

Abstract
Purpose:
To review the recent literature and describe the authors’ experience with congenital upper eyelid coloboma.
Methods:
In this review, we will summarize the embryologic and etiopathogenetic bases of congenital upper eyelid coloboma, and study the published clinical reports. We will also attempt to briefly shed some light on the rarer syndromic curiosities associated with upper eyelid coloboma.
Results:
Congenital upper eyelid colobomas are one of the few nontraumatic oculoplastic emergencies that may occasionally present in the first few days of life with a corneal ulcer and may even present with impending perforation. They can present with or without corneopalpebral adhesions, may be isolated findings or a part of a larger spectrum of congenital anomalies as in the case of Fraser syndrome or Goldenhar syndrome, or could be associated with other rare curiosities that could challenge the clinician with a huge diagnostic dilemma.
Conclusions:
Existing literature dealing with congenital colobomas of the upper eyelid is fraught with nosologic problems, confusing etiologies, and overlapping clinical features. We attempted to clarify the salient clinical features, outline the management principles, and until a time in the not-so-distant future where advances in molecular genetic testing would help redefine the etiology and the diverse clinical spectrum of genetic diseases associated with upper eyelid colobomas, we propose a simplified classification scheme based on the relation of the coloboma to the cornea, the presence or absence of systemic features, and all the syndromic and nonsyndromic associations of congenital coloboma of the upper eyelid known today.
In this review, the authors will describe the pathogenesis of upper eyelid coloboma, suggest a new simplified classification system, describe the clinical picture in detail, clarify the various syndromic associations of upper eyelid coloboma, and lay out the basic surgical principles of management.
Coloboma (plural colobomas or colobomata), which is derived from the Greek word Κολόβωμα, implies a mutilated, curtailed structure; a hole; or a defect in a tissue,1,2 and is a term that is almost exclusively used in Ophthalmology.1 First described in the iris in 1673 by Bartholin the younger, ocular coloboma could involve any layer of the eye3 and usually result from defective closure of the embryonic fissure.3 Eyelid colobomas, however, can be unilateral or bilateral, can be symmetrical or asymmetrical, can involve animals or humans, and may or may not be associated with other ocular or facial deformities.
Although both upper and lower eyelid colobomas may have a significant cosmetic blemish, congenital colobomas of the upper eyelid, in particular, may threaten vision at a very early age and require prompt management. The classic congenital upper eyelid defect includes a shortage of conjunctiva, tarsal plate, orbicularis muscle, and skin, which leaves the cornea unprotected, resulting in possible exposure keratopathy. Even after closing the defect, close monitoring of visual function is of paramount importance because of the very high risk of amblyopia.
NOMENCLATURE
Both cryptophthalmos (CO) and its syndromic counterpart Fraser syndrome (FS) bear different confusing names in the literature: congenital upper eyelid coloboma and CO, Meyer-Schwickerath syndrome, corneopalpebral adhesions (CPA), FS, corneopalpebral synechiae, abortive CO, congenital symblepharon, isolated CO, syndromic and nonsyndromic CO, FRAS/FREM complex disorders, cryptophthalmia, cryptophthalmos, congenital ankyloblepharon, Fraser-Francois syndrome, FS CO, or Ullrich-Feichtiger syndrome.4–7 Goldenhar syndrome (GS), however, has also been called by at least 16 different names and to name but a few of them: oculoauriculovertebral spectrum, oculoauriculovertebral syndrome, oculoauriculovertebral dysplasia, facioauriculovertebral anomaly syndrome, first branchial arch syndrome, lateral facial dysplasia, or hemifacial microsomia.8,9
We recently advocated the use of the term cryptophthalmos–coloboma–symblepharon anomaly instead of CO;7 however, for the purpose of the current discussion, we have retained the use of the terms CO, FS, and GS throughout this review until a single orthonym is universally adopted for each of these disorders.10
EMBRYOLOGICAL AND FETAL DEVELOPMENT OF THE EYELIDS
Embryonic and fetal eyelid development has been extensively studied elsewhere,11–19 but the process of eyelid fusion is of particular relevance for the current discussion. At the beginning of stage 22 (week 8, 54–56 days, 23–28 mm), flattened periderm cells on the eyelid surface undergo a morphogenetic transition into cuboidal epithelium, proliferate, and migrate centripetally toward each other, and thus begin the remarkable process of eyelid fusion.13,20 When a connection is established between both eyelids, the periderm cells flatten again to form a continuous sheet ultimately covering the cornea; therefore, the process of eyelid fusion involves 2 coordinated yet distinct processes: epithelial cell migration and proliferation of the epithelium at the migrating edge.14–19,21 In mammals, only the periderm and epidermal layers are involved in eyelid fusion, while eyelid mesenchyme remains distinct in preparation for future separation.14 The exact timing when separation is complete is difficult to deduce from the literature, but in general separation is complete around the sixth or seventh month.4,12,22,23 Recently, apoptosis has been identified in the rat model as a possible key event (though not the only event) in eyelid separation.24At the molecular level, regulation of eyelid development requires a bidirectional mesenchyme–periderm interaction, which is regulated by several signaling pathways.21,25–28 To better understand the phenotypic logic behind congenital upper eyelid colobomas and CO/FS in particular, these signaling pathways and their effector genes should be further scrutinized in detail.
CLASSIFICATION SCHEMAS
In general, eyelid colobomas fall under the rubric of craniofacial clefting.29 In 1976, Tessier30 listed 15 facial cleft types categorized into 4 groups based purely on their anatomical location. Unfortunately, Tessier classification is strictly an anatomical classification, describes both soft tissue and bony clefts, is descriptive of the entire face and not just the eyelids, is absolutely lacking with regard to syndromic colobomas associated with systemic features, and most importantly is more commonly associated with lower eyelid colobomas than their upper eyelid counterparts. The authors attempting to classify upper eyelid colobomas have traditionally focused on CO/FS more than on GS and usually ignored simple colobomas (SCs) that belie any classification schema.29–35
We can foresee in the not too distant future a categorization of upper eyelid colobomas based on genetic or molecular signatures of each particular anomaly, but until the unique genetic bases of these misleadingly similar phenotypes (FS versus GS versus nonsyndromic varieties) are completely understood, we will adopt a simple classification scheme solely for the purpose of the present discussion.
-
I. Isolated coloboma
-
A. Coloboma associated with CPA (CO)
-
a. Complete
i. No discernable eyelid differentiation, and the eyes are completely covered with skin
-
b. Incomplete
i. A skin fold devoid of tarsus covers the medial aspect of the palpebral aperture
ii. Significant CPA
iii. Lower fornix and lateral upper eyelids usually spared
-
c. Abortive type/congenital symblepharon variant (CSV)
i. True coloboma of variable sizes with a diverse range of CPA
ii. Lower fornix and lateral upper eyelids usually spared
-
-
B. SC
a. Upper eyelid coloboma in isolation not associated with CPA
-
-
II. Syndromic variants
A. FS
B. GS/oculoauriculovertebral spectrum (GS)
-
C. Rare syndromes
a. Manitoba oculotrichoanal syndrome
b. Ablepharon-macrostomia syndrome
c. Nasopalpebral lipoma-coloboma syndrome
d. Amniotic band sequence
e. Oculoectodermal syndrome
-
f. Neurocutaneous syndromes
i. Encephalocraniocutaneous lipomatosis
ii. Delleman syndrome or oculocerebrocutaneous syndrome
iii. Linear nevus sebaceous syndrome
g. CHARGE syndrome
ETIOLOGY AND PATHOGENESIS
To this date, the network of genetic mutations responsible for upper eyelid colobomas is largely unknown, and possible environmental factors or mechanical events during pregnancy may contribute to the development.
Isolated Coloboma.
Coloboma Associated With Corneo plapebral Adhesions (CO).
Most cases of CO are sporadic with unknown etiology probably resulting from a de novo mutation,36 but familial cases have been reported,36–43 and a case could be made that CO is an arrested form of FS based on the fact that within some families with FS, there are family members with isolated CO.38
Although Thomas et al.37 erroneously suggested that the familial cases of isolated CO are of dominant inheritance, it was recently argued that these cases follow an autosomal recessive pattern of inheritance.38 Another possibility is that CO is a disruption anomaly because 2 mothers in our case series were possibly exposed to a teratogen in early pregnancy: the first was accidentally exposed to x-ray irradiation with her elder son and the second was a nail technician with long-term exposure to nail glue and nail gels.
Simple Coloboma.
Isolated SC without CPA and without any systemic features represent an enigma. Whether they are an arrested form of GS remains to be determined.
Syndromic Variants.
Fraser Syndrome.
An insight into genetics of FS has been greatly increased with the study of a certain family of mice mutants termed “bleb mice,” because the shared defect in both conditions is a profound loss of adhesion of epidermis to the underlying basement membrane that results in abnormal epidermal blistering in the mouse embryo.44 In normal mouse embryos, a group of closely related proteins called the FRAS/FREM protein family are universally present in all basement membranes throughout the body and contribute to embryonic epithelial–mesenchymal integrity and tight adhesions during embryogenesis.45–48 When this intricate process of epithelial–mesenchymal protein trafficking is interrupted by a genetic knockout either experimentally (Fras1−/− mice), or in bleb mice, or in humans with a deficiency in chromosome 4q21 or 13q13.1, which encodes FRAS1 or FREM2, respectively,49 the most common resulting phenotype is composed of renal anomalies, abnormal fusion of the digits and the eyelids in addition to subcutaneous hemorrhagic blisters in mice. Mutations in either of these 2 autosomal recessive genes were demonstrated in 50% of patients with FS; however, other genes have been implicated recently.49–52
As we shall see later, the phenotypic features of FS are extremely variable and pleiotropic, and several other distinct syndromes such as Manitoba oculotrichoanal syndrome and ablepharon-macrostomia syndrome may overlap FS and may suffer mutations in the FRAS/FREM complex.38,53–56
Goldenhar Syndrome.
Goldenhar syndrome is usually sporadic; however, familial instances have been demonstrated suggesting an autosomal dominant or autosomal recessive inheritance.57 Various chromosomal abnormalities have been found in GS such as trisomy 7, 9, or 22; mosaicisms; deletions at chromosome 18q or 22q; or unbalanced translocations between chromosomes 5 and 8.57–61 A long list of possible teratogenic factors causing GS includes smoking; cocaine use during pregnancy; diabetic embryopathy; primidone, retinoic acid, or thalidomide use during pregnancy; and an unknown toxin use during the Gulf war.,8,62,63
EPIDEMIOLOGY
Isolated Coloboma.
Coloboma Associated With Corneo plapebral Adhesions (CO).
In their comprehensive review in 1986, Thomas et al.37 reported a total of 27 patients with isolated CO with equal sex distribution of whom 16 were sporadic and 11 were familial. A history of consanguinity was demonstrated in 12.5% of patients. A close examination of the 27 nonsyndromic cases recorded by Thomas et al. shows that they were in error because the same 3 family members published by Coover39–41 in 3 separate articles apparently belong to the same family presented by Magruder42 10 years later bringing the actual number to 24. A major review article published in 2002 mentions an additional 29 cases.38 Several additional reports have appeared in the literature afterward,36,43,64–66 and a review of our own medical records revealed 17 patients with isolated CO without any systemic findings.
The exact number of nonsyndromic CO patients reported in the literature is difficult to determine because unfortunately 2 major reviews in the literature focused on FS alone and disregarded isolated CO,65,66 and the largest 3 published case series in the ophthalmic literature made no attempt to differentiate isolated CO from syndromic cases.4,32,33
Simple Coloboma.
To the best of our knowledge, no epidemiologic data are available. Reports of SCs without CPA are sparse, and a literature review would give the erroneous impression that CO is more prevalent, which is merely an error in reporting because the striking features of CO make it more palatable for publishing. Nouby33 reported 5 patients with SC, while another article reported an additional 15.67 The superior fornix is vaguely referred to in the latter article as “adequate,” and it is not clear whether the superior fornix in these 15 patients was completely free or not because in our experience, patients with CO may have a variable yet constant degree of CPA, which may be minimally visible on casual observation. In our own practice, we have only encountered 2 patients with SC without CPA or any systemic defining features.
Syndromic Variants.
Fraser Syndrome.
Fraser syndrome is a very rare syndrome with fewer than 300 cases described in the literature so far.65 It has an estimated prevalence that varies between 0.20 and 0.43 per 100,000 liveborn infants.65,68 Because of the potentially life-threatening respiratory defects of FS, the reported incidence among stillbirths is higher, 11.06 per 100,000.69
Reported rates of consanguineous marriages varied from 15% to 49%,37,38,65 and reported gender differences are also variable. A recent European study has demonstrated considerable heterogeneity in prevalence rates depending on geographic distribution.37,38,65 In our own case series, 3 out of 5 patients with FS were descendants of the same geographic location in Egypt (Upper Egypt) where consanguineous marriage is the rule.
