

Abstract
Dialkylglycine decarboxylase (DGD) is an unusual pyridoxal phosphate dependent enzyme that catalyzes decarboxylation in the first and transamination in the second half-reaction of its ping-pong catalytic cycle. Directed evolution was employed to alter the substrate specificity of DGD from 2-aminoisobutyrate (AIB) to 1-aminocyclohexane-1-carboxylate (AC6C). Four rounds of directed evolution led to the identification of several mutants, with clones in the final rounds containing five persistent mutations. The best clones show ~2.5-fold decrease in KM and ~2-fold increase in kcat, giving a modest ~5-fold increase in catalytic efficiency for AC6C. Additional rounds of directed evolution did not improve catalytic activity toward AC6C. Only one (S306F) of the five persistent mutations is close to the active site. S306F was observed in all 33 clones except one, and the mutation is shown to stabilize the enzyme toward denaturation. The other four persistent mutations are near the surface of the enzyme. The S306F mutation and the distal mutations all have significant effects on the kinetic parameters for AIB and AC6C. Molecular dynamics simulations suggest that the mutations alter the conformational landscape of the enzyme, favoring a more open active site conformation that facilitates the reactivity of the larger substrate. We speculate that the small increases in kcat/KM for AC6C are due to two constraints. The first is the mechanistic requirement for catalyzing oxidative decarboxylation via a concerted decarboxylation/proton transfer transition state. The second is that DGD must catalyze transamination at the same active site in the second half-reaction of the ping-pong catalytic cycle.
Keywords: Pyridoxal phosphate, Dialkylglycine decarboxylase, Directed evolution, Genetic selection, Conformational change, Stereoelectronic effect
1. Introduction
Dialkylglycine decarboxylase (DGD) is a pyridoxal phosphate (PLP) dependent enzyme, first isolated from the soil bacterium Pseudomomas cepacia [1,2]. It catalyzes two different reactions – decarboxylation-dependent transamination of 2-aminoisobutyrate (AIB; α-methylalanine) to generate the pyridoxamine phosphate enzyme, followed by transamination of pyruvate to l-alanine to regenerate the PLP enzyme – at the same active site. In the first half-reaction of the ping-pong kinetic mechanism (Scheme 1A), DGD catalyzes decarboxylation of AIB to form CO2 and acetone with the amino group of AIB transferred to the cofactor to form pyridoxamine phosphate. In the second half-reaction (Scheme 1B), the amino group is transferred to an α-keto acid (pyruvate, the preferred in vitro substrate and presumed in vivo substrate, is shown) to form an l-amino acid product and regenerate the PLP cofactor.
Scheme 1.

Half-reactions of the DGD catalyzed ping-pong mechanism.
It is fundamentally important to understand the mechanisms by which reaction and substrate specificity are controlled by PLP-dependent enzymes, given their broad role in the nitrogen metabolism of all organisms [3–9]. DGD is especially interesting in this context because of its unusual dual (decarboxylation and transamination) reaction specificity. A functional active site model for DGD was proposed by Toney et al. [10] based on the X-ray structure of DGD and previous kinetic studies [11]. In this model (Fig. 1), the active site of DGD is described by three binding subsites (A, B, and C), each of which differs in specificity and function. The A subsite is the locus of bond making and breaking for both decarboxylation and transamination. The B subsite is capable of binding both aliphatic groups (AIB in decarboxylation), or carboxylate groups (pyruvate in transamination). The C subsite is sterically restrictive, binding small alkyl groups. Catalytic efficiency decreases sharply as larger side chains occupy the C subsite [11]. This steric restriction is thought to be responsible for the poor efficiency of DGD with bulky substrates such as 1-amino-1-cyclopentanecarboxylic acid (AC5C) and 1-amino-1-cyclohexanecarboxylic acid (AC6C) (Fig. 2) [11]. The C subsite reduces the binding affinity for AC5C and AC6C and their bound forms are thought to be misaligned for optimal catalysis thereby diminishing favorable stereoelectronic effects (Fig. 1).
Fig. 1.
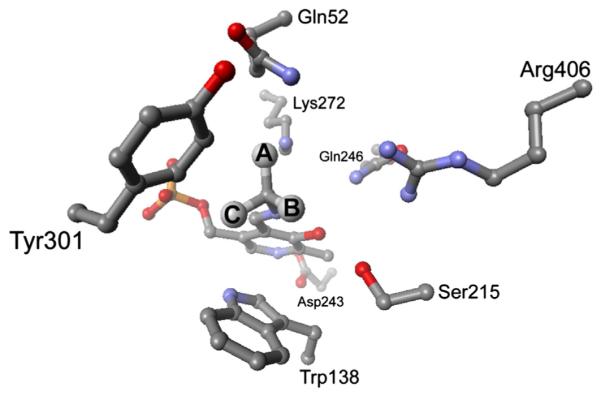
Model of the DGD active site. The three subsites discussed in the text are labeled as A, B, and C. The alignment of the Cα-CO2− bond with the p orbitals of the conjugated π system (Schiff base and pyridine ring) when it is in the A subsite stabilizes negative charge as it develops on Cα in the transition state. This is the stereoelectronic effect referred to in the text.
Fig. 2.
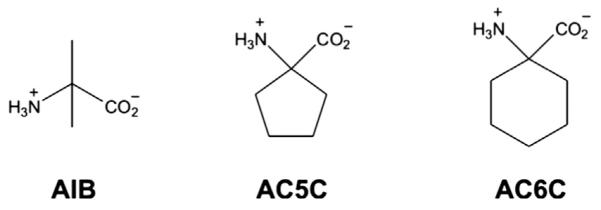
DGD substrates. AIB is the natural substrate of DGD and AC6C was the target substrate here.
The primary means by which the PLP-enzymes control reaction specificity is stereoelectronic [8,9,12]. Stereoelectronic effects are maximized when the labile bond is parallel to the p orbitals of the conjugated π system (Schiff base and pyridine ring). This, in turn, maximizes the rate of bond scission by stabilizing developing negative charge in the transition state through delocalization into the conjugated π system of PLP. Evidence for stereoelectronic effects controlling DGD catalysis includes kinetic studies with alternate substrates [11], X-ray crystallography studies with phosphonate inhibitors [13], and studies performed with DGD mutants [14,15].
Here, we report directed evolution of DGD substrate specificity to examine the ability of the catalytically bifunctional active site to adapt to a new, larger substrate in the decarboxylation half-reaction while maintaining specificity in the transamination half-reaction for pyruvate, the presumed in vivo substrate. A synthetic gene optimized for Escherichia coli expression was constructed. Error-prone PCR and gene shuffling mutagenesis techniques were combined with a genetic selection to isolate DGD mutants with altered specificity for the non-native substrate AC6C. Several mutants with increasing catalytic efficiency were purified and analyzed kinetically. The best mutants isolated show a modest ~5-fold increase in kcat/KM for AC6C compared to the WT enzyme. This modest increase is interpreted as a result of the requirement for a concerted decarboxylation/proton transfer transition state to enforce oxidative decarboxylation, and the requirement for maintaining high efficiency for the small substrate pyruvate in the transamination half-reaction.
