm using a microplate reader. When cell sensitivity was assessed by growth-inhibition assays based on cell counting, exponentially growing cells were seeded in duplicate in six-well plates at 19000–25500 cells/cm2. After 24 h, cells were exposed to different concentrations of drugs for 72 h, and cells were counted at the end of treatment. Before counting, culture medium was removed, and adherent cells were harvested using trypsin and counted with a cell counter (Beckman Coulter, S.p.A., Milan, Italy). IC50 is defined as the concentration causing a 50% inhibition of cell growth as compared to control. The resistance index is the ratio between the IC50 of resistant and sensitive cells. For clonogenic assays, 2 × 104 cells were seeded in 3 mL of medium in a 6-well plate. After 24 h of incubation, cells were drug treated for a 24 h period. Then 250 or 2500 cells were seeded into 10 cm dishes and allowed to grow for 10–14 days to form colonies. These were then fixed by methanol and stained by 0.1% crystal violet. Colonies consisting of more than 50 cells were counted. Plating efficiency and surviving fraction were determined for each drug.
Results and Discussion
Nucleolar localization of TriplatinNC in human breast (MCF-7) carcinoma cells was previously described using nanoscale secondary ion mass spectrometry (nanoSIMS) of a 15N-labeled sample.15 Because polyarginines localize in the nucleolus, given the competitive inhibition of TAMRA-R9-heparan sulfate binding by PPCs, we examined their influence on TAMRA-R9 nucleolar localization. The pattern of intracellular localization of TAMRA-R9 in HCT116 cells is shown in Figure 2A. For competitive inhibition studies, HCT116 cells were pretreated with 10 μM TriplatinNC (4 × IC50, Table S1, Supporting Information), 10 μM BBR3464, or 10 μM cisplatin for 10 min. Then 1 μM of TAMRA-R9 was added to the media and the effects of each compound on the dye’s localization were observed by confocal microscopy. As we have previously reported,17 Tamra-R9 readily enters cells after cisplatin treatment (Figure 2A, upper right panel), but significantly less Tarma-R9 enters the cells after treatment with BBR3464 (Figure 2A, lower left panel) and TriplatinNC (Figure 2A, lower right panel), as evidenced by diminution of the intrinsic fluorescence signal of the labeled peptide. TriplatinNC, but not BBR3464 (or cisplatin), competes with the polyarginine for localization to the nucleolus.
Figure 2.
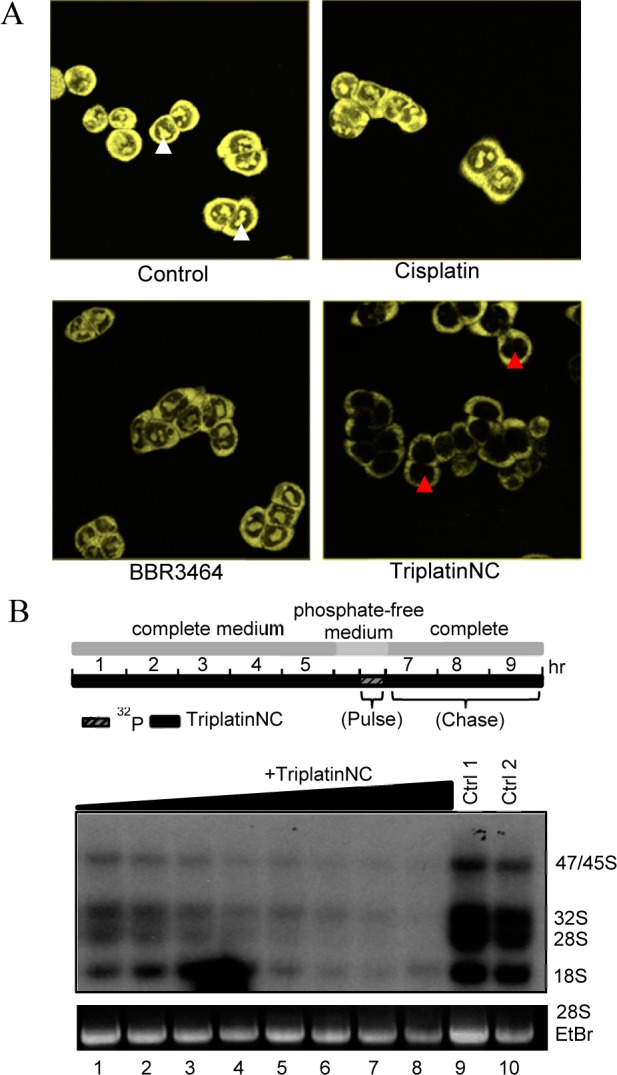
(A) Polyarginine competition assay; cells were pretreated with 10 μM cisplatin, BBR3464, or TriplatinNC, followed by 1 μM Tamra-R9 after 10 min. White arrows; localization of Tamra-R9 to nucleolar region. Red arrows; nucleolar region (no R9 localization). A representative of three independent experiments is shown. (B) (Top) Experimental scheme for 32P-metabolic rRNA labeling and drug treatment in HCT116 cells. (Bottom) Lanes 1–8 were treated with 0.78, 1.56, 3.13, 6.25, 12.5, 25, 50, and 100 μM TriplatinNC, respectively. Lanes 9 and 10 are untreated control samples. A representative of three independent experiments is shown.
In principle, because the nucleolus is not membrane-enclosed, any soluble molecule can diffuse in and out of the nucleolar compartment. Therefore, it is generally accepted that retention of a molecule within the nucleolus must occur as a direct interaction with its components, the two most obvious examples for platinum compounds being rRNA and DNA.36
We next asked if TriplatinNC has an effect on the transcription rate of rDNA in the nucleolus, which could occur by binding to the rDNA promoter/gene, thereby preventing initiation or elongation of the rDNA transcript (pre-rRNA). Second, we asked whether TriplatinNC affects the downstream processing of precursor pre-rRNA into mature rRNAs by binding directly to pre-rRNA or small nucleolar RNAs (snoRNAs) involved in its cleavage.
