

Abstract
Objective
Spinal muscular atrophy (SMA) is the number 1 genetic killer of young children. It is caused by mutation or deletion of the survival motor neuron 1 (SMN1) gene. Although SMA is primarily a motor neuron disease, metabolism abnormalities such as metabolic acidosis, abnormal fatty acid metabolism, hyperlipidemia, and hyperglycemia have been reported in SMA patients. We thus initiated an in-depth analysis of glucose metabolism in SMA.
Methods
Glucose metabolism and pancreas development were investigated in the Smn2B/− intermediate SMA mouse model and type I SMA patients.
Results
Here, we demonstrate in an SMA mouse model a dramatic cell fate imbalance within pancreatic islets, with a predominance of glucagon-producing α cells at the expense of insulin-producing β cells. These SMA mice display fasting hyperglycemia, hyperglucagonemia, and glucose resistance. We demonstrate similar abnormalities in pancreatic islets from deceased children with the severe infantile form of SMA in association with supportive evidence of glucose intolerance in at least a subset of such children.
Interpretation
Our results indicate that defects in glucose metabolism may play an important contributory role in SMA pathogenesis.
Spinal muscular atrophy (SMA) is a neuromuscular disorder affecting 1 in 6,000 to 10,000 live births. SMA is a leading inherited cause of death in children <2 years of age.1,2 Homozygous deletions and/or mutations of the survival motor neuron 1 (SMN1) gene are responsible for >95% of cases of this devastating autosomal recessive disease.1,3 Pathological and clinical manifestations of SMA are largely due to loss of motor neurons in the spinal cord and brainstem, resulting in generalized muscular atrophy, weakness, respiratory insufficiency, and in >50% of cases, death in infancy or early childhood.2
The SMN gene is essential for survival, and a complete depletion of the protein results in early developmental lethality.4 In humans, the loss of the telomeric SMN1 gene is compensated for by the presence of the duplicated centromeric SMN2 gene.3 Although the 2 genes differ by several nucleotides, the functional difference lies within a C to T substitution at position 6 of exon 7 in SMN2.5 This silent mutation causes aberrant splicing of the SMN2 gene and the production of an unstable protein, termed SMNΔ7, due to the excision and loss of exon 7.3,5 This alteration in splicing most likely results from the loss of an exon splice enhancer and/or the gain of an exon splice silencer.3,6,7 Although the major product of the SMN2 gene is the SMNΔ7 protein, the full-length SMN protein is still produced in small quantities (~10%).3 As such, SMA severity is heavily modulated by the number of SMN2 copies present in patients.3,8
Although the pathological hallmark of SMA is motor neuron loss, recent reports have identified additional defects in the muscle,9 at the neuromuscular junction,10 and in the heart.11–13 In the present work, we report for the first time glucose metabolism and pancreatic developmental defects in an SMA mouse model and human SMA patients. Our results demonstrate a progressive loss of the insulin-producing β cells and a corresponding increase in the number of the glucagon-producing α cells in pancreatic islets. This altered cell fate is accompanied by glucose clearance defects in the SMA mice during fasting and following an acute glucose tolerance test. In addition, pathology specimens from human SMA type I infants demonstrate similar abnormalities in pancreatic islet cell morphology and distribution. These observations in the SMA mouse model, and the corresponding parallels in human tissues, indicate that these findings may indeed be relevant to the human condition.
These novel findings have 2 major implications. First, the observed defects in glucose metabolism and pancreatic development may contribute additional stressors, which impact motor neuron loss, muscle function, and survival in SMA. Second, as SMA patients are living longer due to improved assistive technology, further studies to assess the metabolic consequences of impaired glucose metabolism and its potential impact on their clinical course will be of utmost importance.
Materials and Methods
Antibodies
The primary antibodies used were as follows: guinea pig antiinsulin (1:50; Dako, Carpinteria, CA), mouse antiglucagon (1:200; Abcam, Cambridge, MA), rabbit anti-Ki67 (1:50; Leica Microsystems, Bannockburn, IL), rabbit anti–phospho-AKT (p- AKT; Ser 473; 1:500; Cell Signaling Technology, Danvers, MA), and rabbit anti-AKT (1:500; Cell Signaling Technology). The secondary antibodies used were as follows: donkey anti– guinea-pig biotin-SP conjugated (1:200; Jackson ImmunoResearch, West Grove, PA), streptavidin-Cy3 conjugated (1:600; Jackson ImmunoResearch), Alexa Fluor 488 goat antimouse (1:500; Molecular Probes, Eugene, OR), Alexa Fluor 555 goat antirabbit (1:500; Molecular Probes), and horseradish peroxidase- conjugated goat antirabbit immunoglobulin G (1:5,000; Bio-Rad Laboratories, Hercules, CA).