2. Materials and methods
2.1. Materials
Oligonucleotides were obtained from Invitrogen. The Pt-Taq polymerase was from Invitrogen. A 2.5 mM dNTP PCR mix was from Applied Biosystems. PfuTurbo polymerase and the E. coli BL21(DE3)-Gold cells were from Stratagene. The pProEX-HtaB plasmid was from Life Technologies. The pBTac plasmid was from Professor John Keller (Department of Chemistry, University of Alaska) [16]. DNA purification kits were from QIAGEN. Centri-spin 20 size exclusion columns were from Princeton Separations. T4 DNA ligase and all restriction endonucleases were from New England Biolabs. AIB, sodium pyruvate and all antibiotics were from Sigma-Aldrich. AC6C and AC5C were from Acros. NADPH was from Roche. All other media components were from Fisher Scientific. All PCR was carried out using or an Applied Biosystems GeneAmp PCR System 2700 thermocycler. Bacterial transformations were carried out using a Bio-Rad Micropulser electroporation apparatus.
2.2. Gene synthesis
The sequence of a synthetic dgdA gene, designated dgdAecx (“ecx” for E. coli expression), was designed using only the preferred codon usage for strongly expressed E. coli genes [17]. The DGD amino acid sequence (taken from PDB entry 1DKA) was reverse translated into the synthetic gene using a table of codons that contained a single entry for each amino acid type. The entry for each was the most frequently employed codon in highly expressed E. coli genes. The resulting synthetic DNA sequence was analyzed for restriction enzyme sites to ensure that cut sites for BamHI and SpeI (used for subsequent cloning steps) were absent. Following the design of the dgdAecx gene, DNAWorks was used to generate the sequences of the 54 oligonucleotides used for gene assembly [18]. The terminal primers contained BamHI and SpeI for cloning into pProEX-HtaB as follows (cut sites shown in bold; dgdAecx start codon in italics): DGDecx.Fwd1, 5′-TCAGGGCGCCATGGGATCCATGTCTCTGAACGA-3′ and DGDecx.Rev1, 5′-GAAAGCGGCCGCGACTAGTTTACAGAGCACGT-3′.
The gene assembly of dgdAecx was based on previously reported gene assembly protocols [19,20]. Oligonucleotides were dissolved in deionized water to a final concentration of 10 μM. A small volume (10 μL) of each oligonucleotide stock solution was combined to form a single oligonucleotide mix. The volume of this mixture was adjusted to 666 μL to give a final concentration of 0.15 μM for each oligonucleotide. Ten microliters of the oligonucleotide mix was used as template in a 50 μL PCR with PfuTurbo polymerase and no external primers. The assembly PCR parameters were as follows: 94 °C for 2 min, 25 cycles of [94 °C for 45 s, 55 °C for 45 s, 72 °C for min], followed by 72 °C for 7 min and a hold at 4 °C. The assembled product was amplified by using 5 μL of the PCR assembly reaction as template in a 100 μL PCR reaction with PfuTurbo polymerase, including 0.3 μM of each terminal oligonucleotide as primers. The purified assembly product was digested with BamHI and SpeI and ligated into the pProEX-HtaB plasmid and transformed into BL21-Gold (DE3) cells via electroporation. Transformants were washed once with NH4Cl-free M9-glucose medium, resuspended in NH4Cl-free M9-glucose medium, and plated onto AIB selection plates (NH4Cl-free M9 with 2% w/v glucose, 100 mM AIB, 12 μg/mL Tet, and 50 μg/mL Amp). After 3–5 days of incubation at 37 °C, colonies were isolated, and plasmid DNA was purified, analyzed by restriction digestion and gel electrophoresis, and sequenced. A plasmid containing an error-free dgdAecx gene was chosen for future work and designated pEX-DGDecx.
2.3. Gene cloning and plasmid constructions
The dgdAecx gene was cloned into the pBTac plasmid to form the pBT-DGDecx construct. PCR primers were used to add the EcoRI and HindIII restriction sites, and the primer sequences are as follows: DGDecx.5′.EcoRI.pBT, 5′-GGAAACAGAATTCTATGTCTCTGAACGACGACGC-3′ and DGDecx.6′.HindIII.pBT, 5′-AAAACAGAAGCTTTTACAGAGCACGTTCGATAGCC-3′. The pBT-DGDecx construct contains no leader sequence fused to the dgdAecx gene. Expression is initiated by IPTG induction of a Tac promoter, and the primers were designed such that the resulting mRNA transcript begins with the start codon of dgdAecx.
2.4. DGD evolution
Error-prone PCR (epPCR) was performed based upon the methods described previously [21,22]. DNA shuffling was performed as described by Stemmer [23,24] and modified by Lorimer and Pastan [25]. Library creation was performed, with minor modifications, as described previously [26]. For the first round of evolution, the WT dgdAecx gene was amplified by epPCR, followed by DNA shuffling to produce further combinations of mutations. The shuffled fragments were ligated into pBTac and used to transform BL21-Gold (DE3) cells via electroporation. Following a fifteen minute recovery period in SOC medium (without shaking), the transformants were washed once with NH4Cl-free M9-glucose medium, resuspended in NH4Cl-free M9-glucose medium, and plated onto A6C6 selection plates. The plates were incubated at 37 °C in loosely covered plastic boxes containing a beaker of water to ensure plate hydration. The number of clones plated in Round I was ~3 × 105. These mutagenic procedures introduced 2–10 point mutations (an average of ~4) per round, as determined by DNA sequencing of randomly chosen clones.
The formulation of the selection plates was: NH4Cl-free M9-glucose, 20 or 1 mM AC6C, 12 μg/mL Tet, 50 μg/mL Amp, and 50 μM IPTG. After ~3 weeks of incubation, two colonies were found on the plates. Both were streaked onto LB-Amp-Tet plates. Plasmid DNA was purified from these colonies, sequenced, and used to transform fresh BL21-Gold (DE3) cells to confirm the growth phenotype.
For the second round of evolution, a 1:1 mix of the two Round I plasmids were used as template for epPCR. Round II was performed in the same manner as Round I. The number of clones plated onto AC6C selection plates was ~1.5 × 105. After ~2 weeks of incubation at 37 °C, a single colony was found on the plates. The DNA from this colony was isolated, sequenced, and used as the starting DNA for Round III. This clone contained the two Round I mutations (N96S and S306F) combined together as well as an additional mutation (L221P).