To determine whether TriplatinNC affects the rate of rRNA transcription or processing in vivo, HCT116 cells were treated with different drug concentrations for 5 h, and then metabolically labeled according to the scheme depicted in Figure 2B. The 47S rRNA precursor transcript is sequentially cleaved to yield the mature 28S, 18S, and 5.8S rRNA. As cells are pulsed with 32P-radiolabeled ortho-phosphate for 30 min followed by a 3 h chase, the abundance of newly formed 47S precursors, 32S intermediate cleavage products, and mature 28S and 18S rRNA are sufficiently labeled for visualization by autoradiography.6 It was evident that treatment of cells with TriplatinNC inhibits the production rate of 47S rRNA precursor transcripts in a dose-dependent manner (Figure 2B, bottom). It does not appear that TriplatinNC affects the rate of 47S rRNA processing as the abundance of 32S, 28S, and 18S rRNA decrease proportionally to that of the precursor. These data suggest that rDNA may be a defining interaction involved in the antiproliferative activity of TriplatinNC. The ability of TriplatinNC to fully inhibit in vitro transcription (Figure S1, Supporting Information) and, specifically, the transcription factor, TATA binding protein (TBP), binding to its ognate DNA consensus sequence in a dose-dependent manner is consistent with this hypothesis (Figure S2, Supporting Information). In the latter case, this is the first example of a noncovalent platinum–drug interaction, which occurs at remarkably low concentration, to inhibit the association of a transcription factor, i.e., TBP to DNA. The inhibition of transcriptional activity occurs at markedly lower concentration than naturally occurring spermine (23; Figure S2, Supporting Information).
The transcriptional activity of rRNA genes has been reported to change within the cell cycle. rRNA transcription levels are highest in S and G2 phases, nonexistent in mitosis, and rebounding in G1.37−39 Therefore, it was important to consider whether the inhibitory effect of TriplatinNC on the rate of rRNA transcription was direct or if rRNA transcription levels were merely downregulated as an indirect effect of changes within the cell cycle. For this purpose, HCT116 cells treated with TriplatinNC were subjected to cell cycle analysis by flow cytometry (Figure 3A). In cells treated with 20 μM TriplatinNC (IC90) for 6 h, only modest changes occurred within the cell cycle. The population of cells in G1 decreased slightly from 37% to 30% compared to untreated control cells, whereas the population of cells within S + G2 increased slightly from 63% to 70%. These results imply that the disruption of rRNA transcription is an early event of cellular treatment with TriplatinNC and does not result from changes in the cell cycle. In fact, there was no increase in the population of cells in G1 (when rRNA levels are lower).
Figure 3.

(A) Cell cycle analysis; HCT116 cells treated for 6, 24, and 48 h with 20 μM TriplatinNC. Values are derived from Modfit software analysis of histograms (excluding sub-G1) of three repeat experiments combined. (B) Quantitative PCR analysis; p53 and p21 cDNA expression after 24 h treatment with cisplatin and TriplatinNC. Values are derived from two repeat experiments combined. (C) Western blot analysis; p53, p21, and p27 protein expression after treatment with 20 μM TriplatinNC for 3, 6, 12, and 24 h. β-Actin is used as a loading control. A representative of three independent experiments is shown.
The signaling pathway leading to cell cycle arrest after exposure to antitumor agents has been studied in detail.40 Central to this pathway is the stabilization of p53 protein by serine/threonine kinases, followed by transactivation of the cyclin-dependent kinase (CDK) inhibitor, p21. Increased protein levels of p21 inhibit CDK activities resulting in cell cycle arrest. This pathway is induced by cisplatin, which has been shown to arrest cells at the G2-checkpoint as an attempt to repair DNA damage before cells enter mitosis.41,42 In agreement with these studies, HCT116 cells treated with 20 μM cisplatin (IC90) were shown to induce accumulation in S-phase at 24 h and finally in G2 at 48 h (Figure S3, Supporting Information). Treatment of HCT116 cells with 20 μM TriplatinNC, however, induced an arrest in G1 at 24 h continuing to 48 h. The increase in the number of cells in G1 was mostly at the expense of the proportion of cells undergoing DNA replication in S-phase, which decreased 52% at 24 h and 77% at 48 h compared to the control (Figure 3A).
The mRNA expression of p53 and p21 were found to follow the same trend for both cisplatin and TriplatinNC treatments (Figure 3B). Modest increases in p53 transcripts were observed, but this is not unusual, as an increase in p53 protein is typically a result of increased half-life through decrease in proteasomal degradation, rather than increase in transcript levels.43 As expected, after treatment with TriplatinNC, there was a substantial stabilization of p53 protein levels leading into the G1 arrest at 12 and 24 h (Figure 3C). p21 mRNA levels increase 5-fold and 8-fold after treatment with cisplatin and TriplatinNC, respectively. However, p21 protein levels in TriplatinNC-treated samples were not concomitantly upregulated in the classical manner. Despite the increase in p21 mRNA expression, protein levels declined. Furthermore, the protein expression levels of p27, another CDK inhibitor with the potential to cause cell cycle arrest,44 also decreased. These data suggest that the G1 arrest induced by TriplatinNC may not depend on classical signaling events. The fact that TriplatinNC induces G1 arrest in the HCT116 isogenic cell lines lacking either p53 or p21 (Figure S4, Supporting Information) further emphasizes this point. The set of experiments described above allowed us to speculate that TriplatinNC may disrupt the cell cycle at G1 because S phase has high requirements for rRNA production45 and that in TriplatinNC-treated cells quantities of ribosomes for protein translation may be insufficient.