Animal Models
The Smn2B/− mice were established in our laboratory and maintained in our animal facility on a C57BL/6 × CD1 hybrid background.14 The wild-type (WT) mice are also on C57BL/6 × CD1 hybrid background. The 2B mutation consists of a substitution of 3 nucleotides in the exon splicing enhancer of exon 7.15 The Smn knockout allele was previously described by Schrank et al.16 Smn+/− mice were obtained from The Jackson Laboratory. All animal procedures were performed in accordance with institutional guidelines (Animal Care and Veterinary Services and Ethics, University of Ottawa).
Human Samples
Pediatric control pancreatic samples were collected by K.J.S. and K.M. in a deidentified fashion from children who died for reasons other than SMA. For the type I SMA pancreatic samples, informed consent was obtained from the parents of the patients to perform a research autopsy under IRB 8751, reviewed and approved by the University of Utah Institutional Review Board. The autopsies were performed in collaboration with pathologists at each of the US institutions involved (Primary Children’s Medical Center and the Medical Examiner’s Office, Salt Lake City, UT; Cook Children’s Medical Center, Ft Worth, TX; Children’s Hospital of Atlanta Scottish Rite, Atlanta, GA). Ages for the human pancreatic samples were as follows: 4, 5, 7, 15, and 36 months for the controls samples and 18 (#177), 35 (#195), 28 (#206), 33 (#217), 15 (#251), and 7 months (#351) for the SMA samples.
Immunohistochemistry of Pancreas Sections
Whole pancreata were dissected out of P7, P10, P15, and P21 mice and placed overnight in 4% paraformaldehyde. Tissues were embedded in paraffin and cut at a thickness of 4μm. For immunohistochemistry, pancreas sections were first deparaffinized in xylene (3 × 10 minutes), fixed in 100% ethanol (2 × 10 minutes), rehydrated in 95% and 75% ethanol (5 seconds each), and placed for 5 minutes in 1M Tris-HCl pH 7.5. The pancreas sections were then incubated for 2 hours at room temperature (RT) in blocking solution (Tris-buffered L-lysine saline [10% NaN3], 20% goat serum, 0.3% Triton X-100). This was followed by an overnight incubation at 4°C with the primary antibodies. Subsequently, sections were washed 3× with phosphate- buffered saline (PBS), incubated 1 hour at RT with the secondary antibodies, and then washed 3× with PBS. Hoechst (1:1,000) was added to the last PBS wash, followed by the slides being mounted in Fluorescent Mounting Medium (Dako). Images were taken with a Zeiss (Thornwood, NY) confocal microscope, with a ×20 objective, equipped with filters suitable for fluorescein isothiocyanate (FITC)/Cy3/Hoechst fluorescence.
Immunoblot Analysis
Equal amounts of protein extracts from liver samples of random fed P21 mice were separated by electrophoresis on 10% sodium dodecyl sulfate–polyacrylamide gels and blotted onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membranes were blocked in 5% nonfat milk in TBST (10mM Tris-HCl pH 8.0, 150mM NaCl, and 0.1% Tween 20; Sigma, St Louis, MO), and incubated overnight at 4°C with the first primary antibody. The membranes were then incubated at room temperature with the secondary antibody for 1 hour, followed by 3 TBST washes. Signals were visualized using the ECL or the ECL Plus detection kit (Amersham, Piscataway, NJ).
TUNEL Assay
Whole pancreata were removed from P10 mice and immediately flash frozen over liquid nitrogen in a 1:1 solution of OCT (Tissue Tek) and 30% sucrose, and stored at −80°C until they were sectioned at 10μm thickness using a cryostat. Sections were processed for cell death detection by TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; Roche, Indianapolis, IN), following the manufacturer’s instructions. After costaining for insulin, the slides were mounted in Fluorescent Mounting Medium (Dako), and images were taken with a Zeiss confocal microscope, with a ×20 objective, equipped with filters suitable for FITC/Cy3/Hoechst fluorescence.
Glucose Tests
All blood glucose readings were done using the OneTouch Ultra2 glucometer (LifeScan, Milpitas, CA). Random fed P7, P15, and P21 mice were evaluated at the same time of day for nonfasting glucose levels. For fasting glucose levels, P7, P15, and P21 mice were fasted for 6 hours, at which point glucose levels were read. A maximum of 6 hours was allowed for fasting either due to the age of the mice (P7 and P15) or to the severe diseased state of the SMA mice (P21).
The intraperitoneal (IP) glucose tolerance test (IPGTT)17 was performed on P15 and P21 mice fasted for 6 hours. They were then administered a 20% glucose solution (2g glucose/kg body weight) through IP injection. Glucose levels were measured at 0, 15, 30, 90, and 120 minutes in blood samples collected from the tail vein.
Measurement of Insulin and Glucagon Serum Levels
Serum was collected from random fed P7, P15, and P21 mice as well as P21 mice 30 minutes post-IPGTT. Insulin and glucagon levels were measured using the mouse Bio-Plex Pro diabetes magnetic bead-based immunoassay (Bio-Rad) following the manufacturer’s instructions. Data acquisition and analysis were done using the Bio-Plex 200 Luminex-based reader and the accompanying Bio-Plex Manager software.