Goldenhar Syndrome.
Epidemiologic data are sparse and inconsistent with estimates ranging from 1/3,500 to 3.8/100,000 live births. The best guess is around 1/5,600 live births.8,70
CLINICAL PICTURE
Isolated Colobomas.
Colobomas Associated With Corneo plapebral Adhesions.
The clinical features of CO vary from a SC with minimal CPA to a truly “hidden” eye or complete CO. We usually find it hardly convincing to categorically embrace François31 subclassification of cases with CO into complete CO (cryptophthalmie typique), incomplete CO (formes incompletes, atypique, or partielles), and CSV (formes abortive), all of which may coexist in both eyes of the same patient (Figs. 2A and 3B) or in siblings.71 We believe that the clinical spectrum of CO represents a continuum probably resulting from the same genetic defect; however, for the purpose of the current discussion, we still tried to distinguish the various clinical types, although we agree with Gupta and Sen72 that CO may not uniformly lend itself to a rigid clinical classification schema at least until the genetic bases are well defined.
FIG. 2.
Incomplete cryptophthalmos. Various clinical presentations of incomplete cryptophthalmos. A,B, Abnormal skin fold in the medial part of the upper eyelid adhering to the underlying cornea. Normal eyelids remnants could be seen lateral (A,B) and medial to the skin fold, and the upper punctum was also preserved (B). In both examples, the eye is quite, with a small coloboma and minimal keratopathy. Note the bifid nose ipsilateral to the defect in A. The other eye of the patient in A is in Figure 3A. C, A more severe case with the skin fold extending to and fusing with the lower eyelid. The medial part of the upper and lower eyelids are abnormal and no puncti are present, but the lateral part of the eyelid is preserved. D, Surgical division of the eyelid fold clearly illustrates the cornea showing through the thin skin fold, which is completely devoid of tarsus (*) in contrast to the normal lateral part of the eyelid where the tarsus is preserved (short arrow). Also, check Figure 1B in Tawfik.7
FIG. 3.
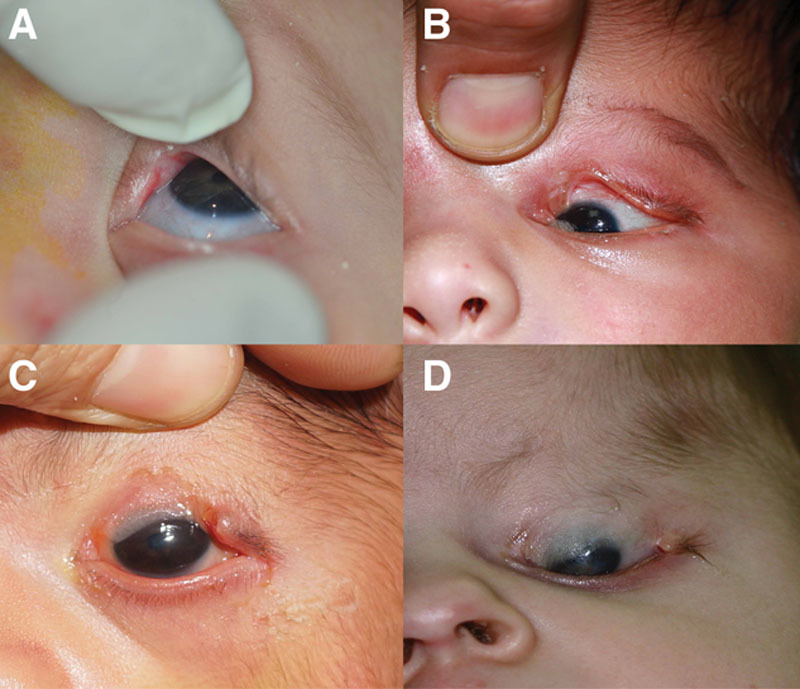
Congenital symblepharon variant. The clinical spectrum of congenital symblepharon variant ranges from an insignificant coloboma with minimal corneopalpebral adhesions that is triangular in shape (A) to a more extensive coloboma that is quadrangular in shape (B). C,D, Larger defects are not uncommon and may involve almost the entire upper eyelid sparing only a small tongue of normal tissue in the lateral part with extensive adhesions to the underlying cornea, which may involve up to three-fourths of the cornea.
Complete CO.
In these patients, the forehead skin extends over the globe and onto the cheek without any discernable differentiation of eyelids except occasionally where a dimple or scar is seen at the site of presumed fusion of both eyelids although as we will discuss later, this dimpling is associated with a different phenotype of CO.36,73
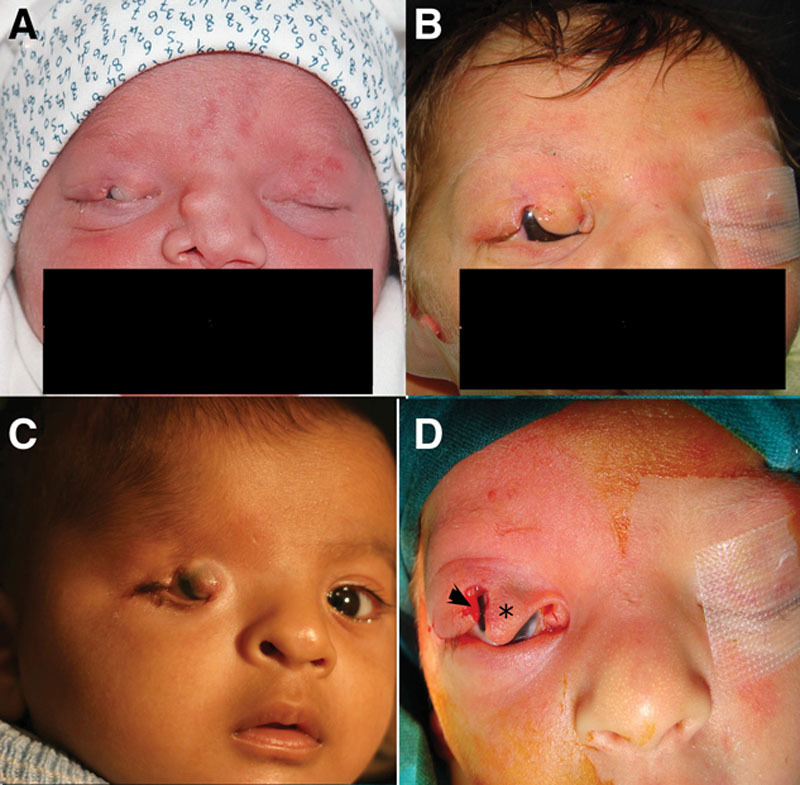
The ocular structures in complete CO are usually atrophic and grossly disorganized, but the eye may be normal in size or even enlarged, may occasionally present as a cyst (Fig. 1D),33,36,73,74 and may occasionally express a reaction to light.73
FIG. 1.
Complete cryptophthalmos. Various clinical presentations of complete cryptophthalmos. A, Typical complete cryptophthalmos with total absence of the eyelids, eyebrow, eyebrow hairs, and a centrally located normal sized globe. B, A medially displaced globe with total absence of the eyebrow and eyebrow hairs except laterally. A skin tag is seen lateral to the globe. Note ipsilateral absence of the nasal cavity. C, A failed previous reconstruction attempt in a patient with complete cryptophthalmos showing an enlarged proptotic centrally located globe with total absence of the eyebrow ipsilateral to the defect and a contralateral tongue of hair replacing the eyebrow—underlies the difficulty in managing these patients. A previous cleft lip repair is also seen ipsilateral to the cryptophthalmic eye. Note the contralateral microphthalmic globe. D, CT scan of the same patient showing the globe replaced by a figure-of-eight cystic lesion to which normally sized extraocular muscles are attached posteriorly. Note the widening of the superior orbital fissure and the defect in the greater wing of the sphenoid. (*) This globe did not react to light shown through the skin in contrast to the contralateral microphthalmic globe that showed some reaction to light.
The extraocular muscles may be of normal size for age or may be ill defined, with limited ocular motility, which could be seen and felt with palpation.31,74 The globe may be central, but may be displaced horizontally or vertically (Fig. 1A–C).
The eyebrows are seldom normally developed,36 and more commonly the entire eyebrow, its medial 2/3 or its lateral 1/3, is absent.32,73,74 A tongue of hair, which is not yet quite distinct at birth, may be seen extending from the scalp hair into the lateral 1/3 of the eyebrow.32,74 In our experience, alopecia of the eyebrows may occur anywhere and may not follow a specific pattern (Figs. 1–4). In complete CO, the conjunctiva is wholly absent and the skin is completely adherent to the anterior surface of sclera and cornea by fibrous tissue. The condition may be unilateral or bilateral.74
FIG. 4.
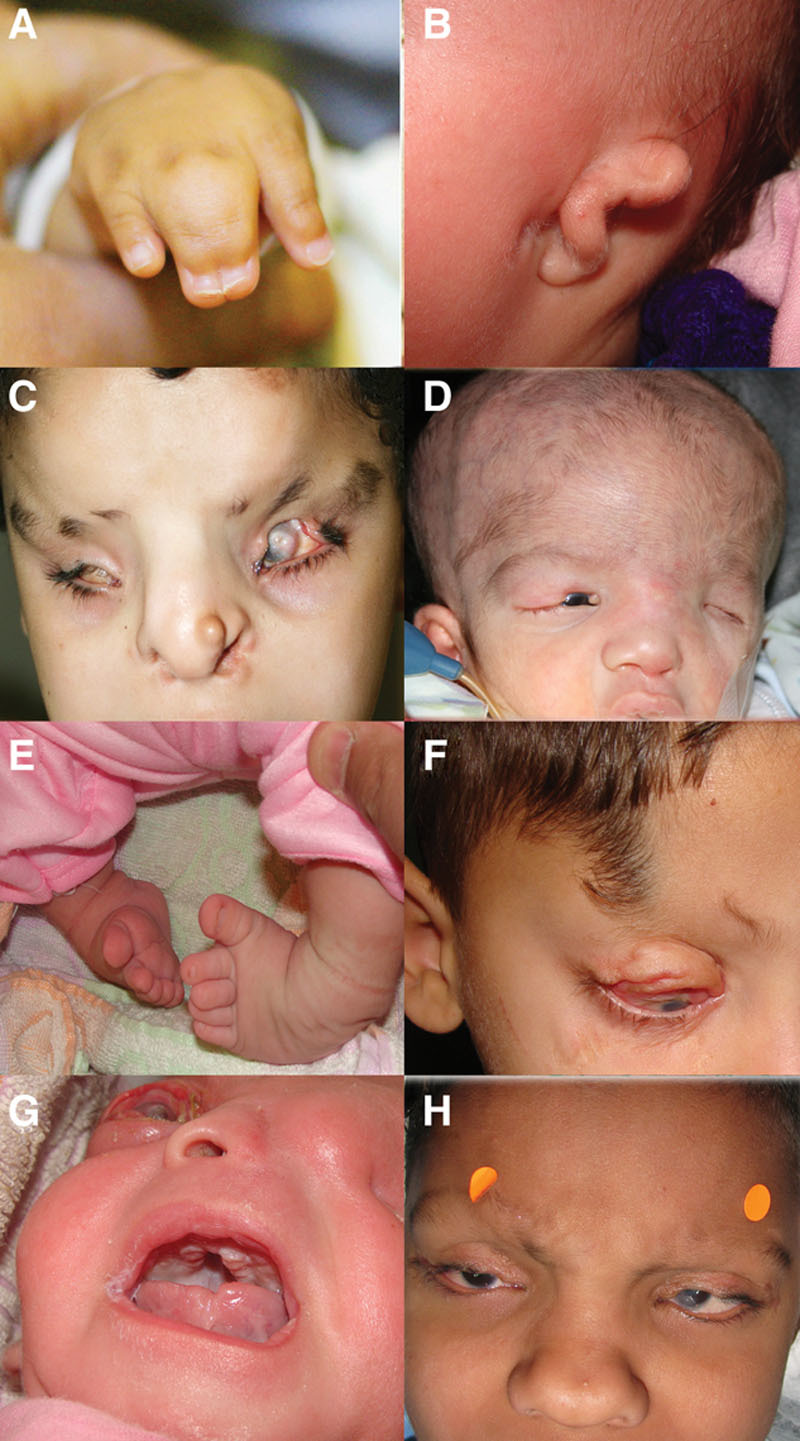
Systemic features of Fraser syndrome. A, Digital malformations. Syndactyly of the fingers, which is the most common nonocular feature of Fraser syndrome. B, Ear malformations. Low-set, malformed, and posteriorly rotated ears. C, Musculoskeletal abnormalities. A furrow in the forehead corresponding to a groove in the frontal bone with an overlying abnormal brow hair pattern. Also, note the bifid nose. D, Cerebral malformations. Macrocephaly due to hydrocephalus. Fraser syndrome is compatible with normal cranial development but both microcephaly and macrocephaly have been reported.14,145 Note contralateral hypoplastic left nostril and microphthalmia. E, Musculoskeletal abnormalities. Talipes varus. F, An abnormal tongue of hair extending from the temple to the eyebrow is not uncommon in Fraser syndrome, and is considered by some authors to be a minor feature of the disease.38 G, High arched palate. H, Nasal malformations. A broad nose and a depressed broad nasal bridge are common nasal abnormalities in Fraser syndrome and a minor feature of the disease.