This hypothesis was tested by synchronizing the cells at the G0/G1 phase, when rRNA levels are low, and then allowing the cells to enter the cell cycle together. As shown in Figure 4, HCT116 cells were synchronized in low serum for 96 h and released into the cell cycle by the addition of serum with or without cisplatin or TriplatinNC to the media. In control cells, progression into S phase was clearly observed at 8, 12, and 16 h after release. As compared to the control, cells treated with cisplatin progressed slowly into S phase, as evidenced at 16 and 20 h after release. However, cells treated with TriplatinNC showed a striking and persistent arrest in G1. Thus, TriplatinNC-treated cells could not proceed into S and G2 phases.
Figure 4.

Cell synchronization and flow cytometry; greater than 85% of HCT116 cells were synchronized in G0/G1 by serum starvation. The cells were released into serum-containing medium with or without cisplatin or TriplatinNC. Cell cycle analysis was performed at the time points indicated. A representative of two independent experiments is shown.
Since cells treated while in early G1 immediately arrest in G1, we can speculate that the majority of cells treated while in S and G2 phases successfully progress through mitosis to re-enter G1, thereby explaining the increase in G1 at 24 h (Figure 3A). It was surprising, therefore, to observe by confocal microscopy that 28.3 ± 4.1% of cells treated with TriplatinNC for 24 h contained DNA that appeared compacted or condensed (Figures 5A and S5, Supporting Information). Condensed DNA is indicative of cells in mitosis, yet an accumulation of cells in G2/M had not been previously observed. To evaluate this seeming discrepancy, the percentages of cells in mitosis (prophase, metaphase, anaphase, and telophase) were counted by confocal microscopy. Each phase was identified through staining DNA with DAPI and labeling two proteins that change dynamically according to the cell cycle, β-tubulin and nucleophosmin (Figure 5B, top panel). When cells were treated with TriplatinNC for 1, 3, 6, or 24 h, no greater than ∼6% of them were in mitosis at any time point. Furthermore, the cells containing condensed DNA were distinct from cells in mitosis, lacking cytoplasmic staining of nucleophosmin or microtubule formation (Figure S5, Supporting Information). In preparation of slides for microscopy, mitotic cells are more loosely attached than cells in interphase, and the mitotic index can be underestimated. For verification, HCT116 cells were immunolabeled with a mitosis-specific phospho-histone H3 (ser 10) antibody after treatment with 20 μM TriplatinNC and quantified by flow cytometry (Figure 5C). Again, the number of cells in mitosis does not change significantly. Taken together our data suggest that the observed DNA compaction events are not related to mitosis.
Figure 5.
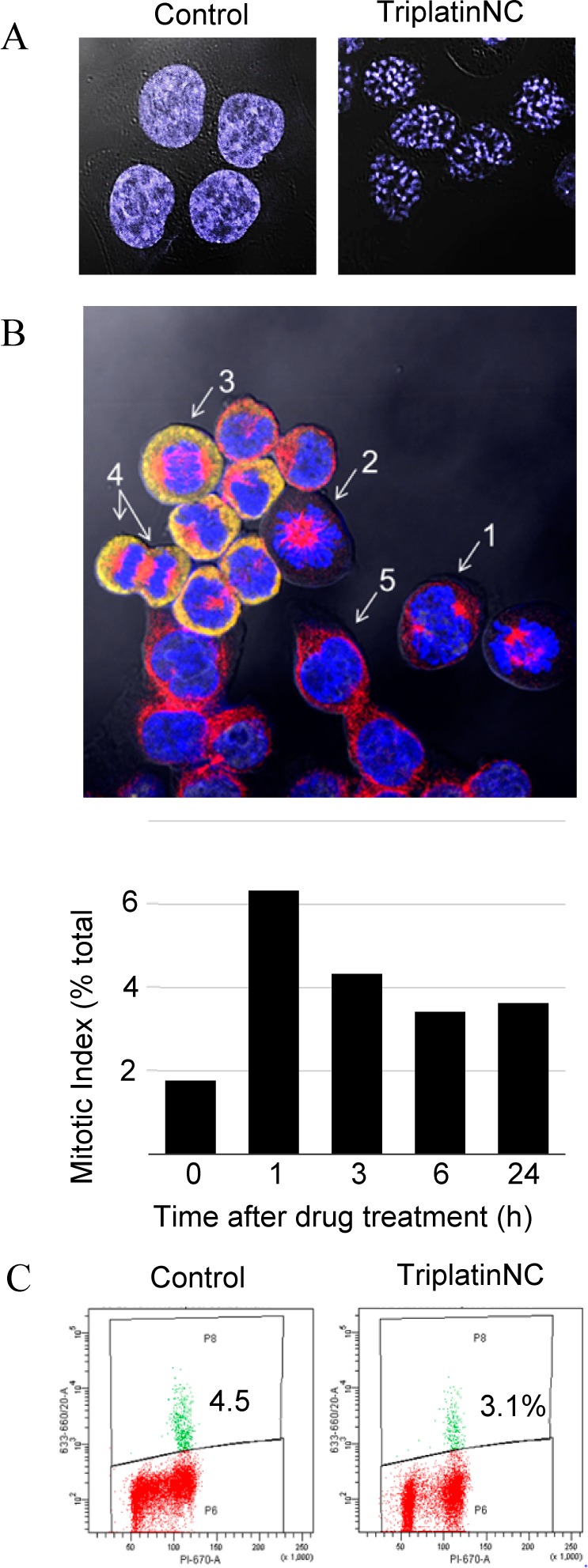
(A) Confocal microscopy; HCT116 cells were stained with DAPI after treatment with 20 μM TriplatinNC for 24 h and visualized by confocal microscopy. (B) (Top) Mitotic index assay; image of (1) early pro-metaphase, (2) late pro-metaphase, (3) anaphase, (4) late anaphase/telophase, and (5) interphase as determined by DAPI DNA stain (blue); β-tubulin (red); and nucleophosmin/B23 (yellow) immunostaining. (Bottom) The mitotic index was derived as the number of cells in all mitotic phases combined (P + M + A + T) and divided by the total number of cells. n > 500 cells per time treatment (>1000 cells counted total for two repeat experiments). (C) Mitotic checkpoint assay; HCT116 were treated with or without 20 μM TriplatinNC, fixed, and incubated with phospho-histone H3 (Ser10), followed by antirabbit Alexa 647 secondary antibody and PI staining. Ten thousand events were analyzed by flow cytometry. Shown is a representative of two independent experiments.