For the third round of evolution, the Round II plasmid was amplified by epPCR, and shuffled in a 1:1 ratio with WT dgdAecx amplified via epPCR. The shuffled fragments were ligated into the pBTac plasmid to produce the Round III library. All other aspects of Round III were identical to the first two rounds. The number of clones plated onto AC6C selection plates was ~1.5 × 105. After one week of incubation at 37 °C, numerous colonies were observed growing on the AC6C selection plates. The largest fifteen colonies were harvested and DNA from these colonies was isolated, sequenced, and used as the starting DNA for Round IV.
Round IV evolution was carried out with non-mutagenic (high-fidelity) conditions to avoid the incorporation of new mutations, as described previously [27]. Round III plasmids were mixed in equal ratios, and this mix was used as a template for regular PCR. The resulting fragments were shuffled in a 1:1 ratio with WT dgdAecx. All other aspects of Round IV were identical to Round III. The number of clones plated onto AC6C selection plates was approximately 1.5 × 105. After ~1 week of incubation at 37 °C, numerous colonies were observed growing on the AC6C selection plates. The largest fifteen colonies were isolated and DNA from these colonies was isolated and sequenced. Subsequent rounds of evolution did not lead to faster growing colonies even though WT produces colonies after ~2 days on AIB medium.
2.5. Mutant enzyme purification
Liquid cultures in TB medium were grown at 37 °C, with shaking, to an OD600 = 0.5. The cultures were cooled to ~33 °C and induced with a final concentration of 0.5 mM IPTG. Following induction, the cultures were incubated with shaking at 33 °C for 8–10 h. The cells were pelleted by centrifugation, resuspended in lysis buffer (20 mM TEA-HCl pH 7.8, 50 mM KCl, 20 μM PLP, 1 M ammonium sulfate) and frozen at −80 °C. The slurry was thawed on ice and sonicated in the presence of 0.5 mg/mL lysozyme. The cell debris was removed via centrifugation at 14,000 rpm (40 min; SS–34 rotor), and the supernatant was brought to 2.2 M of ammonium sulfate slowly over 30–60 min at 4 °C, followed by stirring for one hour at 4 °C. The precipitated protein was collected via centrifugation. The protein pellet was dissolved and dialyzed into loading buffer (20 mM TEA-HCl pH 7.8, 20 μM PLP). The dialyzed solution was loaded onto a 50 mL Q Sepharose Fast Flow anion exchange column (Pharmacia) and eluted with a 100–400 mM of KCL gradient in loading buffer. The purest fractions, as judged by SDS-PAGE, were pooled and dialyzed into 50 mM of TEA-succinate pH 7.8, 50 mM of dipotassium succinate, and 20 mM of PLP. Following dialysis the protein solution was flash frozen and stored at −80 °C. Protein concentration was determined with the DC (Lowry) protein assay kit from Bio-Rad with IgG as a standard.
2.6. Kinetic analysis
The decarboxylation of AIB, AC6C, and AC5C was followed by coupling the reaction to 2-OADH as described previously [11]. Briefly, the decrease in NADPH absorbance at 340 nm that occurs when acetone (from AIB) is reduced to 2-propanol, cyclohexanone (from AC6C) is reduced to cyclohexanol, or cyclopentanone (from AC5C) is reduced to cyclopentanol was monitored. Initial rates were measured over a range of substrate concentrations, and the data were fitted to the Michaelis–Menten equation directly by nonlinear regression to obtain values of kcat and KM. All experiments were performed in 100 mM of TEA-succinate pH 7.4, 100 mM of dipotassium succinate, and 50 μM of PLP. Pyruvate (5 mM) was used as the α-keto acid substrate when AIB was varied and AIB (100 mM) was used as the amino acid substrate when pyruvate was varied. Experiments were performed at 25 °C.
The value of kcat/KM for a half-reaction in a ping-pong kinetic mechanism is independent of the other half-reaction [28]. Therefore, the values of kcat/KM for the amino acid substrates reported in Table 2 are for the decarboxylation half-reaction, independent of the properties of the transamination half-reaction. This is similarly true for the transamination half-reaction and the values of kcat/KM for pyruvate.
Table 2.
Steady-state kinetic parameters for the DGD wild-type and mutant enzymes.a
| AIB |
AC6C |
AC5C |
Pyruvate |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| KM |
kcat |
kcat/KM |
KM |
kcat |
kcat/KM |
KM |
kcat |
kcat/KM |
kcat/KM |
|
| (mM) | (s−1) | (M−1 s−1) | (mM) | (s−1) | (M−1 s−1) | (mM) | (s−1) | (M−1 s−1) | (M−1 s−1) | |
| WT | 2.5 (0.3) | 10.2 (0.3) | 4100 | 25.3 (1.8) | 0.78 (0.02) | 31 | 4.7 (1.2) | 0.56 (0.04) | 120 | 51,000 |
| RIIc1 | 1.9 (0.3) | 1.00 (0.03) | 530 | 7.3 (0.9) | 0.36 (0.02) | 49 | 2.1 (0.4) | 0.24 (0.01) | 110 | 23,000 |
| RIIIc1 | 2.4 (0.2) | 2.59 (0.05) | 1100 | 4.7 (0.3) | 0.51 (0.01) | 110 | 2.3 (0.4) | 0.4 (0.02) | 170 | 31,000 |
| RIIIc3 | 2.7 (0.3) | 2.6 (0.07) | 1000 | 8.9 (1.0) | 0.66 (0.03) | 74 | 35,000 | |||
| RIIIc6 | 2.1 (0.4) | 0.67 (0.03) | 320 | 6.6 (0.6) | 0.25 (0.01) | 38 | 13,000 | |||
| RIIIc12 | 1.9 (0.2) | 2.3 (0.05) | 1200 | 8.2 (1.2) | 0.88 (0.05) | 110 | 41,000 | |||
| RIVc15 | 2.8 (0.3) | 3.6 (0.12) | 1300 | 10.5 (0.9) | 1.49 (0.05) | 140 | 1.2 (0.1) | 0.46 (0.01) | 380 | 23,000 |
Standard errors are given in parentheses. Errors on kcat/KM for pyruvate are ~15%.
Heat inactivation studies were performed by incubating enzyme stock solution at 55 °C in a thermocycler, removing 10 μL of aliquots over time for use in initial rate assays (700 μL total volume) at 25 °C. The incubation and reaction conditions were as described above for initial rate assays.
2.7. Molecular dynamics simulations
Molecular dynamics simulations were carried out with YASARA version 13.9.8 [29–31]. The PDB entry 1D7V was used as the starting structure [32]. The five mutations found in RIVc15 were introduced into both subunits of the dimer. In one active site, the external aldimine intermediate formed between PLP and AIB was built using the existing PPL–AIB inhibitor as a guide for placement. In the active site of the other subunit, the external aldimine intermediate that formed between PLP and AC6C was built using the existing PPL–AIB inhibitor as a guide. A box 100 × 75 × 75 Å was used as the periodic simulation cell. The AMBER03 forcefield was used and the YASARA macro “md_run” was employed to set up calculations by filling the cell with water, calculating pKa values and protonation states at pH 7.4, adding ions, etc. The simulations were allowed to equilibrate for 500 ps at 50 °C and then slow cooled over the following 500 ps. Simulations were repeated five times for the RIVc15 and WT that had been prepared identically [32].