Histopathologically, the eyelids are replaced by undifferentiated fibrous tissue, with total absence of the tarsus, meibomian glands, cilia, and lacrimal glands,31,33,74 but the orbicularis oculi and the levator muscles are usually preserved, and the extraocular muscles may be present or absent.31 The conjunctiva is usually absent in histologic sections, and the cornea is usually replaced with “intertwining collagen bundles” with no epithelial or endothelial lining.31,74 While the anterior segment might be malformed, the choroid, retina, and optic nerves appear to be normally formed.74
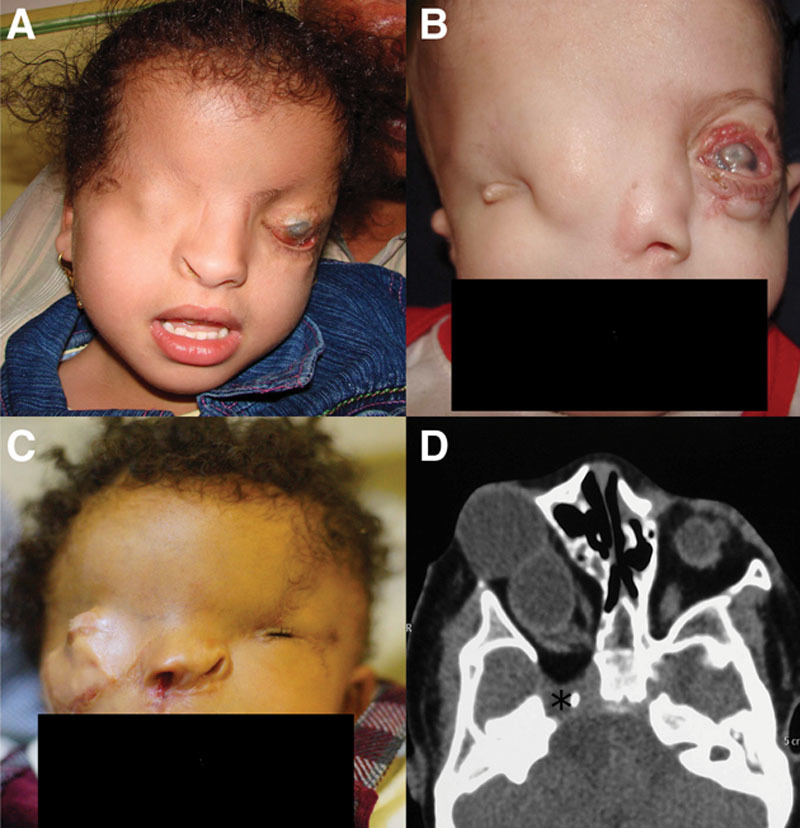
There appears to be a distinct form of familial (autosomal dominant), bilateral isolated CO that was reported only 3 times in the literature called autosomal dominant CO.36,39–42 The palpebral features are quite distinct from typical complete CO in that the eyebrows are completely normal and the eyelids appear grossly as if they have normally developed, but later fused together and to the globe. Additionally, these patients usually have Peters anomaly, a feature not documented in classic complete CO (Fig. 6D).36
FIG. 6.
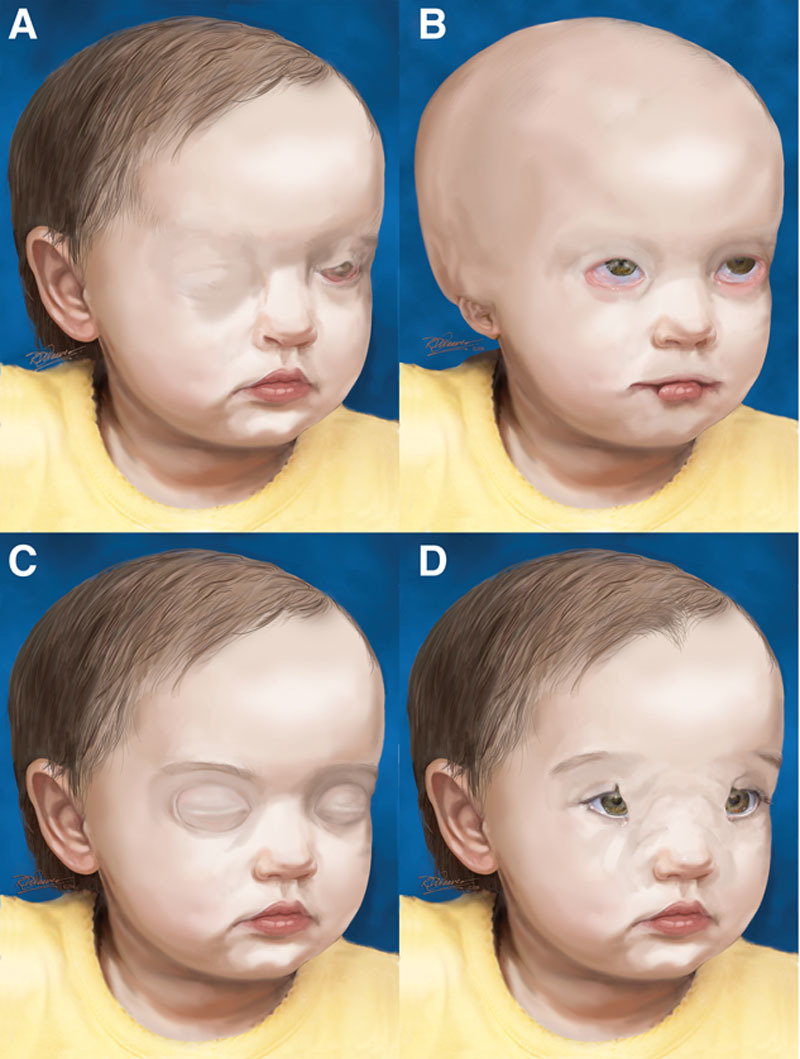
Facial and palpebral features of the rare syndromic associations of upper eyelid colobomas. A, Manitoba oculotrichoanal syndrome. Features not quite dissimilar from Fraser syndrome with right complete cryptophthalmos and left congenital symblepharon variant, with an abnormal tongue of hair from the eyebrow. B, Ablepharon-macrostomia syndrome. Note that the eyelid deficiency (shortening) that is more commonly microblepharon rather than ablepharon35 also involves the lower eyelids.33 Also note the abnormally disfigured right ear and the slightly enlarged mouth. Alopecia or more commonly sparse hair is not an unusual finding.33 C, Autosomal dominant cryptophthalmos. Note the central horizontal eyelid dimpling which corresponds to the area of eyelid attachment to the globe and absence of eyebrow abnormalities.43 This is considered a different phenotype with features that are somewhat distinct from the typical findings of CO/FS.36 D, Nasopalpebral lipoma-coloboma syndrome. Please note the lower eyelid coloboma which is partly concealed by the lipoma. Also note the abnormal brow pattern. Modified with permission from ref145.
Incomplete CO.
A subset of CO patients has an ill-defined upper eyelid or an incomplete skin fold devoid of tarsus covering almost the entire cornea. This fold is completely fused to the underlying keratinized cornea along the entire length and width of the fold, and may or may not extend downward to fuse with the lower eyelid. This abnormal fold does not cover the entire eye; hence, there is the partial or incomplete designation in François clinical observations. These patients usually have an insignificant upper eyelid coloboma because the colobomatous part of the eyelid is partly replaced by the skin fold (Fig. 2A,B).
In his original description of incomplete CO, François31 describes a rudimentary presence of the lateral eyelid structures and conjunctival fornix, which is almost a constant feature in incomplete CO and in CSV even in its most severe forms,32 and contrary to previous authors who maintained that no normal eyelid tissue could be observed medial to the coloboma,32,74 we of this series have observed 1 patient in whom a small lip of normal rudimentary upper eyelid was present nasally together with a functioning upper punctum and canaliculus (Fig. 2B). The inferior fornix may or may not be spared depending on the inferior extent of the abnormal skin fold (Fig. 1C). Contrary to François31 description that patients with “incomplete CO” have microphthalmic eyes, we have encountered patients with incomplete CO whose eyes are of normal size echographically. Of the 3 subtypes of nonsyndromic CO in François classification, incomplete or partial CO is the rarest.75
In all, 2 of the 3 patients with incomplete CO in our series were strictly unilateral, and the third patient only had an insignificant notch with minimal CPA in the contralateral upper eyelid (Figs. 2A and 3B). In their detailed literature review of complete and incomplete CO where CSV was specifically excluded, Konrad and associates75 documented only 15 cases of incomplete CO, 13 of whom were unilateral. Subramanian and associates32 also documented unilateral findings in 4 out of 5 patients.
Congenital symblepharon variant.
In CSV (also referred to as abortive CO or partial CO), the upper eyelid skin is deficient and the remnants of it are visibly adherent to the globe, hence the designation “congenital symblepharon.” The lower eyelid is usually spared, and corneal involvement depends on the extent and severity of CPA.32 Subramanian and associates32 classified CSV into mild, moderate, and severe grades based on the extent of the coloboma and the severity of CPA. In our experience, the defect may indeed range from a minor notch in the eyelid margin associated with minimal CPA overlying the coloboma or even adhesions confined to the limbus to patients in whom there is a near-total eyelid colobomatous defect with extensive involvement of the cornea (Figs. 3A–D).
The term CSV is actually a relic of late nineteenth century medicine,76 and should be abandoned as it may erroneously imply that in the other 2 varieties (complete and incomplete CO), the cornea is spared or is at least less affected than in CSV while the opposite is actually true as the extent of CPA (which is a universal finding in all 3 types without which the diagnosis of CO should be called into question) is in fact less in CSV than in complete or incomplete CO where the conjunctival sac may be entirely absent.31,74
Although Nouby33 maintains that these colobomatous defects assume a triangular shape in contrast to the typically quadrangular shape of SCs and colobomas associated with GS, we consider it a futile exercise to try to ascribe a certain shape to these colobomas. They could indeed appear triangular if minimal in size (Fig. 3A), but they assume a relatively quadrangular shape as they grow larger (Fig. 3B), and it is more accurate to describe CO colobomas as “irregular” in shape because the extent of the typically variable CPA governs the final shape of the defect (Fig. 3C,D).
Simple Coloboma.
According to Mustardè,29 these “pure colobomas” are strictly unilateral, have no associated systemic findings, do not usually present with significant keratopathy despite the corneal exposure, and the upper fornix is always well formed with normal depth. The shape of these colobomas is typically quadrangular,33 but we agree with Van Der Meulen77 that it may also assume a triangular configuration, and for reasons unknown, they are almost exclusively confined to the junction of the medial and central parts of the eyelid.77 The actual size of the defect is difficult to ascertain because the edges are pulled in opposite directions by the separated parts of the orbicularis muscle, which is apparently contracting freely because the eyelids in these patients are not tethered to the globe in any way.77 This fact may explain why corneal exposure may be less pronounced than would be expected even with a big eyelid defect.29,78
Syndromic Variants.
Fraser Syndrome.
In 1962, George Fraser first described the syndrome bearing his name,79 and in more recent times, several excellent reviews have appeared in the literature, where criteria for diagnosing FS were established and its extensive clinical variability was demonstrated.37,38,65,68 In 1986, Thomas et al.37 framed the diagnostic criteria for FS. They defined 4 major criteria and 8 minor criteria, and suggested that the diagnosis should be made with 2 major and 1 minor criteria, or 1 major and 4 minor criteria. In 2007, van Haelst et al.68 suggested that FS is better defined if 3 major criteria are confirmed.