Aside from mitosis, condensed chromatin also occurs during senescence, as senescence-associated heterochromatin foci (SAHF).46 In this process, cells grow larger, flatten shape, and express senescence-associated β-galactosidase. When HCT116 cells were treated with 20 μM cisplatin for 96 h, the remaining cells were large, flattened, and showed β-galactosidase activity, i.e., blue precipitates, representing hydrolysis products of X-gal by β-galactosidase (Figure 6A, lower left). TriplatinNC treated cells were small, with very little cytoplasmic content, and did not show evidence of β-galactosidase activity (Figure 6A, lower right). Therefore, we found that the DNA compaction events were not consistent with senescence.
Figure 6.
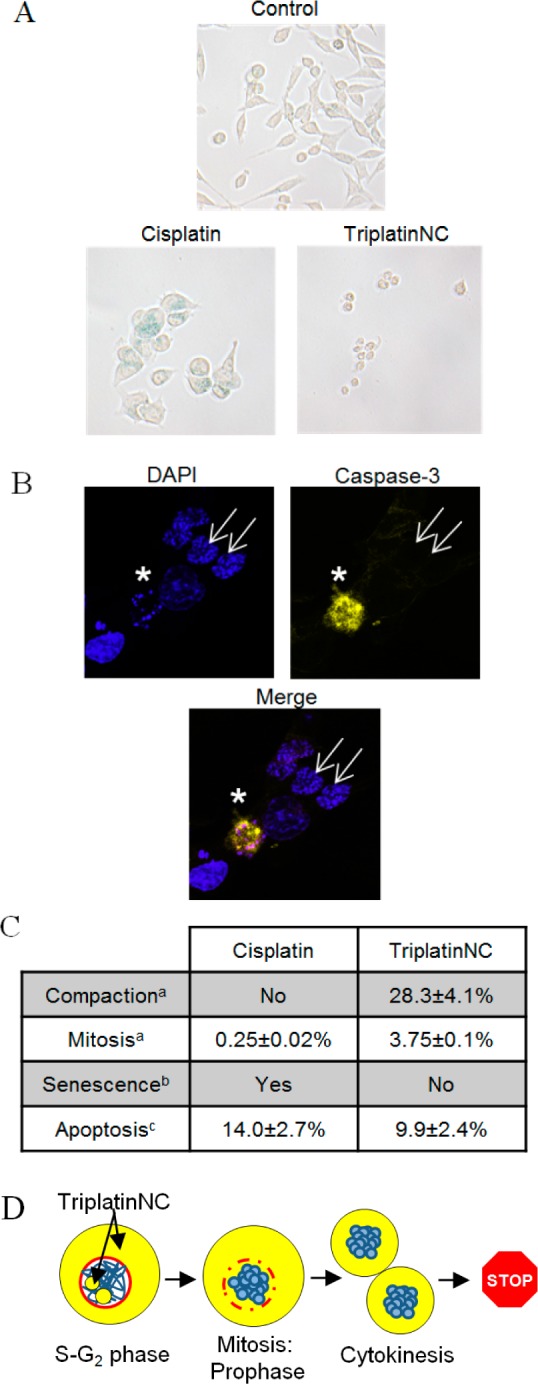
(A) β-Galactosidase activity assay; HCT116 cells were assayed for β-galactosidase activity after treatment with 20 μM cisplatin or TriplatinNC for 96 h. Shown is a representative of two independent experiments. (B) Confocal microscopy; HCT116 cells were treated with 20 μM TriplatinNC for 24 h. Left, top panel; DAPI stained DNA (blue). Right, top panel; cleaved caspase-3 (yellow). Bottom panel; merged DAPI and cleaved caspase-3. Apoptotic cell (white star) containing condensed/fragmented DNA and active caspase-3. Cells with compacted DNA (white arrows) do not contain active caspase-3. (C) Summary; percentage of HCT116 cells undergoing the indicated cellular process after treatment with 20 μM cisplatin or TriplatinNC for 24 h. (D) Schematic; TriplatinNC localizes to the cytoplasm and nucleolus of interphase cells. Cells treated while in S-G2 proceed through mitosis. During prophase, the DNA (blue) condenses and the nuclear membrane (red) disintegrates allowing cytoplasmic pools of TriplatinNC to interact with condensed DNA. Cells undergo cytokinesis; however, the DNA does not decondense or progress through G1. (a) Determined by immunofluorescence: β-tubulin, nucleophosmin/B23, and DAPI DNA staining (Figure 5A,B). (b) Determined by β-galactosidase staining and light microscopy at 200× magnification (panel A). (c) Determined by immunofluorescence; cleaved caspase-3; and DAPI DNA staining. The percentage of cells undergoing apoptosis was determined as the number of cells positive for cleaved caspase-3 divided by the total number of cells. n > 500 cells each for two repeat experiments (Figure 5B).