3. Results and discussion
The genetic selection for improved activity toward AC6C is based on the inability of E. coli cells to grow on 2,2-dialkylamino acids as the sole nitrogen source. The dgdA gene was originally isolated from P. cepacia using such a selection [1,2]. We reasoned that a similar selection could be utilized to evolve DGD to use other non-native substrates, thereby allowing E. coli to grow on novel nitrogen sources. Previous work with DGD identified substrates with which the enzyme exhibited lower activity due the presence of bulky side chains [11]. Among these were AC5C and AC6C. AC5C was found to have an inhibitory effect on E. coli growth on M9-glucose minimal media plates. Additionally, DGD has only 10-fold lower efficiency with AC5C compared to AIB, so AC5C was ruled out as a target substrate for directed evolution. AC6C, on the other hand, is 200-fold less efficient than AIB as a substrate for DGD [11]. AC6C is also well tolerated by E. coli in plates, and is taken up by E. coli as shown by growth inhibition on AIB + AC6C M9-glucose minimal plates. AC6C was therefore chosen as the target substrate for DGD evolution. The complementation-based selection using a randomly mutated DGD gene library was proved effective for improving the activity of DGD toward this non-native amino acid.
A blend of epPCR and DNA shuffling was employed in Rounds I and II to increase combinations of mutations. Round III differed from the first two rounds in that additional epPCR-mutagenized WT dgdAecx was introduced in the shuffling reaction along with epPCR mutagenized positive clones from the previous round. Since Round II only resulted in a single clone, the extra mutagenized dgdAecx was included to provide both extra mutations and wild-type regions of the gene to allow the shuffling of the three mutations within the clone. Round IV evolution was consisted of shuffling of the Round III clones with WT dgdAecx in the shuffling reaction under non-mutagenic conditions. The latter procedure was used to find the optimal combination of mutations that had been incorporated in the first three rounds. The WT dgdAecx was introduced in the shuffling reaction to facilitate concurrent backcrossing to remove non-essential mutations. For each round of evolution, the incubation time allowed before colonies were picked and analyzed was decreased to increase the stringency of the selection. Additional rounds of evolution based on epPCR of RIVc15 did not produce clones with increased fitness as measured by faster growth times on 20 mM of AC6C or growth on 1 mM of AC6C.
The DNA sequences of all 33 clones isolated across four rounds of directed evolution were analyzed. The amino acid mutations in the isolates are presented in Table 1 along with the corresponding WT amino acid residues. All 33 clones have between one and five mutations representing 13 amino acid positions. The different combinations of these thirteen mutations give rise to 20 different types of mutant proteins, five of which are observed more than once. Of the individual mutations observed, six occur with high frequency throughout the four rounds of evolution. The S306F mutation is the most common change observed, present in 32 of the 33 clones, and is present as the sole mutation in seven clones. The N96S mutation is present in 16 clones, and is distributed more or less evenly throughout all four rounds of directed evolution. The N203S, R85H, and N12D mutations, present in 15, 10, and eight clones respectively, are concentrated in the later rounds of directed evolution. Finally, the L221P mutation is present in 10 clones; it is, however, concentrated in the first three rounds of evolution, and almost completely lost during Round IV.
Table 1.
Amino acid substitutions in the mutant DGD enzymes.
| Residue | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| 12 | 23 | 28 | 60 | 69 | 85 | 96 | 203 | 221 | 306 | 329 | 387 | 402 | |
| DGD WT | N | T | I | C | G | R | N | N | L | S | R | M | G |
| RIc1a,b | F | ||||||||||||
| RIc2 | S | ||||||||||||
| RIIc1 | S | P | F | ||||||||||
| RIIIc1 | F | ||||||||||||
| RIIIc2 | S | S | F | ||||||||||
| RIIIc3 | P | F | |||||||||||
| RIIIc4 | S | P | F | R | S | ||||||||
| RIIIc5 | I | P | F | ||||||||||
| RIIIc6 | D | P | F | ||||||||||
| RIIIc7 | F | ||||||||||||
| RIIIc8 | H | S | P | F | |||||||||
| RIIIc9 | S | S | F | ||||||||||
| RIIIc10 | S | P | F | H | |||||||||
| RIIIc11 | P | F | |||||||||||
| RIIIc12 | S | S | F | ||||||||||
| RIIIc13 | D | S | S | P | F | ||||||||
| RIIIc14 | H | S | S | F | |||||||||
| RIIIc15 | V | S | S | F | |||||||||
| RIVc1 | S | F | |||||||||||
| RIVc2 | F | ||||||||||||
| RIVc3 | H | S | S | P | F | ||||||||
| RIVc4 | F | ||||||||||||
| RIVc5 | H | S | F | ||||||||||
| RIVc6 | D | H | F | ||||||||||
| RIVc7 | I | S | F | ||||||||||
| RIVc8 | F | ||||||||||||
| RIVc9 c | D | H | S | S | F | ||||||||
| RIVc10 | D | H | S | S | F | ||||||||
| RIVc11 | F | ||||||||||||
| RIVc12 | D | H | S | S | F | ||||||||
| RIVc13 | S | F | |||||||||||
| RIVc14 | D | H | S | S | F | ||||||||
| RIVc15 | D | H | S | S | F | ||||||||
| FoldX ΔΔG d | |||||||||||||
| X-ray Cryst Structg | 1.2 | 1.2 | 0.52 | 0.40 | 0.03 | 0.63 | −0.56 | 0.70 | 3.53 | −4.7 | 0.52 | −0.07 | 1.8 |
| MD_Sim_An WTf | 1.2 | 0.38 | 0.90 | 0.73 | −1.0 | 0.26 | 0.23 | 0.78 | 1.21 | −0.70 | 1.5 | 1.9 | 4.5 |
| MD_Sim_An RIVc15e | −0.07 | −0.86 | 0.62 | −1.3 | 2.5 | ||||||||
| rvET score (scale = 1–88)h | |||||||||||||
| 52 | 51 | 38 | 39 | 65 | 42 | 61 | 47 | 21 | 11 | 28 | 48 | 31 | |
The mutant names represent the evolution round and clone from which the mutants were isolated. For example, RIc1 refers to the first clone isolated in evolution round I.
The single S306F mutant occurs seven times and is italicized and underlined.
The 12D/R85H/N96S/N203S/S306F mutant occurs five times and is shown in bold.
ΔΔG(Mut-WT) in kcal/mol calculated by FoldX.
Values calculated for mutation reversion with the simulated annealing model of the RIVc15 mutant as the template.
Values calculated with theWT simulated annealing model as the template.
Values calculated with theWT crystal structure as the template.