The major features of FS include CO (85–93%), which is considered the single most common diagnostic anomaly in FS.37,38,65,68 If CO is excluded, 3 other syndromes could share the same features (cutis aplasia-total, Nager acrofacial dysostosis, Pallister–Hall syndrome)68; therefore without a proband or at least a family member with CO, the diagnosis of FS should be called into question.38 Complete CO is the subtype most commonly associated with FS, and CSV is the least.68 A tongue of hair extending from the temple to the eyebrow (Fig. 4F) is encountered in around 35% of FS patients and is considered by some authors to be a minor feature of FS.38,68 We occasionally observe the same finding in patients with isolated CO, and we do not believe it is specific to FS, as this abnormality is simply considered a general indicator of maldevelopment of the upper eyelid region because normal eyelid development suppresses hair growth in the forehead.80
The most common major nonocular criterion of FS is cutaneous syndactyly of the hands and feet, which usually involves fusion of the second, third, and fourth digits with a reported frequency that varies between 62% and 95%,37,38,65,68 followed by ambiguous genitalia (40%–66%).37,38,65,66,68 Urinary tract abnormalities are also quite common (37%–80%), and usually take the form of unilateral or bilateral renal agenesis, renal hypoplasia, ureteral agenesis, or less common bladder abnormalities.37,38,65,68 A positive family history (40%–60%) of the disease is also an important requisite for diagnosing FS.38,68 Respiratory tract abnormalities in the form of laryngeal and/or tracheal stenosis are very frequent findings and are currently considered as one of the major features of FS90, and should be considered when FS patients are subjected to general anesthesia.81,82
Minor features are not infrequent (Fig. 4) and include congenital malformations of the ears, which are found in up to 75% of patients with FS patients (low set, malformed, posteriorly rotated, or dysplastic)68; nasal malformations (~50%), which take the form of a wide nasal bridge, an absent or hypoplastic nasal ala, or a bifid nose32,37,38,65,68; cleft lip/palate; skeletal abnormalities, which may manifest in the skull, ribs, or limbs (Fig. 4C, E); and umbilical anomalies.37,38,65,68 Rarer features include craniofacial malformations, mental retardation, and congenital cardiac malformations.68 The prognosis for life is generally poor. Almost half of FS patients will not survive beyond the first year of life, and only 2% make it beyond the age of 20 years.36,38
Goldenhar Syndrome.
With a causally heterogeneous etiology and with such a wide variability of expression, it comes as no surprise that GS bears 16 different names in the literature, which until recently were thought to represent separate entities.9 Goldenhar syndrome is a pleiotropic condition with a spectrum of cardiac, renal, and skeletal manifestations besides the typical facial and vertebral anomalies. Therefore, the term oculoauriculovertebral spectrum was suggested to be better descriptive of the spectrum of anomalies in GS because names such as hemifacial microsomia and first branchial arch syndrome give the erroneous impression that GS abnormalities are limited to the face only.9,83
There are several conflicting classification schemes and no general consensus on criteria for diagnosis.84–87 Strömland et al.57 proposed that to establish the diagnosis, 2 of the 4 major criteria they suggested have to be met (orocraniofacial, ocular, auricular, and vertebral), but Tasse et al.87 excluded ocular manifestations from the major criteria of GS on statistical bases.
As a syndrome with such diverse clinical features and conflicting reports of laterality and sex predilection,84–87 it is interesting to note that there exists a mandatory feature of the syndrome that is microtia and/or preauricular tags (composed of skin and cartilage), which are present in 95%–100% of patients and may be bilateral in up to half of the patients even if other manifestations are strictly unilateral.57,87 In familial cases, the diagnosis is considered even if preauricular tags are the only manifestation of the disease.87
Flattening of one half of the face causing marked facial asymmetry (hemifacial microsomia) is quite common and second only to ear anomalies,57,87 and is caused by an underdeveloped mandible, maxilla, or zygomatic bone with partial atrophy of the muscles of expression and muscles of mastication.88 Cervical spine abnormalities are the third most common feature of GS, but minor manifestations also occur frequently, and some of them may tend to cluster together.87 These include deafness, facial clefting, facial nerve paralysis, urogenital anomalies, brain anomalies, congenital heart defects, delayed motor development, short stature, delayed speech development, and microcephaly.87
The hallmark of ocular features in GS is an eccentric or limbal epibulbar dermoid (ED), the presence of which should prompt the clinician to look for other ocular anomalies as they are positively correlated with the presence of ED. The most common location is inferotemporal, but nasal limbal choristomas have been reported.88,89
Strikingly more yellowish than ED (because of a substantial deep layer of mature fat cells)90, lipodermoids (dermolipomas) tend to be less frequently associated with GS than ED and are not only limited to the superotemporal quadrant but may also occur inferiorly (Fig. 5A).91 The true prevalence of both is unknown as ED and lipodermoids are usually grouped together in cohort studies in the nonophthalmic literature, but the percentage varies from 12% to 100%.88
FIG. 5.
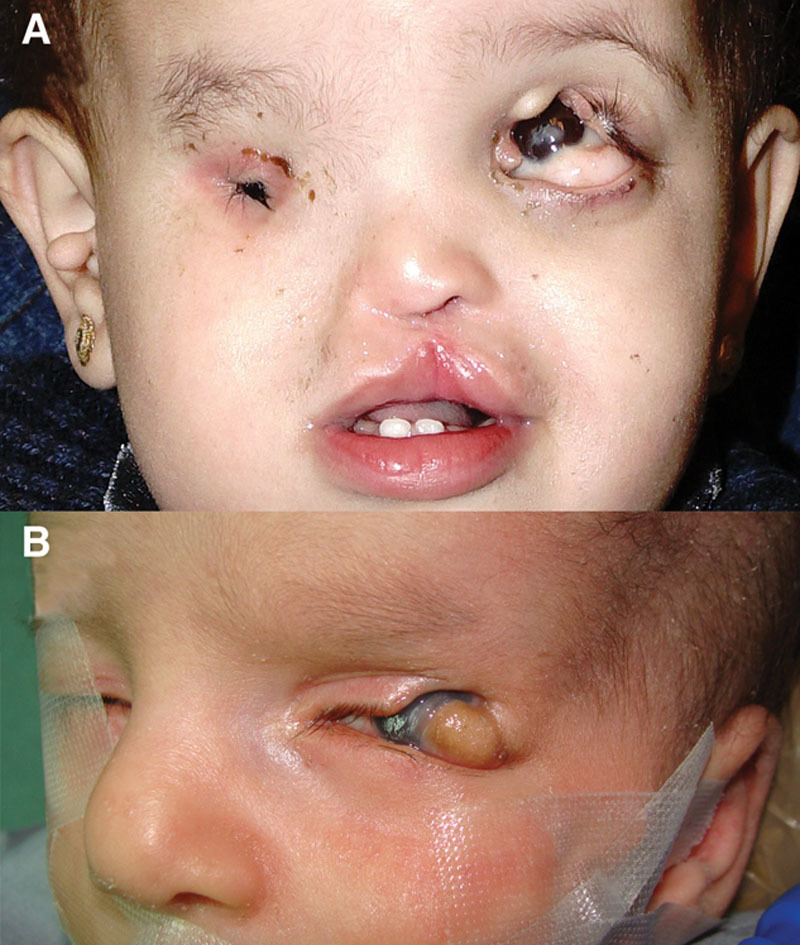
A, Features of Goldenhar syndrome. A rectangular upper eyelid coloboma is seen not attached to the cornea or the bulbar conjunctiva occupying the medial half of the eyelid with an overlying ridge of tissue encroaching on the coloboma. An inferiorly located lipodermoid is also seen. This lipodermoid could be used in reconstruction of the colobomatous eyelid.58 Also note the repaired cleft lip, and the contralateral anophthalmia and preauricular tags. B, An unusual case of lateral coloboma associated with Goldenhar syndrome. Note the strip of tissue extending from the lipodermoid to the colobomatous eyelid defect.
Dermolipomas and ED are more commonly associated with GS than upper eyelid colobomas, the frequency of which tend to increase with increasing severity of GS.87 The reported frequency of upper eyelid colobomas ranges from 6% to 24%.57,87,89,91,92 Usually unilateral and occasionally bilateral,89,92 the most frequent location for these colobomas is at the junction between the medial and central thirds of the upper eyelid.89,93 Although Fries and Katowitz91 make an unsubstantiated claim that the majority of these colobomas are temporal in location, we have only encountered a single case in a temporal location, which is identical to a patient described by Baum and Feingold92 in 1973 (Fig. 5B). When it assumes its typical quadrangular shape, colobomas associated with GS may manifest an overriding ridge or thickening of eyelid tissue, which is sometimes observed encroaching on the superior border of the coloboma and may be confused for a previous surgical scar (Fig. 5A).
Because GS colobomas are typically associated with a limbal dermoid,89 Mustardè29 attributed these colobomas to mechanical pressure from ED but the fact that the usual location of ED (inferotemporal) does not typically coincide with the usual location of the coloboma (junction of medial and central thirds of the eyelid),77 coupled with the fact that it may even occur in the other eye,89,94 in addition to our own observation of the association of small EDs with large upper eyelid colobomas; all belie this hypothesis. By definition, the upper fornix is deep and well formed and these colobomas are not usually attached to the surface of the eye, but a small pedicle may occasionally connect the coloboma to an exceptionally large ED (Fig. 5B).90
Although some authors89,91 mention the lower eyelid as a possible location for eyelid colobomas in GS, we believe that what they observed is actually a shallow, wide depression rather than a true coloboma caused perhaps by mechanical pressure of an ED on the lower eyelid during development rather than a true coloboma.29
Additional ocular features include ptosis (12%), nasolacrimal abnormalities (11%), horizontal phimosis, and strabismus (10%–19%) in the form of esotropia, exotropia, or Duane syndrome.89,91 Microphthalmia and anophthalmia can occur with a reported frequency equal to or even exceeding that of upper eyelid coloboma.57,87 Rarer associations include eyelid skin tags, iris coloboma, tortuous retinal vessels, choroidal colobomas, hypoplastic optic nerve, macular hypoplasia, and a tilted optic disc.91 Unfortunately 3 of the 4 major articles dealing exclusively with the ocular features of GS date back to the seventies and mid-eighties, and the fourth focuses on dermolipoma only.88,89,92,95 A fresh insight into the ocular findings of GS in a pediatrics setting and not in an ophthalmic referral center (where referral bias may skew the results) is warranted.
RARE SYNDROMIC ASSOCIATIONS WITH CONGENITAL UPPER EYELID COLOBOMA
A detailed discussion of the syndromic curiosities associated with upper eyelid coloboma, including Manitoba oculotrichoanal syndrome, ablepharon-macrostomia syndrome, amniotic band sequence, oculoectodermal syndrome, and neurocutaneous syndrome, is beyond the scope of the current discussion and is reviewed elsewhere.34,53–56,96–127 The distinct facial and ocular features of some of these syndromes are outlined in Figure 6.
MANAGEMENT
Upper eyelid coloboma may represent one of the few oculoplastic emergencies that may be faced at a very early age. Operating on very young infants with multiple congenital anomalies37,38,68 with attendant possible anesthetic risks,81,82 dealing with the shocked and grieved parents of a “blind” or a “disfigured” child, coupled with the relative lack of tissue for reconstruction could be a major surgical challenge.34,82 Families should be counseled that several operations may be required, and in cases of CO/FS the visual potential may be poor despite multiple surgeries, and that even in the absence of good visual outcome, cosmetic rehabilitation might not be perfect.128
In discordance with the rest of the text, the management of CO/FS colobomas will be discussed together with the management of SC/GS because the basic management principles are similar. Therefore, we will split the management into 2 sections according to the presence or absence of CPA.
Although several shared reconstruction techniques are employed in either category, they will be dealt with separately. Rather than giving a detailed discussion of the various methods of upper eyelid reconstruction, which is well beyond the realm of this text, we will focus on relevant surgical points that are pertinent to the discussion.
Colobomas Not Associated With CPA (SC/GS).