TriplatinNC has been reported to induce apoptosis,25 and whereas chromatin condensation and DNA fragmentation are hallmarks of apoptosis, the DNA compaction induced by TriplatinNC appeared to be quite different from the appearance of DNA in apoptotic cells. For verification, cells treated with TriplatinNC for 24 h were immunostained with cleaved caspase-3 and visualized by confocal microscopy. Cells containing compacted DNA are shown labeled with white arrows (Figure 6B), and there appeared to be no active caspase-3 in these cells. An apoptotic cell with condensed/fragmented DNA and active caspase-3 is shown in the same frame for comparison.
In summary, it appears that DNA compaction events induced by TriplatinNC are separate from the DNA condensation events of mitosis, senescence, and apoptosis (Figure 6C). From previous studies,15,16 we observed that TriplatinNC is limited to the cytoplasm and nucleolus while cells are in interphase (G1-S-G2). However, during mitosis, the DNA condenses and the nuclear membrane recedes, and the cytoplasmic pools of TriplatinNC are likely exposed to DNA that are otherwise inaccessible (Figure 6D). We hypothesize that TriplatinNC may prevent the decondensation of DNA at this point, leaving the cell suspended between cytokinesis and G1. Although surprising, this would not be completely without precedent, given the efficiency of DNA condensation in vitro.22−24
The advantage of DNA compaction may increase the effectiveness of platinum compounds in apoptotic defective cell lines. Nearly all cancers harbor genetic defects that directly, or indirectly, inhibit proapoptotic or tumor suppressor functions of p53.40 For this reason, we asked whether the absence of p53 affects the ability of TriplatinNC to effectively induce cell cycle arrest and cell death. HCT116 p53+/+ and p53–/– cells were treated with 20 μM cisplatin or TriplatinNC for 12, 24, 48, or 72 h. The ability of cisplatin to inhibit cell growth was significantly limited in the absence of p53 (Figure 7A, upper panel), as shown by MTT assays. However, the growth inhibitory effects of TriplatinNC was unaffected by the absence of p53 protein (Figure 7A, lower panel). In support of these results, we asked further whether the absence of p53 affects the ability of TriplatinNC to inhibit colony formation or reproductive viability, using the clonogenic survival assay. HCT116 p53+/+ and HCT116 p53–/– cells were treated with 20 μM TriplatinNC for 24, 48, and 72 h (Figures 7B and S6, Supporting Information). The percentage of p53+/+ cells that failed to replicate was determined to be 68.5 ± 7.8% after 24 h, 88 ± 2.8% after 48 h, and 92 ± 9.9% after 72 h. There was no significant difference with or without p53.
Figure 7.

(A) MTT assay; comparison of growth inhibition in HCT116 p53+/+ (lt gray) and p53–/– (dk gray) cells treated with 20 μM cisplatin or TriplatinNC. Percent inhibition is calculated as 1 – (N/N0), where N = treated samples and N0 = untreated control samples. Values are derived from three experiments combined. **p < 0.05 and ***p < 0.005 (B) Clonogenic survival assay; comparison of reproductive viability in HCT116 p53+/+ cells and p53–/– cells treated with 20 μM TriplatinNC for 24 and 48 h. Shown is a representative of four independent experiments; the value of the combined experiments is shown in Figure S7, Supporting Information. (C) Western blot analysis; timecourse analysis of cleaved caspase-9 and -3 protein in HCT116 p53+/+ and p53–/– cells treated with 20 μM TriplatinNC for 5, 15, 30, and 55 h. (D,E) Comparison of cleaved caspase-8 and PARP-1 protein in HCT116 p53+/+ and p53–/– cells treated with 20 μM cisplatin or TriplatinNC at 48 h. β-Actin was used as a loading control. A representative of three independent experiments is shown.
TriplatinNC induces apoptosis in mast cells through activation of the mitochondrial-dependent pathway initiator, procaspase-9, and the downstream effector, procaspase-3.25 In extension of these studies, we examined whether TriplatinNC induced accumulation of the activated forms of procaspase-9 and procaspase-3 in p53+/+ as compared to p53–/– HCT116 isogenic colon carcinoma cell lines. Both cell lines showed a time-dependent increase in active caspase-9 and caspase-3 after treatment with 20 μM TriplatinNC (Figure 7C). Further, it was examined whether caspase-3 may be activated also by the initiator of the mitochondrial-independent pathway of apoptosis, caspase-8.
Caspase-8 has the ability to activate procaspase-3 through both mitochondrial-dependent and -independent apoptotic pathways. In the mitochondria-dependent pathway, caspase-8 cleaves BID to tBID, which translocates to the mitochondria and causes damage by culminating an efflux of death promoting proteins such as cytochrome-C. These events, in turn, lead to activation of procaspase-9, followed by procaspase-3. In the mitochondrial-independent pathway, caspase-8 instead directly activates procaspase-3 and downstream substrates such as PARP-1, eventually leading to cell death.47 In HCT116 cells treated with 20 μM cisplatin for 48 h, the abundance of active caspase-8 (p18) and downstream target, PARP-1,was found to be reduced in cells lacking p53 as compared to the wild-type control (Figure 7D). However, treatment with 20 μM TriplatinNC for 48 h induced similar levels of active caspase-8 and PARP-1 in p53+/+ and p53–/– cells (Figure 7E). Furthermore, TriplatinNC did not induce the cleavage of BID (Figure S7, Supporting Information); therefore, the mitochondrial-dependent and -independent pathways are likely activated independent of each other.48
Loss or mutation of p53 is associated with platinum resistance.49 The platinum-resistant ovarian carcinoma sublines, IGROV-1/Pt1, IGROV-1/CP, and IGROV-1/OHP, were generated by chronic exposure of IGROV-1 (wt p53) to cisplatin or oxaliplatin. The IGROV-1/Pt1 and IGROV-1/OHP sublines were found to be mutants for p53.50 In Table 1, cell sensitivity to TriplatinNC is compared to that of cisplatin in each of these cell lines. The RI values for cisplatin is 12.2-fold higher in platinum-resistant IGROV-1/Pt1 than parental IGROV-1. Importantly, the IC50 values for TriplatinNC in these cell lines are not significantly different. This trend was maintained in other platinum-resistant cell lines generated from a second ovarian carcinoma cell line, A2780, lung carcinoma cell lines, H460 and A549, and the osteosarcoma cell line, U2-OS.