Real value evolutionary trace score calculated as described in Ref. [36]
WT and six mutants that represent well the diversity of mutations present were purified and their activities were measured (Table 2). The kinetic parameters were measured here for WT agree with those reported previously [33]. All mutants exhibit a decrease in efficiency (kcat/KM) for AIB of approximately 3–13-fold. The decrease is primarily due to lower kcat with AIB, rather than higher KM. This is notable for clones RIIc1 and RIIIc6, both of which have >10-fold decrease in kcat compared to WT. The remaining four mutants show 3–4-fold decreases in kcat. Previous substrate evolution work also reported losses in activity or binding for the native substrate [34,35].
There is a 2–5-fold increase in kcat/KM with AC6C for most mutants. The RIIIc1 and RIIIc3 mutants both have lower kcat but also have decreased KM values, with a net increase in kcat/KM. For example, the RIIIc1 mutant has ~2-fold decrease in kcat but ~6-fold decrease in KM, giving a 3-fold increase in kcat/KM for RIIIc1 toward AC6C. Two mutants (RIIIc12 and RIVc15) exhibit both an increase in kcat and a decrease in KM, leading to ~3-fold increase in kcat/KM for RIIIc12 and ~5-fold increase for RIVc15. The RIVc15 mutant also showed a 3-fold increase in kcat/KM toward AC5C compared to WT DGD.
From the engineering point of view, one of the most interesting and most frustrating results is that DGD activity toward AC6C could only be increased ~5-fold after multiple rounds of directed evolution; the best mutants give colonies in 5 days on AC6C even though WT gives colonies in 2 days when grown on AIB. Nevertheless, this was detectable in the selection as a significant growth advantage, with the mutants allowing colony formation significantly faster than WT on this substrate. Importantly, the mutants isolated were all re-transformed into fresh E. coli cells and their growth phenotypes were confirmed, allowing us to exclude the possibility that mutations in genes other than DGD provided the observed selective advantage. No mutations in the 5′ control elements of the expression vector were observed. Additionally, the persistence of five DGD mutations through the mutagenesis and selection rounds additionally indicates that the growth advantages observed are due to alterations in the protein sequence of DGD. It is possible that a further increase in activity could be found with much larger libraries that contain a greater number of mutations and/or different combinations of mutations, but the enormous size of simple epPCR-based libraries impedes fully exploring them in transformation-based selections. For example, a library containing one mutation at each of the 433 residues in DGD, with all combinations of five mutations per protein present in the library, has ~1011 members, while that with four mutations per protein has ~109 members.
S306F is present in 32 of the 33 isolates, and is the only mutation in seven of these. Thus, S306F provides a significant selective advantage for E. coli survival on AC6C as a sole nitrogen source. The kinetic parameters of the S306F single mutant were measured (RIIIc1; Tables 1 and 2). S306F has a 6-fold decrease in KM, which for WT is known to be the true substrate dissociation constant and therefore reflects binding affinity [16]. These observations suggest that S306F is central to the evolution of DGD specificity for AC6C. Table 1 also presents the results of an evolutionary trace analysis of DGD [36]. Ser306 is relatively highly conserved in DGD, with an rvET score of 11 (on a scale of 1–88; lower is more important) and a ranking at the 10th percentile of evolutionary importance. It is the most conserved of all the mutations found, and has the largest effect on catalytic properties and enzyme stability (see below).
The location of the S306F mutation within DGD is intriguing. The structure of DGD is presented in Fig. 3 with positions labeled where mutations were observed. The active site of DGD, showing the detailed location of S306 relative to the active site, is shown in Fig. 4A. Ser306 is part of the monovalent cation binding site near the DGD active site. It does not directly coordinate to either K+ or Na+ but its two neighboring residues, Val305 and Asp307, do. Previously, we showed that smaller monovalent cations such as Li+ and Na+ give a less active enzyme that has an altered C subsite structure, as shown in Fig. 4B [10, 33]. We speculate that the S306F mutation, which exchanges a small for a large side chain at the monovalent cation binding site, also causes a substantial change in the positioning of the β strand containing the C subsite residues L299-T303, allowing AC6C to be better accommodated in the active site.
Fig. 3.

Stereoview of WT labeled at positions where mutations were observed. The residues for the five persistent mutations are shown in ball and stick and labeled in red. Other mutations are shown in stick and labeled in black. PLP is shown as a space-filling structure.
Fig. 4.

(A) Location of the S306F mutation. PDB entry 1D7R is shown with 1-aminocyclopropane carboxylate bound. Different subunits have carbons colored differently. S306, the bound K+ ion, and the 1-aminocyclopropane carboxylate adduct with PLP are shown in space-filling representation. S306F is located at the potassium ion binding site, above the C subsite. (B) Overlay of the 1-aminocyclopropane carboxylate structure (gray) which has K+ bound, and the Na+ bound form of DGD. Note the large differences in structure found at the C subsite, especially at L299 and Y301.
N12D, R85H, N96S, and N203S are surface mutations observed with S306F in the most common mutant isolated in Round IV. All of these residues have relatively low evolutionary importance as determined by the rvET scores (Table 1). Three of these, N12D, R85H, and N96S, are positioned above the S306F mutation (Fig. 3). R85H and N96S are positioned at either end of an α-helix above S306F. N12D is located in the N-terminal α-helix and is within interaction distance of residue 85. The remaining surface mutation, N203S, is located near the dimer–dimer interface, distant from both PLP and the other mutations.
The decrease in kcat due to the S306F mutation (Table 2) is countered by the addition of the surface mutations. When the S306F mutation is accompanied by two of these (N96S and N203S; RIIIc12) kcat doubles relative to the S306F single mutant, and surpasses that of WT. This increase in kcat is accompanied by a 2-fold increase in KM, which results in an unchanged kcat/KM for N96S/N203S/S306F compared to the S306F single mutant. The addition of the other two surface mutations (RIVc15) continues this trend. The N12D/R85H/N96S/N203S/S306F mutant exhibits an even greater increase in kcat; it is 4-fold greater than the S306F mutant and 2-fold greater than WT.
The hypothesis that the mutations alter the conformational landscape of DGD was addressed using molecular dynamics simulations. Both WT and the RIVc15 mutant were simulated with the AIB external aldimine intermediate in one active site and that of AC6C in the other; there is no evidence of interaction between active sites in DGD. Fig. 5 presents the results of these calculations. The structures were simulated at 50 °C for 500 ps and then slow cooled over the following 500 ps. In all repetitions, the RIVc15 mutant led to an open conformation in which the small N-terminal domain is rotated ~5° away from the active site compared to WT. This movement opens the active site for the larger AC6C substrate and allows the external aldimine intermediate to assume a position more conducive to catalysis, by analogy to its similarity to AIB bound to WT. As observed previously in crystallographic studies, the carboxylate of both AIB and AC6C prefers to interact with R406 at the B subsite as seen in Fig. 5B [32]. We hypothesize that this preference for a more open conformation is the origin of the observed increase in AC6C binding affinity by the directed evolution mutants. For catalysis to occur, the carboxylate must be placed in the A subsite, which requires rotation about the Cα-N bond of the external aldimine intermediate. This is discussed further in the context of stereoelectronic effects below. (See Fig. 6.)