For small colobomas without keratopathy where the defect size could be overrated because of the unrestricted pull of the orbicularis muscle,77 several authors recommend watchful observation, deferring management till the ripe age of 2 to 4 years when the eyelids have grown to a more manageable size and more tissue is available for reconstruction, particularly if they have a good Bell phenomenon.29,78,129
For small defects that do require suturing (up to 25%), direct closure may suffice, although for moderately sized defects (25%–50%), where a severing of the upper crus of the lateral canthal tendon is required for satisfactory closure. We do not recommend direct closure even if clinical judgment intraoperatively favors a simpler direct closure approach. This is primarily because the upper crus readily readheres by scar tissue,29 and this could result in early wound dehiscence despite the fact that it was sutured without any undue tension on the edges.130 Therefore, in the context of congenital upper eyelid coloboma, caution is warranted in relying too heavily on the often quoted dictum that when defect edges are brought under “normal” tension, the “real” size of the defect is actually smaller than it appears,129,131 because it overlooks the relative lack of underlying laxity in infants.130 Even if early wound dehiscence does not occur after direct approximation, severe ptosis is observed that may persist for several months29 obstructing the visual axis, which is potentially amblyogenic hindering the functional value of the procedure and is not cosmetically pleasing to the parents. This mechanical ptosis occurs because with simple direct closure in the tight milieu of an infant eyelid, the eyelid is placed under severe tension. What further complicates the picture and exacerbates the ptosis is that with direct closure, it is not usually possible to resuture the levator muscle and counter the ptosis.
For larger defects (50% or more of the eyelid), available options include the Cutler-Beard procedure, an eyelid rotational (switch) flap, or a tarsomarginal graft. The classic or still better “modified bridge flap,” where a spacer graft is used beneath the flap, provides excellent donor–recipient match132 but has several disadvantages; it is potentially amblyogenic, which is of real concern in SC/GS where the visual potential is good. Lower eyelid instability and inferior fornix shortening are also of particular concern because they assume that there is an excess of lower forniceal conjunctiva while in fact there is not. Postoperative lower eyelid retraction is a real concern.133 Another concern raised by parents postoperatively is bulkiness of the bridge flap, which tends to persist and does not improve with time, and may require revision prior to school age. An often overlooked source for posterior lamellar reconstruction if a modified bridge flap is chosen in cases of GS is the cartilage in the preauricular skin tags,120 which is composed of keratinizing epithelium surrounding a central core of elastic cartilage,134 and is occasionally large enough to provide sufficient cartilage for posterior lamellar support.
An issue frequently raised by cosmetically concerned parents when the Cutler-Beard procedure is suggested is the lack of eyelashes in the reconstructed area, and for this reason, an eyelid switch or rotational flap may provide a better cosmetic outcome than a bridge flap.29,32,78,135 After careful fashioning of a medially based or a laterally based full thickness eyelid switch or rotational flap on an adequate (5–6 mm) vascular pedicle incorporating the marginal arcade,29 the flap is rotated 180°, secured in a lamellar fashion to the freshened edges of the remnants of the colobomatous eyelid, and is used to fashion a new upper eyelid margin, which is separated 2 to 3 weeks later. Up to 50% of the upper eyelid can be reconstructed with only 25% of the lower eyelid, and the defect in the lower eyelid is easily closed directly although occasionally a rotational cheek flap may be required.4,29,78,135,136
Another option in patients with SC/GS is the use of a tarsomarginal graft covered with an overlying skin-muscle advancement flap, which does not occlude the pupil and may be a better option than a eyelid sharing procedure.131 It is a single-stage procedure that is less demanding in terms of blood supply requirements than a full-thickness composite eyelid graft because it receives additional blood supply from the overlying skin-muscle flap. However, parents are occasionally concerned about harvesting tissue from the contralateral normal side, and therefore, the ipsilateral lower eyelid may be chosen.137 If taken from the contralateral side, it is recommended that to get a good contour, temporal defects should be reconstructed with grafts taken from the nasal eyelid and vice versa.131
Colobomas Associated With CPA (CO/FS).
The management of colobomas associated with CPA (CO/FS) differs from SC/GS in several respects, particularly in the fact that in the latter, no reconstruction of the fornix is required, while in CO/FS, a separate or combined 2-stage reconstruction is usually required. Another valid clinical point to remember is that in contrast to SC/GS where it is not always easy to define the exact size of the defect,29 the size of the defect in CO/FS observed clinically is more accurate because the CPA fix the defect in place and limit the freedom of motility of the orbicularis oculi muscle. In either case, any form of measurement will be largely inaccurate,78 but as a rough estimate, Mustardè29 recommends counting and comparing the number of eyelashes in both eyelids.
It should be remembered that prior to eyelid fusion, the corneal epithelium is fused with the surface ectoderm22, and only after eyelid fusion, do they separate and the cornea takes its normal developmental course. Consequently, by definition, a “patient with CO will not have a normal cornea.” 4 This dictum is not absolute and in our experience, when this “abnormal cornea” causes adhesions to recur after surgical reconstruction of the fornix, they tend to occur in exactly the same area where they were initially observed and they had a tendency to spare the originally “normal” part of the cornea. Thus, it may be better to rephrase by saying that a patient with CO will not have a normal cornea in the area corresponding to the CPA. Accordingly, parents should be informed that complete and incomplete COs are incompatible with normal vision,4 and the visual potential in CSV is dependent on the extent of adhesions at the first presentation.
Complete CO.
Complete CO deserves a special category of its own because technically there is no coloboma, the eye is painless with no risk of impending perforation, and as we mentioned earlier, normal development of the cornea is impossible in the first place and fornix reconstruction for prosthetic shell placement is extremely challenging because the surgeon has the double task of creating a fornix and recreating the eyelids.138 Therefore, Saleh et al.4 preferred deferring these surgeries on the grounds of poor visual, cosmetic, and reconstructive outcomes, and they got disappointing results when forced to operate by the parents on psychological grounds. If surgery is to be undertaken, it should be performed in a stepwise fashion and the first objective is the elusive task of creating a conjunctival sac.4,73 The skin covering the ocular remnants is divided followed by placement of a conformer covered with a mucous membrane (MM) graft. Eyelid reconstruction with stiffening of the posterior lamella and possibly further socket MM grafting could be carried out 1 year later.4 An alternative source for socket reconstruction if MM grafting fails in these difficult cases is the preputial skin in uncircumcised children.139,140
In patients with complete CO, orbitopalpebral cysts are infrequently observed and probably represent a surface ectodermal anomaly rather than a true ocular cyst caused by failure of invagination of the primary optic cup.49,141 These cysts pose a controversial problem. While Subramaniam et al. advocated early intervention at the age of 3 months,139 others raise legitimate concerns that surgical excision is questionable citing communication with the dysplastic globe and light perception vision in these patients.141 Others argue that the cyst contents should be left intact because evacuation of the contents at least at an early age could lead to underdevelopment of the bony orbit with subsequent facial asymmetry.4 In our own case series, we encountered a single FS patient with a unilateral orbitopalpebral cyst after a previous failed attempt at surgical reconstruction elsewhere, and it did not react to light although the contralateral microphthalmic eye did (Fig. 1C,D).
Incomplete CO.
Incomplete CO sometimes poses a challenging problem in trying to explain to the parents why a poor visual potential is expected. To the parents’ eyes, it may look less devastating than either complete CO or CSV because the coloboma looks small as it is fictitiously reduced in size by the skin fold, the diseased part of the cornea is completely concealed, keratopathy is unusual, the eye is painless, and the remaining exposed part of the cornea usually looks normal (Fig. 2). Saleh et al.4 caution against rushing to early surgery, because most of the cornea is keratinized and the prognosis for functional or cosmetic reconstruction is dismal at best. We tend to agree that incomplete CO does not pose a surgical emergency except if parents are devastated by the disfigurement, and in that case, surgery may be necessary on cosmetic bases alone because in our experience, recurrence of adhesions after fornix reconstruction is the rule rather than the exception.
If reconstruction is to be undertaken, an eyelid sharing procedure or an eyelid switch procedure should be used. After severing the CPA, and excision of the abnormal skin fold, the defect is usually more than 50% of the eyelid and would not lend itself to direct approximation. The use of a switch or a bridge flap might actually reduce the recurrence of adhesions, because by transposing “normal” tissue, including palpebral conjunctiva from the normal inferior fornix to its narrow superior counterpart, the upper eyelid and palpebral apertures are widened superiorly.29 Patients with incomplete CO where the abnormal skin fold extends to the lower eyelid may pose a challenging problem, and a Tenzel flap or direct approximation could be attempted. An innovative technique was recently described by Witmer and Slonim142 who used a lateral modified sliding Hughes procedure where a tarsoconjunctival flap is fashioned from the lateral part of the same eyelid, which usually has a substantial and normal tarsal remnant to work with (Fig. 2D). This flap is advanced horizontally and secured to the medial canthal tendon and medial edge of the eyelid, followed by placement of an overlying skin graft. A possible source for skin grafting is the excised skin fold, which covered the cornea.7
With the possible failure of salvaging vision in these eyes and with future prosthetic placement in mind, every attempt should be undertaken at reconstructing the superior fornix even if the visual potential is poor. Significant controversies exist in the literature regarding the timing and type of the graft used for fornix reconstruction. There is no clear consensus whether fornix reconstruction should be carried out concomitantly with eyelid reconstruction or deferred until flap separation and opinions are split between both approaches.4,32,64,71,129,135,138–140,143 Our personal preference is to defer fornix reconstruction to be performed with flap separation because as we mentioned earlier, a switch flap might actually help deepen the fornix by lengthening the eyelid. If such an approach is chosen, the distal end of the severed conjunctiva in the first procedure is sutured in a back fashion with a double-armed 6-0 polyglactin suture to the superior fornix, which is exteriorized and sutured on the skin before the levator muscle is sutured to the tarsus, followed by fornix reconstruction at a later stage.
The second controversial issue is the type of graft used. While some authors had excellent experience with amniotic membrane (AM) grafting,32,64,138 others argued that the AM has a tendency to dry out and contract and is not recommended in CO/FS.4 It should be noted that AM grafts are substrate grafts not substitute grafts144 and require a substantial source of conjunctival holoclones to grow on the surface of the AM, a situation which may be difficult to meet in CO/FS. We have experienced inconsistent results with both AM and MM grafts, and regardless of the type of the graft, CPA may recur in the same area where it was located originally. Therefore, we conclude that the rate limiting step in fornix reconstruction in CO/FS patients is the extent of CPA initially observed before surgical intervention rather than the type or timing of the graft.
Congenital Symblepharon Variant.
Upper eyelid colobomas in this variety are potentially vision threatening and should be promptly treated. In our country and in Europe,65 CO/FS has a predilection to occur in distant geographic locations where close follow up is impossible. Consequently, watchful observation cannot be advocated except in eyes with minimal colobomatous notches and in the absence of keratopathy (Fig. 3A).
The same basic management principles as in incomplete CO apply, and except when the defect is really small where direct suturing could work, we generally favor an eyelid switch flap technique or an eyelid sharing procedure. In our experience, recurrence of CPA occurs more frequently when direct suturing is undertaken. The cause of this is unknown, but it could be theorized that when an eyelid sharing procedure is employed, the area of the cornea that was adherent to the eyelid remnants is no longer in contact with the diseased upper eyelid but rather faces an overlying normal conjunctiva and tarsus from the unaffected lower eyelid, an effect that is further exacerbated if the remnants of the levator muscle are sutured to the edges of the rotated tarsus, thus lifting the abnormal eyelid remnants/flap further away from the damaged area of the cornea.
Obviously, a contralateral tarsomarginal graft is not an option in bilateral cases, and it should not be used even when the other eye is minimally affected, so as not to compromise the visual potential of the “normal” eye.4
When an AM is used for reconstruction (AmnioGraft, Bio-Tissue, Miami, FL, U.S.A.), a 1.5 cm × 1 cm graft is sufficient to cover the entire fornix and the affected part of the cornea after careful superficial keratectomy, although in patients with incomplete CO a 2.5 cm × 2 cm piece is preferable as the defect is larger. The graft is secured with fibrin glue (Fibrogloo, CMCBB, Cairo, Egypt) and not with sutures to reduce irritation and postoperative inflammation, followed by placement of a specially designed symblepharon ring with 2 adjacent superior holes. A double-armed 6-0 polyglactin suture is passed through these holes and the superior fornix, and then advanced full thickness through the upper eyelid to be sutured directly on the surface of the skin without using bolsters.
When an MM is used, it should be thinned as much as possible to ensure a good take. To avoid suture irritation and in an attempt to minimize any possible source of postoperative inflammation, we also fix the MM graft in place with fibrin glue followed by placement of a symblepharon ring. In our hands, recurrence of adhesions is frequently observed although the casual observer would get the calming impression that the fornix looks significantly deeper, which is simply due to eyelid lengthening from the lower eyelid. This is of paramount importance if a penetrating keratoplasty is planned, and should be pointed out to the cornea surgeon very clearly, because even if the future cornea would seem to receive proper protection from the reconstructed eyelid, when this eyelid is carefully lifted off the globe, the adhesions may still be located in the same exact place where they were initially observed.