Table 1. Sensitivity of Platinum-Sensitive and -Resistant Cell Lines to TriplatinNC after 72 h Exposure As Tested by Growth Inhibition Assays; Resistance (RI) Values for TriplatinNC and Cisplatin Are Reporteda.
| cell line | p53 status | TriplatinNC IC50 (μM) | TriplatinNC RI | cisplatin RI |
|---|---|---|---|---|
| IGROV-1 | wt | 3.6 ± 0.7 | ||
| IGROV-1/Pt1 | mutant | 5.1 ± 1.7 | 1.4 | 12.2 |
| IGROV-1/CP | mutant | 2.9 ± 0.7 | 0.79 | 8.3 |
| IGROV-1/OHP | mutant | 7.4 ± 2.3 | 2.0 | 8.2 |
| H460 | wt | 3.9 ± 1.3 | ||
| H460/Pt | wt | 2.0 ± 0.4 | 0.5 | 2.6 |
| A549 | wt | 4.6 ± 0.9 | ||
| A549/Pt | wt | 2.8 ± 0.9 | 0.6 | 4.8 |
| A2780 | wt | 2.7 ± 0.9 | ||
| A2780/CP | wt | 3.4 ± 1.1 | 1.2 | 4.0 |
| U2-OS | wt | 7.0 ± 3.5 | ||
| U2-OS/Pt | wt | 5.5 ± 0.7 | 0.78 | 5.0 |
RI; resistance index. IC50 resistant cells/IC50 sensitive cells.
Finally, proof-of-concept of meaningful antitumor activity of TriplatinNC in vivo has been obtained in a mouse ovarian carcinoma A2008 model; Figure S8, Supporting Information. At a dose of 25 mg/kg (ip), drug efficacy was assessed by measuring the tumor growth inhibition (TGI) on day 10, with a 32% decrease in tumor volume in drug treated animals versus control, with a tumor growth delay index (GDI) indicating a delay of 2.5 days for the tumor to reach 3 times its starting size, and an absolute growth delay (AGD), calculated as median time in days to reach 3 times starting tumor volume, of 21.25 days. The data compares very favorably with that of cisplatin obtained as comparison; Figure S8, Supporting Information.
Summary and Conclusions
The substitution-inert polynuclear platinum complexes characterized by noncovalent binding to biomolecules are emerging as a distinct subset of the wide and diverse polynuclear platinum chemotype.20,28 Unfortunately, pharmacokinetic issues and a relatively narrow therapeutic index, coupled to complications from drug company takeovers and realignment, stalled the only compound of this class that has undergone clinical evaluation, BBR3464 (in Phase II clinical trials), a not uncommon fate for anticancer chemotherapy.51 The results presented here show that TriplatinNC is endowed with very interesting biological properties. Replacement of Pt–Cl by substitution-inert ligands such as NH3 or the “dangling” amine −H2N(CH2)nNH3+ prevents metabolic deactivation, and TriplatinNC fills the criteria for a second generation drug candidate. In fact, the complex is cytotoxic at micromolar concentrations in a wide range of tumor cell lines, and its cytotoxicity is unaffected by serum degradation.16,17,25
The phosphate clamp is a third mode of ligand–DNA binding, discrete from intercalation and minor-groove binding.19,20 The distinguishing features from extracellular studies is that of high DNA binding affinity, some selectivity for minor groove A-T-rich sequences and very effective condensation of both tRNA and DNA.22−24 The results discussed here suggest that TriplatinNC arrests the cells in two different ways, both leading to cell death. Cells treated while in G1 undergo cell cycle arrest (by inhibition of rRNA), while cells treated in S-G2 undergo DNA compaction. The data show that the nucleic acid compaction events observed in cells cannot be associated with mitosis, senescence, or apoptosis, presenting to our knowledge a unique biological (cellular) consequence for a platinum agent or, indeed, any chemotherapeutic drug. The efficient nucleolar localization is followed by a rapid decrease in rRNA production leading to an immediate G1 arrest. While other platinum complexes are implicated in inhibition of RNA polymerase-I-mediated rRNA synthesis,52 the downstream effects of TriplatinNC are unique. Hitherto all clinical platinum candidates have been assumed to require covalent Pt–DNA binding to manifest meaningful antitumor properties. Overall, the biological activity of TriplatinNC reflects reduced metabolic deactivation (substitution-inert compound not reactive to sulfur nucleophiles), high cellular accumulation, and novel consequences of high-affinity noncovalent DNA binding, producing a new profile and a further shift in the structure–activity paradigms for antitumor complexes.
Acknowledgments
This work was supported by a grant NIH R01CA78754 and in part by a grant from the Fondazione Guido Berlucchi. Microscopy was performed at the VCU Department of Neurobiology and Anatomy Microscopy Facility, supported, in part, with funding from NIH-NINDS Center core grant 5P30NS047463.