Fig. 5.

Overlay of the structures of WT and the RIVc15 mutant resulting from simulated annealing MD. Both structures contained the external aldimine intermediate of AIB in one active site and that for AC6C in the other. (A) Cartoon view of the structures highlighting the difference in the degree of small domain closure, which is observed in both subunits. The WT structure is shown in mauve with the small domain highlighted in red. The RIVc15 mutant is shown in gray with the small domain highlighted in blue. (B) Active site view of the AC6C external aldimine. The external aldimine positioning in the RIVc15 active site is similar to that of AIB in the WT active site.
Fig. 6.
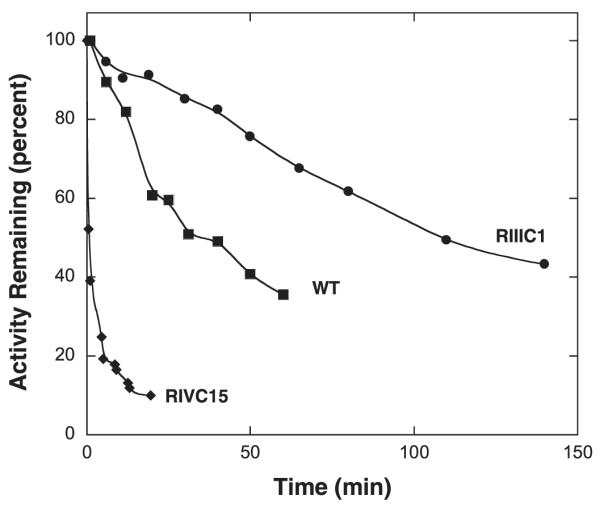
The kinetics of activity loss for enzymes incubated at 55 °C. Data for WT, RIIIc1, and RIVc15 are shown. Enzyme stock solutions were held at 55 °C and 10 μL aliquots were removed and assayed for activity at 25 °C after various times of incubation.
The effects of the mutations on the stability of DGD were estimated computationally. A large number of methods are available and their performances have been reviewed recently [37–39]. The available methods generally have mediocre performance when based on a single structure, but performance improves substantially when ensembles of structures are used instead to model the dynamic context of mutations [40,41]. The software to model ensembles of structures was not available to us. Therefore, we modeled the context dependence of calculated free energy changes using three structures: the X-ray crystal structure of DGD (PDB ID 1DKA), the final simulated annealing structure of WT with AIB and AC6C external aldimines, and the final simulated annealing structure of RIVc15 with AIB and AC6C external aldimines. The FoldX plugin for YASARA was employed with FoldX version 3.0 beta 6, since this program generally performs as well or better than the others [42]. The results are reported in Table 1.
The error in ΔΔG calculations by FoldX has been estimated to be ~0.8 kcal/mol [39,43]. Therefore, values greater than this in magnitude are highlighted as a guide in Table 1. Of the five mutations that persist in round four of the selection, two have statistically significant predicted effects on the stability of WT DGD: N12D based on both the X-ray structure and MD model, and S306F based on the X-ray structure. N12D is predicted to be destabilizing for WT. Surprisingly given the large structural change in the side chain, S306F is predicted to be stabilizing, which was confirmed experimentally (see below).
Reversions of the mutations were analyzed based on the RIVc15 simulated annealing MD structure. In principle, calculations on forward mutations with the WT MD model show how a single mutation will alter the stability of the WT structure, while calculations on reverse mutations with the RIVc15 MD model show how removal of a single mutation alters the stability of the mutant structure. There is a contextual difference, so the effect of a mutation does not need to be identical in the two calculations. The S203N reversion is predicted to stabilize the RIVc15 model while the F306S reversion is predicted to destabilize it. The most significant prediction from these calculations is that changing the smaller serine at position 306 into a larger phenylalanine generally stabilizes the enzyme and selectively stabilizes the more open structure of the RIVc15 simulated annealing model. Offsetting the effects of other destabilizing mutations may be a factor in the persistence of S306F in all four rounds of directed evolution.
The sums of the effects of the five persistent mutations, either calculated individually then added up or combined together as a single penta-mutant protein, were also calculated. The effect of the mutations on DGD stability in the context of the WT MD model calculated individually then summed up is + 1.8 kcal/mol (i.e., destabilization of the WT conformation; + 1.2 kcal/mol if only the statistically significant values are included). Comparatively, the effect of the five reversion mutations in the context of the RIVc15 MD model calculated individually then summed up is +0.9 kcal/mol (i.e., destabilization of the RIVc15 conformation; also +1.2 kcal/mol if only the statistically significant values are included). Simultaneous incorporation of the five persistent mutations into these MD models gives FoldX calculated stability changes of +0.9 kcal/mol for the forward mutations (i.e. destabilization of the more closed WT MD model) and +0.8 kcal/mol for the reversions (i.e. destabilization of the more open RIVCc15 conformation). The overall predicted effect of the five persistent mutations is selective stabilization of the more open RIVc15 conformation over the more closed WT conformation by ~1.5 kcal/mol.
To provide experimental support for these calculations, the kinetics of activity loss of three enzymes (WT, RIIIc1 which only has the S306F mutation, and RIVc15 which has the five persistent mutations) was measured. The half-life of RIIIc1 at 55 °C is ~4-fold longer than WT at this temperature, while that for RIVc15 is ~30-fold shorter than WT. Thus, the S306F mutation stabilizes the enzyme as predicted by FoldX, while the other four persistent mutations destabilize it. The positive effect the S306F mutation on both AC6C activity and stability likely accounts for its nearly uniform presence in the clones isolated.
The observed changes in kcat can be rationalized within the model of stereoelectronic effects for PLP enzymes proposed by Dunathan [12] and the DGD active site model proposed by Toney et al. (Fig. 1) [10]. Catalytically optimal positioning of the carboxylate bond is parallel to the π-bond system, which corresponds to the carboxylate in the A subsite. Steric clashes at the C subsite affect both the affinity of DGD for AC6C and the positioning of the Cα-carboxylate bond relative to the conjugated π-system of the external aldimine intermediate. We speculate that the S306F is largely responsible for the increase in affinity of DGD for AC6C (since KM decreases ~5-fold and substrate binding to DGD has been shown to be a rapid equilibrium process [16]) by altering the energetics of the domain movement discussed above, concomitantly leading to a decrease in kcat by altering the equilibrium between the carboxylate being positioned at the A vs. the B subsite. The addition of the surface mutations must modify, statically or dynamically, this equilibrium to allow the Cα–CO2− bond to spend a greater fraction of time in the A subsite thereby increasing kcat through stereoelectronic optimization.