CONCLUSION
In the near future, advances in molecular genetic testing will help redefine the etiopathogenesis and the diverse clinical spectrum of genetic diseases associated with upper eyelid colobomas and will undoubtedly unravel the diagnostic dilemmas and phenotypic overlap that we have discussed in this review. It would also help families with afflicted individuals make better family planning decisions and may even offer gene therapy obviating the need of the oft frustrating surgical approaches. We sincerely hope that in a decade or less, this review article will be obsolete.
METHODS OF LITERATURE SEARCH
English and world medical literatures were searched from 1880 up through May 2014 using Google search and PubMed for papers staring from 1950 till the present, and for earlier papers, we used the Index-Catalogue (Library of the Surgeon-General’s Office 1880–1961) that could be accessed at http://indexcat.nlm.nih.gov/vivisimo/cgi-bin/query-meta?v:project=indexcat&v:sources=indexcat&sortby=ID. We used the following keywords: upper eyelid coloboma, colobomata, Fraser syndrome, cryptophthalmos, cryptophthalmia, cryptophthalmos, Goldenhar syndrome, oculoauriculovertebral spectrum, HFM, epibulbar dermoid, lipodermoid, dermolipoma, ocular choristoma, amniotic band sequence, neurocutaneous syndromes, oculoectodermal syndrome, nasopalpebral lipoma-coloboma syndrome, Manitoba oculotrichoanal syndrome, and ablepharon-macrostomia syndrome. Articles in all languages were considered. For articles in French, we carried out the translation ourselves (with extreme difficulty) and for German articles, a professional medical translator was hired. Important references in these articles were retrieved and reviewed. Every single reference was retrieved with the exception of 1 reference that was apparently a motion picture, and we specifically mentioned the source we cited it from. As a general rule, articles recovered on Google search that were not on PubMed were usually excluded if not published in peer-reviewed journals except a few papers that were deemed of exceptional quality.
Footnotes
The authors have no financial or conflicts of interest in the study.
REFERENCES
- 1.Anderson KN, Anderson LE, Glanze WD. In: Mosby’s Medical, Nursing and Allied Health Dictionary. 4th ed. St. Louis: Mosby; 1994. Coloboma. p. 3613. [Google Scholar]
- 2.Dorland WN, Taylor EJ. Dorland’s Illustrated Medical Dictionary. 27th ed. Philadelphia, PA: WB Saunders; 1988. p. 359. [Google Scholar]
- 3.Onwochei BC, Simon JW, Bateman JB, et al. Ocular colobomata. Surv Ophthalmol. 2000;45:175–94. doi: 10.1016/s0039-6257(00)00151-x. [DOI] [PubMed] [Google Scholar]
- 4.Saleh GM, Hussain B, Verity DH, et al. A surgical strategy for the correction of Fraser syndrome cryptophthalmos. Ophthalmology. 2009;116:1707–12. doi: 10.1016/j.ophtha.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Pavlakis E, Chiotaki R, Chalepakis G. The role of Fras1/Frem proteins in the structure and function of basement membrane. Int J Biochem Cell Biol. 2011;43:487–95. doi: 10.1016/j.biocel.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 6.Pao KY, Levin AV. Genetics in oculoplastics. In: Black EH, Nesi FA, Gladstone G, editors. In: Smith and Nesi’s Ophthalmic Plastic and Reconstructive Surgery. 3rd ed. New York: Springer; 2012. pp. 1249–63. [Google Scholar]
- 7.Tawfik HA. Re: “Cryptophthalmos: reconstructive techniques–expanded classification of congenital symblepharon variant”. Ophthal Plast Reconstr Surg. 2013;29:505–6. doi: 10.1097/IOP.0000000000000043. [DOI] [PubMed] [Google Scholar]
- 8.Hartsfield JK. Review of the etiologic heterogeneity of the oculo-auriculo-vertebral spectrum (Hemifacial Microsomia). Orthod Craniofac Res. 2007;10:121–8. doi: 10.1111/j.1601-6343.2007.00391.x. [DOI] [PubMed] [Google Scholar]
- 9.Cohen MM, Jr, Rollnick BR, Kaye CI. Oculoauriculovertebral spectrum: an updated critique. Cleft Palate J. 1989;26:276–86. [PubMed] [Google Scholar]
- 10.Kahana A, Elner VM. The meaning of diagnoses in orbital disease. Ophthal Plast Reconstr Surg. 2013;29:347–8. doi: 10.1097/IOP.0b013e3182a65004. [DOI] [PubMed] [Google Scholar]
- 11.Pearson AA. The development of the eyelids. Part I. External features. J Anat. 1980;130:33–42. [PMC free article] [PubMed] [Google Scholar]
- 12.Byun TH, Kim JT, Park HW, et al. Timetable for upper eyelid development in staged human embryos and fetuses. Anat Rec (Hoboken) 2011;294:789–96. doi: 10.1002/ar.21366. [DOI] [PubMed] [Google Scholar]
- 13.Alazami AM, Shaheen R, Alzahrani F, et al. FREM1 mutations cause bifid nose, renal agenesis, and anorectal malformations syndrome. Am J Hum Genet. 2009;85:414–8. doi: 10.1016/j.ajhg.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Z, Bhandari A, Mannik J, et al. Grainyhead-like factor Get1/Grhl3 regulates formation of the epidermal leading edge during eyelid closure. Dev Biol. 2008;319:56–67. doi: 10.1016/j.ydbio.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao H, Ono K, Kurose H, et al. Exogenous FGF10 can rescue an eye-open at birth phenotype of Fgf10-null mice by activating activin and TGFalpha-EGFR signaling. Dev Growth Differ. 2006;48:339–46. doi: 10.1111/j.1440-169X.2006.00869.x. [DOI] [PubMed] [Google Scholar]
- 16.Findlater GS, McDougall RD, Kaufman MH. Eyelid development, fusion and subsequent reopening in the mouse. J Anat. 1993;183(pt 1):121–9. [PMC free article] [PubMed] [Google Scholar]
- 17.Martin P, Parkhurst SM. Parallels between tissue repair and embryo morphogenesis. Development. 2004;131:3021–34. doi: 10.1242/dev.01253. [DOI] [PubMed] [Google Scholar]
- 18.Shirakata Y, Kimura R, Nanba D, et al. Heparin-binding EGF-like growth factor accelerates keratinocyte migration in skin wound healing. J Cell Sci. 2005;118(pt 11):2363–70. doi: 10.1242/jcs.02346. [DOI] [PubMed] [Google Scholar]
- 19.Ohuchi H. Wakayama Symposium: epithelial-mesenchymal interactions in eyelid development. Ocul Surf. 2012;10:212–6. doi: 10.1016/j.jtos.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Tao H, Shimizu M, Kusumoto R, et al. A dual role of FGF10 in proliferation and coordinated migration of epithelial leading edge cells during mouse eyelid development. Development. 2005;132:3217–30. doi: 10.1242/dev.01892. [DOI] [PubMed] [Google Scholar]
- 21.Mine N, Iwamoto R, Mekada E. HB-EGF promotes epithelial cell migration in eyelid development. Development. 2005;132:4317–26. doi: 10.1242/dev.02030. [DOI] [PubMed] [Google Scholar]
- 22.Sevel D. A reappraisal of the development of the eyelids. Eye (Lond) 1988;2(pt 2):123–9. doi: 10.1038/eye.1988.25. [DOI] [PubMed] [Google Scholar]
- 23.Ozanics V, Jakobiec FA. Prenatal development of the eye and its adnexa. In: Duane TD, Jaeger EA, editors. In: Biomedical Foundation of Ophthalmology. Philadelphia, PA: Harper and Row; 1983. pp. 73–5. [Google Scholar]
- 24.Mohamed YH, Gong H, Amemiya T. Role of apoptosis in eyelid development. Exp Eye Res. 2003;76:115–23. doi: 10.1016/s0014-4835(02)00269-5. [DOI] [PubMed] [Google Scholar]
- 25.Xia Y, Karin M. The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol. 2004;14:94–101. doi: 10.1016/j.tcb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 26.Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- 27.Jin C, Chen J, Meng Q, et al. Deciphering gene expression program of MAP3K1 in mouse eyelid morphogenesis. Dev Biol. 2013;374:96–107. doi: 10.1016/j.ydbio.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Wang W, Hayashi Y, et al. A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J. 2003;22:4443–54. doi: 10.1093/emboj/cdg440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mustardè JC. Congenital soft tissue deformities. In: Black EH, Nesi FA, Gladstone G, editors. In: Smith and Nesi’s Ophthalmic Plastic and Reconstructive Surgery. 3rd ed. New York: Springer; 2012. pp. 1085–102. [Google Scholar]
- 30.Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J Maxillofac Surg. 1976;4:69–92. doi: 10.1016/s0301-0503(76)80013-6. [DOI] [PubMed] [Google Scholar]
- 31.François J. Syndrome malformatif avec cryptophtalmie. Ophthalmologica. 1965;150:215–8. doi: 10.1159/000304848. [DOI] [PubMed] [Google Scholar]
- 32.Subramanian N, Iyer G, Srinivasan B. Cryptophthalmos: reconstructive techniques–expanded classification of congenital symblepharon variant. Ophthal Plast Reconstr Surg. 2013;29:243–8. doi: 10.1097/IOP.0b013e3182895683. [DOI] [PubMed] [Google Scholar]
- 33.Nouby G. Congenital upper eyelid coloboma and cryptophthalmos. Ophthal Plast Reconstr Surg. 2002;18:373–7. doi: 10.1097/00002341-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Jackson IT, Shaw KE, del Pinal Matorras F. A new feature of the ablepharon macrostomia syndrome: zygomatic arch absence. Br J Plast Surg. 1988;41:410–6. doi: 10.1016/0007-1226(88)90084-7. [DOI] [PubMed] [Google Scholar]
- 35.Hornblass A, Reifler DM. Ablepharon macrostomia syndrome. Am J Ophthalmol. 1985;99:552–6. doi: 10.1016/s0002-9394(14)77956-5. [DOI] [PubMed] [Google Scholar]
- 36.Egier D, Orton R, Allen L, et al. Bilateral complete isolated cryptophthalmos: a case report. Ophthalmic Genet. 2005;26:185–9. doi: 10.1080/13816810500374557. [DOI] [PubMed] [Google Scholar]
- 37.Thomas IT, Frias JL, Felix V, et al. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25:85–98. doi: 10.1002/ajmg.1320250111. [DOI] [PubMed] [Google Scholar]
- 38.Slavotinek AM, Tifft CJ. Fraser syndrome and cryptophthalmos: review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. J Med Genet. 2002;39:623–33. doi: 10.1136/jmg.39.9.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coover DH. Two cases of cryptophthalmia. Ophthalmoloscope. 1910;8:259–261. [Google Scholar]
- 40.Coover DH. Cryptophthalmia. JAMA. 1910;55:370–371. [Google Scholar]
- 41.Coover DH. Cryptophthalmia. Ophthalmoloscope. 1915;13:586. [Google Scholar]
- 42.Magruder AC. Cryptophthalmos. Am J Ophthalmol. 1921;l4:48. [Google Scholar]
- 43.Saal HM, Traboulsi EI, Gavaris P, et al. Dominant syndrome with isolated cryptophthalmos and ocular anomalies. Am J Med Genet. 1992;43:785–8. doi: 10.1002/ajmg.1320430505. [DOI] [PubMed] [Google Scholar]
- 44.Smyth I, Scambler P. The genetics of Fraser syndrome and the blebs mouse mutants. Hum Mol Genet. 2005;14:R269–74. doi: 10.1093/hmg/ddi262. [DOI] [PubMed] [Google Scholar]
- 45.Petrou P, Chiotaki R, Dalezios Y, et al. Overlapping and divergent localization of Frem1 and Fras1 and its functional implications during mouse embryonic development. Exp Cell Res. 2007;313:910–20. doi: 10.1016/j.yexcr.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Vrontou S, Petrou P, Meyer BI, et al. Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat Genet. 2003;34:209–14. doi: 10.1038/ng1168. [DOI] [PubMed] [Google Scholar]