Glossary
Abbreviations
- PPC
polynuclear platinum complex
- GAG
glycosaminoglycans
- nanoSIMS
nanoscale secondary ion mass spectrometry
- TAMRA-R9
nona-arginine peptide labeled with 5-(and 6-)-carboxytetramethylrhodamine
Supporting Information Available
Additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- White R. J. RNA polymerases I and III, growth control and cancer. Nat. Rev. Mol. Cell Biol. 2005, 6, 69–78. [DOI] [PubMed] [Google Scholar]
- Ruggero D.; Pandolfi P. P. Does the ribosome translate cancer?. Nat. Rev. Cancer 2003, 3, 179–192. [DOI] [PubMed] [Google Scholar]
- Thomas G. An encore for ribosome biogenesis in the control of cell proliferation. Nat. Cell Biol. 2000, 2, E71–2. [DOI] [PubMed] [Google Scholar]
- Raska I.; Koberna K.; Malinsky J.; Fidlerova H.; Masata M. The nucleolus and transcription of ribosomal genes. Biol. Cell. 2004, 96, 579–594. [DOI] [PubMed] [Google Scholar]
- Drygin D.; Rice W. G.; Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [DOI] [PubMed] [Google Scholar]
- Burger K.; Muhl B.; Harasim T.; Rohrmoser M.; Malamoussi A.; Orban M.; Kellner M.; Gruber-Eber A.; Kremmer E.; Holzel M.; Eick D. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Lippard S. J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discovery 2005, 4, 307–320. [DOI] [PubMed] [Google Scholar]
- Longley D. B.; Harkin D. P.; Johnston P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [DOI] [PubMed] [Google Scholar]
- Khynriam D.; Prasad S. B. Cisplatin-induced genotoxic effects and endogenous glutathione levels in mice bearing ascites Dalton’s lymphoma. Mutat. Res. 2003, 526, 9–18. [DOI] [PubMed] [Google Scholar]
- Emmott E.; Hiscox J. A. Nucleolar targeting: the hub of the matter. EMBO Rep. 2009, 10, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C. V.; Lee W. M. Nuclear and nucleolar targeting sequences of c-erb-A, c-myb, N-myc, p53, HSP70, and HIV tat proteins. J. Biol. Chem. 1989, 264, 18019–18023. [PubMed] [Google Scholar]
- Futaki S.; Suzuki T.; Ohashi W.; Yagami T.; Tanaka S.; Ueda K.; Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [DOI] [PubMed] [Google Scholar]
- Martin R. M.; Tunnemann G.; Leonhardt H.; Cardoso M. C. Nucleolar marker for living cells. Histochem. Cell Biol. 2007, 127, 243–251. [DOI] [PubMed] [Google Scholar]
- Mitchell D.; Steinman L.; Kim D.; Fathman C.; Rothbard J. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Peptide Res. 2000, 56, 318–325. [DOI] [PubMed] [Google Scholar]
- Wedlock L. E.; Kilburn M. R.; Liu R.; Shaw J. A.; Berners-Price S. J.; Farrell N. P. NanoSIMS multi-element imaging reveals internalisation and nucleolar targeting for a highly-charged polynuclear platinum compound. Chem. Commun. 2013, 49, 6944–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti B. T.; Peterson E. J.; Kabolizadeh P.; Martínez A.; Kipping R.; Farrell N. P. Effects of noncovalent platinum drug–protein interactions on drug efficacy: use of fluorescent conjugates as probes for drug metabolism. Mol. Pharmaceutics 2011, 8, 940–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva H.; Frézard F.; Peterson E. J.; Kabolizadeh P.; Ryan J. J.; Farrell N. P. Heparan sulfate proteoglycan-mediated entry pathway for charged tri-platinum compounds: differential cellular accumulation mechanisms for platinum. Mol. Pharmaceutics 2012, 9, 1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangrum J. B.; Engelmann B. J.; Peterson E. J.; Ryan J. J.; Berners-Price S. J.; Farrell N. P. A new approach to glycan targeting: enzyme inhibition by oligosaccharide metalloshielding. Chem. Commun. 2014, 50, 4056–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeda S.; Moulaei T.; Woods K. K.; Chikuma M.; Farrell N. P.; Williams L. D. A third mode of DNA binding: phosphate clamps by a polynuclear platinum complex. J. Am. Chem. Soc. 2006, 128, 16092–16103. [DOI] [PubMed] [Google Scholar]
- Mangrum J. B.; Farrell N. P. Excursions in polynuclear platinum DNA binding. Chem. Commun. 2010, 46, 6640–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon I.; Lee J.; Andrade J.; de Gennes P.; Hermens H. Arginine-mediated RNA recognition: the arginine fork. Science 1991, 252, 1167–1171. [DOI] [PubMed] [Google Scholar]
- Malina J.; Farrell N. P.; Brabec V. DNA condensing effects and sequence selectivity of DNA binding of antitumor noncovalent polynuclear platinum complexes. Inorg. Chem. 2014, 53, 1662–1671. [DOI] [PubMed] [Google Scholar]
- Malina J.; Farrell N. P.; Brabec V. Noncovalent trinuclear platinum complexes efficiently condense/aggregate nucleic acids and inhibit enzymatic activity. Angew. Chem. 2014, 10.1002/anie.201408012R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisecaru A.; Molphy Z.; Kipping R. G.; Peterson E. J.; Kellett A.; Farrell N. P.. The phosphate clamp: dynamic nucleic acid binding profiles and conformational induction of endonuclease inhibition by cationic Triplatin complexes. Nucleic Acids Res. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A. L.; Ryan J. J.; Farrell N. Biological consequences of trinuclear platinum complexes: comparison of [{trans-PtCl(NH3)2}2μ-(trans-Pt(NH3)2(H2N(CH2)6NH2)2)]4+ (BBR 3464) with its noncovalent congeners. Mol. Pharmacol. 2006, 69, 666–672. [DOI] [PubMed] [Google Scholar]