It is somewhat surprising that multiple rounds of directed evolution beyond those reported here (even under more stringent conditions) were unable to produce colonies that grew faster. We speculate that the constraints imposed by the requirement that DGD maintain specificity for oxidative decarboxylation with concerted CO2 loss and proton transfer to C4’ of PLP, instead of the usual Cα protonation catalyzed by most PLP dependent decarboxylases. We have shown that in DGD the loss of CO2 from Cα and proton transfer from Lys272 to C4’ occurs in a concerted process that is sensitive to alteration in substrate structure [16]. It might be that a limited set of mutations in DGD is compatible with enforcing this concerted decarboxylation/proton transfer transition state with AC6C, which produces the PMP enzyme form that donates the nitrogen to pyruvate to form l-Ala. There is also the constraint imposed by the second, transamination half-reaction; adaptation to a larger decarboxylation substrate while maintaining activity with pyruvate (the presumed in vivo substrate) may limit the extent of possible adaptation. The kcat/KM values for the mutants in Table 2 are all significantly larger than those for the AC6C, suggesting that this constraint is not limiting in the present experiments. Another possibility is that a different amino acid in vivo competes with AC6C and that the mutations observed here are a compromise between minimizing this competition and maximizing activity with AC6C, thereby maximizing the flux of amino groups from AC6C to l-Ala under the growth conditions. Given the large number amines and amino acids in E. coli as potential competitors, this possibility should not be excluded.
The results presented here reaffirm the power of directed evolution in finding less-than-obvious mutations that increase enzyme fitness, and the difficulty of rational protein engineering when mutations distal to the active site are important. In the case of DGD, it would be impossible to predict rationally the mutations found here given that the obvious substrate-contacting residues (e.g., Trp138, Met141, Tyr301) are not mutated in the isolated clones. Other examples of distal mutations significantly affecting catalytic activity are found in the literature. Schramm and coworkers have shown that distal mutations in purine nucleoside phosphorylase can have substantial effects on catalytic rates and transition state structures [44–46]. In work from our laboratory, the Janus algorithm was used to predict mutations in aspartate aminotransferase that convert it into a tyrosine aminotransferase, and many of these were removed from the active site [47]. During the evolution of aspartate aminotransferase substrate specificity to that of valine aminotransferase, Oue et al. found that multiple mutations distant from the active site (>10 Å) caused significant change in the active site and substrate specificity [48]. Sacchi et al. found that a cluster of mutations on the surface of D-amino acid oxidase were critical for evolution of substrate specificity [49]. Similar results have also been found by Iffland et al. with cytochrome c [50], Spiller et al. with p-nitrobenzyl esterase [51]. These very interesting results point to a general and important role for amino acids far removed from enzyme active site in catalytic activity that remains an important, and poorly understood and unexplored area of enzymology.
Footnotes
This work was supported by grant GM54779 from the National Institutes of Health.
References
- [1].Bailey GB, Dempsey WB. Purification and properties of an α-dialkyl amino acid transaminase. Biochemistry. 1967;6:1526–1533. [Google Scholar]
- [2].Aaslestad HG, Larson AD. Bacterial metabolism of 2-methylalanine. J. Bacteriol. 1964;88:1296–1303. doi: 10.1128/jb.88.5.1296-1303.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hayashi H, Wada H, Yoshimura T, Esaki N, Soda K. Recent topics in pyridoxal 5′-phosphate enzyme studies. Annu. Rev. Biochem. 1990;59:87–110. doi: 10.1146/annurev.bi.59.070190.000511. [DOI] [PubMed] [Google Scholar]
- [4].John RA. Pyridoxal phosphate-dependent enzymes. Biochim. Biophys. Acta. 1995;1248:81–96. doi: 10.1016/0167-4838(95)00025-p. [DOI] [PubMed] [Google Scholar]
- [5].Mehta PK, Christen P. The molecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 2000;74:129–184. doi: 10.1002/9780470123201.ch4. [DOI] [PubMed] [Google Scholar]
- [6].Percudani R, Peracchi A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003;4:850–854. doi: 10.1038/sj.embor.embor914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- [8].Toney MD. Reaction specificity in pyridoxal phosphate enzymes. Arch. Biochem. Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- [9].Toney MD. Controlling reaction specificity in pyridoxal phosphate enzymes. Biochim. Biophys. Acta. 2011;1814:1407–1418. doi: 10.1016/j.bbapap.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Toney MD, Hohenester E, Keller JW, Jansonius JN. Structural and mechanistic analysis of two refined crystal structures of the pyridoxal phosphate-dependent enzyme dialkylglycine decarboxylase. J. Mol. Biol. 1995;245:151–179. doi: 10.1006/jmbi.1994.0014. [DOI] [PubMed] [Google Scholar]
- [11].Sun S, Zabinski RF, Toney MD. Reactions of alternate substrates demonstrate stereoelectronic control of reactivity in dialkylglycine decarboxylase. Biochemistry. 1998;37:3865–3875. doi: 10.1021/bi972055s. [DOI] [PubMed] [Google Scholar]
- [12].Dunathan HC. Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc. Natl. Acad. Sci. U. S. A. 1966;55:712–716. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu W, Rogers CJ, Fisher AJ, Toney MD. Aminophosphonate inhibitors of dialkylglycine decarboxylase: structural basis for slow binding inhibition. Biochemistry. 2002;41:12320–12328. doi: 10.1021/bi026318g. [DOI] [PubMed] [Google Scholar]
- [14].Fogle EJ, Liu W, Woon ST, Keller JW, Toney MD. Role of q52 in catalysis of decarboxylation and transamination in dialkylglycine decarboxylase. Biochemistry. 2005;44:16392–16404. doi: 10.1021/bi051475b. [DOI] [PubMed] [Google Scholar]
- [15].Fogle EJ, Toney MD. Mutational analysis of substrate interactions with the active site of dialkylglycine decarboxylase. Biochemistry-Us. 2010;49:6485–6493. doi: 10.1021/bi100648w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou X, Jin X, Medhekar R, Chen X, Dieckmann T, Toney MD. Rapid kinetic and isotopic studies on dialkylglycine decarboxylase. Biochemistry. 2001;40:1367–1377. doi: 10.1021/bi001237a. [DOI] [PubMed] [Google Scholar]
- [17].Grosjean H, Fiers W. Preferential codon usage in prokaryotic genes: the optimal codon–anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene. 1982;18:199–209. doi: 10.1016/0378-1119(82)90157-3. [DOI] [PubMed] [Google Scholar]