- 47.Kiyozumi D, Osada A, Sugimoto N, et al. Identification of a novel cell-adhesive protein spatiotemporally expressed in the basement membrane of mouse developing hair follicle. Exp Cell Res. 2005;306:9–23. doi: 10.1016/j.yexcr.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 48.Smyth I, Du X, Taylor MS, et al. The extracellular matrix gene Frem1 is essential for the normal adhesion of the embryonic epidermis. Proc Natl Acad Sci U S A. 2004;101:13560–5. doi: 10.1073/pnas.0402760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mocan MC, Ozgen B, Irkec M. Bilateral orbito-palpebral cysts in a case of cryptophthalmos associated with Fraser syndrome. J AAPOS. 2008;12:210–1. doi: 10.1016/j.jaapos.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Hoefele J, Wilhelm C, Schiesser M, et al. Expanding the mutation spectrum for Fraser syndrome: identification of a novel heterozygous deletion in FRAS1. Gene. 2013;520:194–7. doi: 10.1016/j.gene.2013.02.031. [DOI] [PubMed] [Google Scholar]
- 51.Vogel MJ, van Zon P, Brueton L, et al. Mutations in GRIP1 cause Fraser syndrome. J Med Genet. 2012;49:303–6. doi: 10.1136/jmedgenet-2011-100590. [DOI] [PubMed] [Google Scholar]
- 52.Carney TJ, Feitosa NM, Sonntag C, et al. Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel Fraser syndrome disease genes. PLoS Genet. 2010;6:e1000907. doi: 10.1371/journal.pgen.1000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Marles SL, Greenberg CR, et al. Manitoba Oculotrichoanal (MOTA) syndrome: report of eight new cases. Am J Med Genet A. 2007;143:853–7. doi: 10.1002/ajmg.a.31446. [DOI] [PubMed] [Google Scholar]
- 54.Slavotinek AM, Baranzini SE, Schanze D, et al. Manitoba-oculo-tricho-anal (MOTA) syndrome is caused by mutations in FREM1. J Med Genet. 2011;48:375–82. doi: 10.1136/jmg.2011.089631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cavalcanti DP, Matejas V, Luquetti D, et al. Fraser and Ablepharon macrostomia phenotypes: concurrence in one family and association with mutated FRAS1. Am J Med Genet A. 2007;143:241–7. doi: 10.1002/ajmg.a.31426. [DOI] [PubMed] [Google Scholar]
- 56.Schanze D, Harakalova M, Stevens CA, et al. Ablepharon macrostomia syndrome: a distinct genetic entity clinically related to the group of FRAS–FREM complex disorders. Am J Med Genet A. 2013;161:3012–7. doi: 10.1002/ajmg.a.36119. [DOI] [PubMed] [Google Scholar]
- 57.Strömland K, Miller M, Sjögreen L, et al. Oculo-auriculo-vertebral spectrum: associated anomalies, functional deficits and possible developmental risk factors. Am J Med Genet A. 2007;143:1317–25. doi: 10.1002/ajmg.a.31769. [DOI] [PubMed] [Google Scholar]
- 58.Josifova DJ, Patton MA, Marks K. Oculoauriculovertebral spectrum phenotype caused by an unbalanced t(5;8)(p15.31;p23.1) rearrangement. Clin Dysmorphol. 2004;13:151–3. doi: 10.1097/01.mcd.0000126138.37196.26. [DOI] [PubMed] [Google Scholar]
- 59.Hodes ME, Gleiser S, DeRosa GP, et al. Trisomy 7 mosaicism and manifestations of Goldenhar syndrome with unilateral radial hypoplasia. J Craniofac Genet Dev Biol. 1981;1:49–55. [PubMed] [Google Scholar]
- 60.Wilson GN, Barr M., Jr Trisomy 9 mosaicism: another etiology for the manifestations of Goldenhar syndrome. J Craniofac Genet Dev Biol. 1983;3:313–6. [PubMed] [Google Scholar]
- 61.Pridjian G, Gill WL, Shapira E. Goldenhar sequence and mosaic trisomy 22. Am J Med Genet. 1995;59:411–3. doi: 10.1002/ajmg.1320590402. [DOI] [PubMed] [Google Scholar]
- 62.Ritchey ML, Norbeck J, Huang C, et al. Urologic manifestations of Goldenhar syndrome. Urology. 1994;43:88–91. doi: 10.1016/s0090-4295(94)80273-4. [DOI] [PubMed] [Google Scholar]
- 63.Lessick M, Vasa R, Israel J. Severe manifestations of oculoauriculovertebral spectrum in a cocaine exposed infant. J Med Genet. 1991;28:803–4. doi: 10.1136/jmg.28.11.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fung AT, Martin P, Petsoglou C, et al. Repair of isolated abortive cryptophthalmos with lower eyelid switch flap and amniotic membrane graft. Ophthal Plast Reconstr Surg. 2009;25:158–61. doi: 10.1097/IOP.0b013e31819aaafb. [DOI] [PubMed] [Google Scholar]
- 65.Barisic I, Odak L, Loane M, et al. Fraser syndrome: epidemiological study in a European population. Am J Med Genet A. 2013;161:1012–8. doi: 10.1002/ajmg.a.35839. [DOI] [PubMed] [Google Scholar]
- 66.Gattuso J, Patton MA, Baraitser M. The clinical spectrum of the Fraser syndrome: report of three new cases and review. J Med Genet. 1987;24:549–55. doi: 10.1136/jmg.24.9.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grover AK, Chaudhuri Z, Malik S, et al. Congenital eyelid colobomas in 51 patients. J Pediatr Ophthalmol Strabismus. 2009;46:151–9. doi: 10.3928/01913913-20090505-06. [DOI] [PubMed] [Google Scholar]
- 68.van Haelst MM, Scambler PJ, Hennekam RC Fraser Syndrome Collaboration Group. Fraser syndrome: a clinical study of 59 cases and evaluation of diagnostic criteria. Am J Med Genet A. 2007;143:3194–203. doi: 10.1002/ajmg.a.31951. [DOI] [PubMed] [Google Scholar]
- 69.Martínez-Frías ML, Bermejo E, Sánchez Otero T, et al. Sclerocornea, hypertelorism, syndactyly, and ambiguous genitalia. Am J Med Genet. 1994;49:195–7. doi: 10.1002/ajmg.1320490206. [DOI] [PubMed] [Google Scholar]
- 70.Barisic I, Odak L, Loane M, et al. Prevalence, prenatal diagnosis and clinical features of oculo-auriculo-vertebral spectrum: a registry-based study in Europe. Eur J Hum Genet. 2014;22:1026–33. doi: 10.1038/ejhg.2013.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weng CJ. Surgical reconstruction in cryptophthalmos. Br J Plast Surg. 1998;51:17–21. doi: 10.1054/bjps.1997.0167. [DOI] [PubMed] [Google Scholar]
- 72.Gupta VP, Sen DK. Unilateral cryptophthalmos. Indian J Ophthalmol. 1990;38:97–9. [PubMed] [Google Scholar]
- 73.Kanhere S, Phadke V, Mathew A, et al. Cryptophthalmos. Indian J Pediatr. 1999;66:805–8. doi: 10.1007/BF02726274. [DOI] [PubMed] [Google Scholar]
- 74.Gupta SP, Saxena RC. Cryptophthalmos. Br J Ophthalmol. 1962;46:629–32. doi: 10.1136/bjo.46.10.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Konrad H, Merriam JC, Jones IS. Rehabilitation of a child with partial unilateral cryptophthalmos and multiple congenital anomalies. Trans Am Ophthalmol Soc. 1995;93:219–40. doi: 10.1016/s0002-9394(14)70557-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blessig E. Fall einer seltenen Missbildung der Augen: symblepharon totale congenitum palp sup oc dextri, ankyloblepharon totale congenitum kryptophthalmos oc sinistri. Klin Monatsbl Augenheilkd. 1900;38:652–62. [Google Scholar]
- 77.Van der Meulen JC. Palpebral anomalies. In: Van der Meulen JC, Gruss JS, editors. In: Ocular Plastic Surgery. 1st ed. Barcelona: Mosby-Wolfe; 1996. pp. 103–16. [Google Scholar]
- 78.Collin JR. Congenital upper lid coloboma. Aust N Z J Ophthalmol. 1986;14:313–7. doi: 10.1111/j.1442-9071.1986.tb00465.x. [DOI] [PubMed] [Google Scholar]
- 79.Fraser GR. Our genetical load: a review of some aspects of genetic variation. Ann Hum Genet. 1962;25:387–415. [Google Scholar]
- 80.Roizenblatt J, Wajntal A, Diament AJ. Median cleft face syndrome or frontonasal dysplasia: a case report with associated kidney malformation. J Pediatr Ophthalmol Strabismus. 1979;16:16–20. doi: 10.3928/0191-3913-19790101-05. [DOI] [PubMed] [Google Scholar]
- 81.Saito M, Higuchi A, Kamitani K, et al. [Anesthetic management of a patient with a cryptophthalmos syndactyly syndrome and subglottic stenosis]. Masui. 1994;43:415–7. [PubMed] [Google Scholar]
- 82.Jagtap SR, Malde AD, Pantvaidya SH. Anaesthetic considerations in a patient with Fraser syndrome. Anaesthesia. 1995;50:39–41. doi: 10.1111/j.1365-2044.1995.tb04512.x. [DOI] [PubMed] [Google Scholar]
- 83.Cohen MM., Jr Malformations of the craniofacial region: evolutionary, embryonic, genetic, and clinical perspectives. Am J Med Genet. 2002;115:245–68. doi: 10.1002/ajmg.10982. [DOI] [PubMed] [Google Scholar]
- 84.Cousley RR. A comparison of two classification systems for hemifacial microsomia. Br J Oral Maxillofac Surg. 1993;31:78–82. doi: 10.1016/0266-4356(93)90165-s. [DOI] [PubMed] [Google Scholar]
- 85.Vento AR, LaBrie RA, Mulliken JB. The O.M.E.N.S. classification of hemifacial microsomia. Cleft Palate Craniofac J. 1991;28:68–76. doi: 10.1597/1545-1569_1991_028_0068_tomens_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 86.Rollnick BR, Kaye CI, Nagatoshi K, et al. Oculoauriculovertebral dysplasia and variants: phenotypic characteristics of 294 patients. Am J Med Genet. 1987;26:361–75. doi: 10.1002/ajmg.1320260215. [DOI] [PubMed] [Google Scholar]
- 87.Tasse C, Böhringer S, Fischer S, et al. Oculo-auriculo-vertebral spectrum (OAVS): clinical evaluation and severity scoring of 53 patients and proposal for a new classification. Eur J Med Genet. 2005;48:397–411. doi: 10.1016/j.ejmg.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 88.Khong JJ, Hardy TG, McNab AA. Prevalence of oculo-auriculo-vertebral spectrum in dermolipoma. Ophthalmology. 2013;120:1529–32. doi: 10.1016/j.ophtha.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 89.Mansour AM, Wang F, Henkind P, et al. Ocular findings in the facioauriculovertebral sequence (Goldenhar-Gorlin syndrome). Am J Ophthalmol. 1985;100:555–9. doi: 10.1016/0002-9394(85)90681-6. [DOI] [PubMed] [Google Scholar]
- 90.Jakobiec FA, Pineda R, Rivera R, et al. Epicorneal polypoidal lipodermoid: lack of association of central corneal lesions with Goldenhar syndrome verified with a review of the literature. Surv Ophthalmol. 2010;55:78–84. doi: 10.1016/j.survophthal.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 91.Fries PD, Katowitz JA. Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol. 1990;35:87–119. doi: 10.1016/0039-6257(90)90067-6. [DOI] [PubMed] [Google Scholar]
- 92.Baum JL, Feingold M. Ocular aspects of Goldenhar’s syndrome. Am J Ophthalmol. 1973;75:250–7. doi: 10.1016/0002-9394(73)91020-9. [DOI] [PubMed] [Google Scholar]
- 93.Brief Timeline of Historical Developments on the Eye and its Embryology. Available at http://embryology.med.unsw.edu.au/embryology/index.php?title=2012_Group_Project_1#cite_ref. Accessed December 14, 2013. [Google Scholar]
- 94.Maconnachie E. A study of digit fusion in the mouse embryo. J Embryol Exp Morphol. 1979;49:259–76. [PubMed] [Google Scholar]
- 95.Margolis S, Aleksic S, Charles N, et al. Retinal and optic nerve findings in Goldenhar-Gorlin syndrome. Ophthalmology. 1984;91:1327–33. doi: 10.1016/s0161-6420(84)34147-1. [DOI] [PubMed] [Google Scholar]