- Harris A. L.; Yang X.; Hegmans A.; Povirk L.; Ryan J. J.; Kelland L.; Farrell N. P. Synthesis, characterization, and cytotoxicity of a novel highly charged trinuclear platinum compound. Enhancement of cellular uptake with charge. Inorg. Chem. 2005, 44, 9598–9600. [DOI] [PubMed] [Google Scholar]
- Kauffman G. B.; Cowan D. O.; Slusarczuk G.; Kirschner S. cis-and trans-Dichlorodi-ammineplatinum (II). Inorg. Synth. 2007, 7, 239–245. [Google Scholar]
- Farrell N. Polynuclear platinum drugs. Met. Ions Biol. Syst. 2004, 42, 251–296. [PubMed] [Google Scholar]
- Perego P.; Giarola M.; Righetti S. C.; Supino R.; Caserini C.; Delia D.; Pierotti M. A.; Miyashita T.; Reed J. C.; Zunino F. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996, 56, 556–562. [PubMed] [Google Scholar]
- Benedetti V.; Perego P.; Luca Beretta G.; Corna E.; Tinelli S.; Righetti S. C.; Leone R.; Apostoli P.; Lanzi C.; Zunino F. Modulation of survival pathways in ovarian carcinoma cell lines resistant to platinum compounds. Mol. Cancer Ther. 2008, 7, 679–687. [DOI] [PubMed] [Google Scholar]
- Cossa G.; Lanzi C.; Cassinelli G.; Carenini N.; Arrighetti N.; Gatti L.; Corna E.; Zunino F.; Zaffaroni N.; Perego P. Differential outcome of MEK1/2 inhibitor-platinum combinations in platinum-sensitive and-resistant ovarian carcinoma cells. Cancer Lett. 2014, 347, 212–224. [DOI] [PubMed] [Google Scholar]
- Gatti L.; Cossa G.; Tinelli S.; Carenini N.; Arrighetti N.; Pennati M.; Cominetti D.; De Cesare M.; Zunino F.; Zaffaroni N.; Perego P. Improved apoptotic cell death in drug-resistant non-small-cell lung cancer cells by tumor necrosis factor-related apoptosis-inducing ligand-based treatment. J. Pharmacol. Exp. Ther. 2014, 348, 360–371. [DOI] [PubMed] [Google Scholar]
- Perego P.; Gatti L.; Righetti S. C.; Beretta G. L.; Carenini N.; Corna E.; Bo L. D.; Tinelli S.; Colangelo D.; Leone R. Development of resistance to a trinuclear platinum complex in ovarian carcinoma cells. Int. J. Cancer 2003, 105, 617–624. [DOI] [PubMed] [Google Scholar]
- Perego P.; Casati G.; Gambetta R. A.; Soranzo C.; Zunino F. Effect of modulation of protein kinase C activity on cisplatin cytotoxicity in cisplatin-resistant and cisplatin-sensitive human osteosarcoma cells. Cancer Lett. 1993, 72, 53–58. [DOI] [PubMed] [Google Scholar]
- Menon V. R.; Peterson E. J.; Valerie K.; Farrell N. P.; Povirk L. F. Ligand modulation of a dinuclear platinum compound leads to mechanistic differences in cell cycle progression and arrest. Biochem. Pharmacol. 2013, 86, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M.; Mendes-Soares L.; Campos I. To be or not to be in the nucleolus. Nat. Cell Biol. 2000, 2, E107–E112. [DOI] [PubMed] [Google Scholar]
- Cully M. J.; Leevers S. J. RNA interference pinpoints regulators of cell size and the cell cycle. Genome Biol. 2006, 7, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J.; Grummt I. Cell cycle-dependent regulation of RNA polymerase I transcription: the nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6096–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi C. P.; Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry W. S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [DOI] [PubMed] [Google Scholar]
- Voland C.; Bord A.; Peleraux A.; Penarier G.; Carriere D.; Galiegue S.; Cvitkovic E.; Jbilo O.; Casellas P. Repression of cell cycle-related proteins by oxaliplatin but not cisplatin in human colon cancer cells. Mol. Cancer Ther. 2006, 5, 2149–2157. [DOI] [PubMed] [Google Scholar]
- Pani E.; Stojic L.; El-Shemerly M.; Jiricny J.; Ferrari S. Mismatch repair status and the response of human cells to cisplatin. Cell Cycle 2007, 6, 1796–1802. [DOI] [PubMed] [Google Scholar]
- Giaccia A. J.; Kastan M. B. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Toyoshima H.; Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [DOI] [PubMed] [Google Scholar]
- Derenzini M.; Montanaro L.; Chillà A.; Tosti E.; Vici M.; Barbieri S.; Govoni M.; Mazzini G.; Treré D. Key role of the achievement of an appropriate ribosomal RNA complement for G1-S phase transition in H4-II-E-C3 rat hepatoma cells. J. Cell. Physiol. 2005, 202, 483–491. [DOI] [PubMed] [Google Scholar]
- Kuilman T.; Michaloglou C.; Mooi W. J.; Peeper D. S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl S. J.; Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [DOI] [PubMed] [Google Scholar]
- Korsmeyer S.; Wei M.; Saito M. T.; Weiler S.; Oh K.; Schlesinger P. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [DOI] [PubMed] [Google Scholar]
- Righetti S. C.; Della Torre G.; Pilotti S.; Menard S.; Ottone F.; Colnaghi M. I.; Pierotti M. A.; Lavarino C.; Cornarotti M.; Oriana S.; Bohm S.; Bresciani G. L.; Spatti G.; Zunino F. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res. 1996, 56, 689–693. [PubMed] [Google Scholar]
- Perego P.; Giarola M.; Righetti S. C.; Supino R.; Caserini C.; Delia D.; Pierotti M. A.; Miyashita T.; Reed J. C.; Zunino F. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996, 56, 556–562. [PubMed] [Google Scholar]
- Farrell N. P. Progress in platinum-derived drug development. Drugs Future 2012, 37, 795–806. [Google Scholar]
- Pickard A. J.; Bierbach U. The cell’s nucleolus: an emerging target for chemotherapeutic intervention. ChemMedChem 2013, 8, 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.