- [18].Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- [20].Withers-Martinez C, Carpenter EP, Hackett F, Ely B, Sajid M, Grainger M, Blackman MJ. PCR-based gene synthesis as an efficient approach for expression of the A + T-rich malaria genome. Protein Eng. 1999;12:1113–1120. doi: 10.1093/protein/12.12.1113. [DOI] [PubMed] [Google Scholar]
- [21].Cadwell RC, Joyce GF. Mutagenic PCR. PCR Methods Appl. 1994;3:S136–S140. doi: 10.1101/gr.3.6.s136. [DOI] [PubMed] [Google Scholar]
- [22].Cirino PC, Mayer KM, Umeno D. Generating mutant libraries using error-prone PCR. Methods Mol. Biol. 2003;231:3–9. doi: 10.1385/1-59259-395-X:3. [DOI] [PubMed] [Google Scholar]
- [23].Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- [24].Stemmer WP. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. U. S. A. 1994;91:10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lorimer IA, Pastan I. Random recombination of antibody single chain Fv sequences after fragmentation with DNaseI in the presence of Mn2+ Nucleic Acids Res. 1995;23:3067–3068. doi: 10.1093/nar/23.15.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tobias AV. Preparing libraries in Escherichia coli. Methods Mol. Biol. 2003;231:11–16. doi: 10.1385/1-59259-395-X:11. [DOI] [PubMed] [Google Scholar]
- [27].Zhao H, Arnold FH. Optimization of DNA shuffling for high fidelity recombination. Nucleic Acids Res. 1997;25:1307–1308. doi: 10.1093/nar/25.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Velick SF, Vavra J. A kinetic and equilibrium analysis of the glutamic oxaloacetate transaminase mechanism. J. Biol. Chem. 1962;237:2109–2122. [PubMed] [Google Scholar]
- [29].Krieger E, Vriend G. YASARA View – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Krieger E, Dunbrack RL, Jr., Hooft RW, Krieger B. Assignment of protonation states in proteins and ligands: combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012;819:405–421. doi: 10.1007/978-1-61779-465-0_25. [DOI] [PubMed] [Google Scholar]
- [31].Krieger E, Joo K, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(Suppl. 9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Malashkevich VN, Strop P, Keller JW, Jansonius JN, Toney MD. Crystal structures of dialkylglycine decarboxylase inhibitor complexes. J. Mol. Biol. 1999;294:193–200. doi: 10.1006/jmbi.1999.3254. [DOI] [PubMed] [Google Scholar]
- [33].Liu W, Toney MD. Kinetic and thermodynamic analysis of the interaction of cations with dialkylglycine decarboxylase. Biochemistry. 2004;43:4998–5010. doi: 10.1021/bi035854l. [DOI] [PubMed] [Google Scholar]
- [34].Yano T, Oue S, Kagamiyama H. Directed evolution of an aspartate aminotransferase with new substrate specificities. Proc. Natl. Acad. Sci. U. S. A. 1998;95:5511–5515. doi: 10.1073/pnas.95.10.5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rothman SC, Kirsch JF. How does an enzyme evolved in vitro compare to naturally occurring homologs possessing the targeted function? Tyrosine aminotransferase from aspartate aminotransferase. J. Mol. Biol. 2003;327:593–608. doi: 10.1016/s0022-2836(03)00095-0. [DOI] [PubMed] [Google Scholar]
- [36].Morgan DH, Kristensen DM, Mittelman D, Lichtarge O. ET viewer: an application for predicting and visualizing functional sites in protein structures. Bioinformatics. 2006;22:2049–2050. doi: 10.1093/bioinformatics/btl285. [DOI] [PubMed] [Google Scholar]
- [37].Khan S, Vihinen M. Performance of protein stability predictors. Hum. Mutat. 2010;31:675–684. doi: 10.1002/humu.21242. [DOI] [PubMed] [Google Scholar]
- [38].Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng. Des. Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- [39].Thiltgen G, Goldstein RA. Assessing predictors of changes in protein stability upon mutation using self-consistency. PLoS One. 2012;7:e46084. doi: 10.1371/journal.pone.0046084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Christensen NJ, Kepp KP. Accurate stabilities of laccase mutants predicted with a modified FoldX protocol. J. Chem. Inf. Model. 2012;52:3028–3042. doi: 10.1021/ci300398z. [DOI] [PubMed] [Google Scholar]
- [41].Benedix A, Becker CM, de Groot BL, Caflisch A, Bockmann RA. Predicting free energy changes using structural ensembles. Nat. Methods. 2009;6:3–4. doi: 10.1038/nmeth0109-3. [DOI] [PubMed] [Google Scholar]
- [42].Van Durme J, Delgado J, Stricher F, Serrano L, Schymkowitz J, Rousseau F. A graphical interface for the FoldX forcefield. Bioinformatics. 2011;27:1711–1712. doi: 10.1093/bioinformatics/btr254. [DOI] [PubMed] [Google Scholar]
- [43].Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- [44].Luo M, Li L, Schramm VL. Remote mutations alter transition-state structure of human purine nucleoside phosphorylase. Biochemistry. 2008;47:2565–2576. doi: 10.1021/bi702133x. [DOI] [PubMed] [Google Scholar]
- [45].Ghanem M, Li L, Wing C, Schramm VL. Altered thermodynamics from remote mutations altering human toward bovine purine nucleoside phosphorylase. Biochemistry. 2008;47:2559–2564. doi: 10.1021/bi702132e. [DOI] [PubMed] [Google Scholar]
- [46].Saen-Oon S, Ghanem M, Schramm VL, Schwartz SD. Remote mutations and active site dynamics correlate with catalytic properties of purine nucleoside phosphorylase. Biophys. J. 2008;94:4078–4088. doi: 10.1529/biophysj.107.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Addington TA, Mertz RW, Siegel JB, Thompson JM, Fisher AJ, Filkov V, Fleischman NM, Suen AA, Zhang C, Toney MD. Janus: prediction and ranking of mutations required for functional interconversion of enzymes. J. Mol. Biol. 2013;425:1378–1389. doi: 10.1016/j.jmb.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Oue S, Okamoto A, Yano T, Kagamiyama H. Redesigning the substrate specificity of an enzyme by cumulative effects of the mutations of non-active site residues. J. Biol. Chem. 1999;274:2344–2349. doi: 10.1074/jbc.274.4.2344. [DOI] [PubMed] [Google Scholar]
- [49].Sacchi S, Rosini E, Molla G, Pilone MS, Pollegioni L. Modulating D-amino acid oxidase substrate specificity: production of an enzyme for analytical determination of all D-amino acids by directed evolution. Protein Eng. Des. Sel. 2004;17:517–525. doi: 10.1093/protein/gzh064. [DOI] [PubMed] [Google Scholar]
- [50].Iffland A, Gendreizig S, Tafelmeyer P, Johnsson K. Changing the substrate specificity of cytochrome c peroxidase using directed evolution. Biochem. Biophys. Res. Commun. 2001;286:126–132. doi: 10.1006/bbrc.2001.5366. [DOI] [PubMed] [Google Scholar]
- [51].Spiller B, Gershenson A, Arnold FH, Stevens RC. A structural view of evolutionary divergence. Proc. Natl. Acad. Sci. U. S. A. 1999;96:12305–12310. doi: 10.1073/pnas.96.22.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]