- 96.Marles SL, Greenberg CR, Persaud TV, et al. New familial syndrome of unilateral upper eyelid coloboma, aberrant anterior hairline pattern, and anal anomalies in Manitoba Indians. Am J Med Genet. 1992;42:793–9. doi: 10.1002/ajmg.1320420609. [DOI] [PubMed] [Google Scholar]
- 97.Yeung A, Amor D, Savarirayan R. Familial upper eyelid coloboma with ipsilateral anterior hairline abnormality: two new reports of MOTA syndrome. Am J Med Genet A. 2009;149:767–9. doi: 10.1002/ajmg.a.32743. [DOI] [PubMed] [Google Scholar]
- 98.McCarthy GT, West CM. Ablepheron macrostomia syndrome. Dev Med Child Neurol. 1977;19:659–63. doi: 10.1111/j.1469-8749.1977.tb07999.x. [DOI] [PubMed] [Google Scholar]
- 99.Brancati F, Mingarelli R, Sarkozy A, et al. Ablepharon-macrostomia syndrome in a 46-year-old woman. Am J Med Genet A. 2004;127:96–8. doi: 10.1002/ajmg.a.20658. [DOI] [PubMed] [Google Scholar]
- 100.Matic A, Komazec J. Amniotic band syndrome. Acta Medica Medianae. 2009;48:44–8. [Google Scholar]
- 101.Isidor B, Baujat G, Le Caignec C, et al. Congenital skin pedicles with or without amniotic band sequence: extending the human phenotype resembling mouse disorganization. Am J Med Genet A. 2009;149:1734–9. doi: 10.1002/ajmg.a.32796. [DOI] [PubMed] [Google Scholar]
- 102.Orioli IM, Ribeiro MG, Castilla EE. Clinical and epidemiological studies of amniotic deformity, adhesion, and mutilation (ADAM) sequence in a South American (ECLAMC) population. Am J Med Genet A. 2003;118:135–45. doi: 10.1002/ajmg.a.10194. [DOI] [PubMed] [Google Scholar]
- 103.Torpin R. Amniochorionic mesoblastic fibrous strings and amniotic bands: associated constricting fetal malformations or fetal death. Am J Obstet Gynecol. 1965;91:65–75. doi: 10.1016/0002-9378(65)90588-0. [DOI] [PubMed] [Google Scholar]
- 104.Robin NH, Abbadi N, McCandless SE, et al. Disorganization in mice and humans and its relation to sporadic birth defects. Am J Med Genet. 1997;73:425–36. doi: 10.1002/(sici)1096-8628(19971231)73:4<425::aid-ajmg11>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 105.Robin NH, Nadeau JH. Disorganization in mice and humans. Am J Med Genet. 2001;101:334–8. [PubMed] [Google Scholar]
- 106.Purandare SM, Ernst L, Medne L, et al. Developmental anomalies with features of disorganization (Ds) and amniotic band sequence (ABS): a report of four cases. Am J Med Genet A. 2009;149:1740–8. doi: 10.1002/ajmg.a.32716. [DOI] [PubMed] [Google Scholar]
- 107.Braude LS, Miller M, Cuttone J. Ocular abnormalities in the amniogenic band syndrome. Br J Ophthalmol. 1981;65:299–303. doi: 10.1136/bjo.65.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schramm C, Rohrbach JM, Reinert S, et al. Amniotic bands as a cause of congenital anterior staphyloma. Graefes Arch Clin Exp Ophthalmol. 2013;251:959–65. doi: 10.1007/s00417-012-2197-z. [DOI] [PubMed] [Google Scholar]
- 109.Miller MT, Deutsch TA, Cronin C, et al. Amniotic bands as a cause of ocular anomalies. Am J Ophthalmol. 1987;104:270–9. doi: 10.1016/0002-9394(87)90416-8. [DOI] [PubMed] [Google Scholar]
- 110.von Hippel E. Kiyptophthalmus congenitus. Graefes Arch Klin Exp Ophthalmol. 1906;63:25–38. [Google Scholar]
- 111.Brown KE, Goldstein SM, Douglas RS, et al. Encephalocrani ocutaneous lipomatosis: a neurocutaneous syndrome. J AAPOS. 2003;7:148–9. doi: 10.1016/mpa.2003.S1091853103000120. [DOI] [PubMed] [Google Scholar]
- 112.Hennekam RC. Scalp lipomas and cerebral malformations: overlap between encephalocraniocutaneous lipomatosis and oculocerebrocutaneous syndrome. Clin Dysmorphol. 1994;3:87–9. [PubMed] [Google Scholar]
- 113.Hunter AG. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: blind men and an elephant or separate syndromes? Am J Med Genet A. 2006;140:709–26. doi: 10.1002/ajmg.a.31149. [DOI] [PubMed] [Google Scholar]
- 114.Almer Z, Vishnevskia-Dai V, Zadok D. Encephalocraniocutaneous lipomatosis: case report and review of the literature. Cornea. 2003;22:389–90. doi: 10.1097/00003226-200305000-00023. [DOI] [PubMed] [Google Scholar]
- 115.Nowaczyk MJ, Mernagh JR, Bourgeois JM, et al. Antenatal and postnatal findings in encephalocraniocutaneous lipomatosis. Am J Med Genet. 2000;91:261–6. [PubMed] [Google Scholar]
- 116.Chacon-Camacho OF, Lopez-Martinez MS, Vázquez J, et al. Nasopalpebral lipoma-coloboma syndrome: clinical, radiological, and histopathological description of a novel sporadic case. Am J Med Genet A. 2013;161:1470–4. doi: 10.1002/ajmg.a.35916. [DOI] [PubMed] [Google Scholar]
- 117.Babu NS, Raviprakash D, Kumar R. Nasopalpebral lipoma coloboma syndrome. Indian J Ophthalmol. 2011;59:379–80. doi: 10.4103/0301-4738.83615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Akarsu AN, Sayli BS. Nasopalpebral lipoma-coloboma syndrome. Clin Genet. 1991;40:342–4. doi: 10.1111/j.1399-0004.1991.tb03106.x. [DOI] [PubMed] [Google Scholar]
- 119.Penchaszadeh VB, Velasquez D, Arrivillaga R. The nasopalpebral lipoma-coloboma syndrome: a new autosomal dominant dysplasia-malformation syndrome with congenital nasopalpebral lipomas, eyelid colobomas, telecanthus, and maxillary hypoplasia. Am J Med Genet. 1982;11:397–410. doi: 10.1002/ajmg.1320110404. [DOI] [PubMed] [Google Scholar]
- 120.Spano A, Piozzi E, Cavallini M, et al. Surgical approach in a rare case of coloboma-choristoma. Br J Plast Surg. 2005;58:732–5. doi: 10.1016/j.bjps.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 121.Bock-Kunz AL, Lyon DB, Singhal VK, et al. Nasopalpebral lipoma-coloboma syndrome. Arch Ophthalmol. 2000;118:1699–701. [PubMed] [Google Scholar]
- 122.Lee TK, Johnson RL, MacDonald IM, et al. A new case of oculoectodermal syndrome. Ophthalmic Genet. 2005;26:131–3. doi: 10.1080/13816810500228811. [DOI] [PubMed] [Google Scholar]
- 123.Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis. 2006;1:34. doi: 10.1186/1750-1172-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.The CHARGE Syndrome Foundation. About CHARGE. Available at http://www.chargesyndrome.org/about-charge.asp. Accessed March 14, 2014.
- 125.Blake KD, Russell-Eggitt IM, Morgan DW, et al. Who’s in CHARGE? Multidisciplinary management of patients with CHARGE association. Arch Dis Child. 1990;65:217–23. doi: 10.1136/adc.65.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lodhi AA, Junejo SA, Khanzada MA, et al. Surgical outcome of 21 patients with congenital upper eyelid coloboma. Int J Ophthalmol. 2010;3:69–72. doi: 10.3980/j.issn.2222-3959.2010.01.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Källén K, Robert E, Castilla EE, et al. Relation between oculo-auriculo-vertebral (OAV) dysplasia and three other non-random associations of malformations (VATER, CHARGE, and OEIS). Am J Med Genet A. 2004;127:26–34. doi: 10.1002/ajmg.a.20643. [DOI] [PubMed] [Google Scholar]
- 128.Araneta MR, Moore CA, Olney RS, et al. Goldenhar syndrome among infants born in military hospitals to Gulf War veterans. Teratology. 1997;56:244–51. doi: 10.1002/(SICI)1096-9926(199710)56:4<244::AID-TERA3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 129.Seah LL, Choo CT, Fong KS. Congenital upper lid colobomas: management and visual outcome. Ophthal Plast Reconstr Surg. 2002;18:190–5. doi: 10.1097/00002341-200205000-00007. [DOI] [PubMed] [Google Scholar]
- 130.Cannon PS, Madge SN, Kakizaki H, et al. Composite grafts in eyelid reconstruction: the complications and outcomes. Br J Ophthalmol. 2011;95:1268–71. doi: 10.1136/bjo.2009.170548. [DOI] [PubMed] [Google Scholar]
- 131.Hoyama E, Limawararut V, Malhotra R, et al. Tarsomarginal graft in upper eyelid coloboma repair. J AAPOS. 2007;11:499–501. doi: 10.1016/j.jaapos.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 132.Morley AM, deSousa JL, Selva D, et al. Techniques of upper eyelid reconstruction. Surv Ophthalmol. 2010;55:256–71. doi: 10.1016/j.survophthal.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 133.Mathijssen IM, van der Meulen JC. Guidelines for reconstruction of the eyelids and canthal regions. J Plast Reconstr Aesthet Surg. 2010;63:1420–33. doi: 10.1016/j.bjps.2009.05.035. [DOI] [PubMed] [Google Scholar]
- 134.Lam J, Dohil M. Multiple accessory tragi and hemifacial microsomia. Pediatr Dermatol. 2007;24:657–8. doi: 10.1111/j.1525-1470.2007.00560.x. [DOI] [PubMed] [Google Scholar]
- 135.Fountain TR, Goldberger S. A case of bilateral cryptophthalmia and euryblepharon with two-stage reconstruction. Ophthal Plast Reconstr Surg. 2001;17:53–5. doi: 10.1097/00002341-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 136.Naik MN, Raizada K. Eyelid switch flap technique for the management of congenital anophthalmos associated with contracted socket. Ophthal Plast Reconstr Surg. 2006;22:476–9. doi: 10.1097/01.iop.0000244971.05669.ce. [DOI] [PubMed] [Google Scholar]
- 137.Werner MS, Olson JJ, Putterman AM. Composite grafting for eyelid reconstruction. Am J Ophthalmol. 1993;116:11–6. doi: 10.1016/s0002-9394(14)71737-4. [DOI] [PubMed] [Google Scholar]
- 138.Stewart JM, David S, Seiff SR. Amniotic membrane graft in the surgical management of cryptophthalmos. Ophthal Plast Reconstr Surg. 2002;18:378–80. doi: 10.1097/00002341-200209000-00011. [DOI] [PubMed] [Google Scholar]
- 139.Subramaniam N, Udhay P, Mahesh L. Prepucial skin graft for forniceal and socket reconstruction in complete cryptophthalmos with congenital cystic eye. Ophthal Plast Reconstr Surg. 2008;24:227–9. doi: 10.1097/IOP.0b013e31816e34c8. [DOI] [PubMed] [Google Scholar]
- 140.Lee JH, Sa HS, Woo K, et al. Prepuce mucosal graft for forniceal and conjunctival sac reconstruction in surgically intractable symblepharon. Ophthal Plast Reconstr Surg. 2011;27:e103–6. doi: 10.1097/IOP.0b013e3181f29e5c. [DOI] [PubMed] [Google Scholar]
- 141.Mocan MC, Ozgen B, Irkec M. Bilateral orbito-palpebral cysts in a case of cryptophthalmos associated with Fraser syndrome. JAAPOS. 2008;12:210–1. doi: 10.1016/j.jaapos.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 142.Witmer MT, Slonim CB. Repair of bilateral upper eyelid colobomas in infants. Eyenet. 2010;1:31–3. [Google Scholar]
- 143.Lessa S, Nanci M, Sebastiá R, et al. Two-stage reconstruction for eyelid deformities in partial cryptophthalmos. Ophthal Plast Reconstr Surg. 2011;27:282–6. doi: 10.1097/IOP.0b013e318201d627. [DOI] [PubMed] [Google Scholar]
- 144.Tawfik HA, Raslan AO, Talib N. Surgical management of acquired socket contracture. Curr Opin Ophthalmol. 2009;20:406–11. doi: 10.1097/ICU.0b013e32832ed85b. [DOI] [PubMed] [Google Scholar]
- 145.Cranicare Foundation. Nasopalpebral lipoma-coloboma syndrome (NLCS) Available at http://www.cranirare.eu/phenotypes/nasopalpebral_lipoma_coloboma/index.php. Accessed May 7, 2014. [Google Scholar]