Pancreatic Islet Isolation
Islets were isolated from anaesthetized P15 mice by perfusion of the pancreatic bile duct with collagenase XI (Sigma). Due to the technical limitations of this technique, P15 was the earliest time point at which it was possible to obtain the quality and quantity of islets required for analysis. The islets were then purified by 2 rounds of manual selection using a dissecting microscope, and cultured in RPMI + 11mM glucose as described in Fu et al.18
Reverse Transcriptase Polymerase Chain Reaction
RNA extraction from P15 islets was done using the RNeasy Mini Kit and QIAshredder (Qiagen, Valencia, CA), following the manufacturer’s instructions. Reverse transcription was performed using equal amounts of total RNA. The following primers were used for reverse transcriptase polymerase chain reaction (RT-PCR) analysis: Pdx1 forward (5′ AGG TCA CCG CAC AAT CTT GCT 3′), Pdx1 reverse (5′ CTT TCC CGA ATG GAA CCG A 3′), Pax6 forward (5′ AAC AAC CTG CCT ATG CAA CC 3′), Pax6 reverse (5′ ACT TGG ACG GGA ACT GAC AC 3′), NeuroD forward (5′ GTC CCA GCC CAC TAC CAA TT 3′), NeuroD reverse (5′ CGG CAC CGG AAG AGA AGA TT 3′), Islet1 forward (5′ ATG ATG GTG GTT TAC AGG CTA AC 3′), Islet1 reverse (5′ TCG ATG CTA CTT CAC TGC CAG 3′), insulin forward (5′ CGA GGC TTC TTC TAC ACA CC 3′), insulin reverse (5′ GAG GGA GCA GAT GCT GGT 3′), glucagon forward (5′ CCA CTC ACA GGG CAC ATT CA 3′), glucagon reverse (5′ GTC CCT GGT GGC AAG ATT GT 3′), actin forward (5′ CCG TCA GGC AGC TCA TAG CTC TTC 3′), and actin reverse (5′ CTG AAC CCT AAG GCC AAC CGT 3′).
Statistical Analysis
All statistical analyses were done with GraphPad Prism software (San Diego, CA). When appropriate, a Student 2-tail t test or a 2-way analysis of variance followed by a Bonferroni multiple comparison test was used. Data were considered significantly different at p < 0.05.
Results
Over the years, published data have indicated that SMA patients can display a host of metabolic abnormalities, particularly in the setting of a catabolic state.19–22 However, in addition to previously reported abnormalities, including metabolic acidosis, abnormal fatty acid metabolism, and hyperlipidemia, we have noted significant hyperglycemia on several occasions when our SMA patients received an infusion of intravenous glucose or underwent prolonged fasting, particularly during a catabolic state associated with illness (unpublished observations, K.J.S. and K.M.). This encouraged us to initiate an in-depth analysis of glucose metabolism in a previously characterized intermediate SMA mouse model, Smn2B/−.14 These mice express approximately 15% residual full-length Smn protein and display the characteristic motor neuron loss, neuromuscular junction defects, and muscular atrophy of SMA, resulting in a median lifespan of 1 month.14,23 Additionally, we looked for evidence in support of such abnormalities in human subjects participating in an ongoing clinical study.
SMA Mice Are Glucose Intolerant
We first compared random fed (non-fasting) and fasting glucose levels of WT and Smn2B/− mice at 3 different time points: postnatal (P) day 7, 15, and 21. These time points represent pre-phenotype (P7), early phenotype (P15), and late phenotype (P21) stages of SMA in this mouse model. At P7 and P15, both WT and Smn2B/− mice had normal glycemia in non-fasting states (Fig 1). As would be expected, the glucose levels in WT mice were significantly decreased following a 6-hour fast. However, fasting Smn2B/− mice were hyperglycemic, suggesting an inability to properly metabolize glucose. At P21, the glucose metabolism defects were amplified by a subpopulation of Smn2B/− mice with higher fasting than non-fasting glucose levels, suggesting a combination of hypoglycemia in the random fed state and an inability to metabolize glucose. Interestingly, the more severe SMNΔ7 SMA mouse model also displayed hypoglycemia upon reaching end stage of the disease.24 At P21, although reduced, the WT fasting glucose levels were not significantly decreased compared to non-fasting levels, probably due to an insufficient fasting period. However, due to the severe diseased state of the SMA mice at P21, a longer fasting period was neither possible nor allowable.
FIGURE 1.

Glucose intolerance in Smn2B/− mice. (A–C) Serum glucose levels in postnatal day (P) 7, P15, and P21 random fed and fasted wild-type (WT; n = 4 [P7], 5 [P15], and 5 [P21]) and Smn2B/− (n = 3 [P7], 5 [P15], and 11 [P21]) mice. At all time points, fasted WT mice showed reduced glucose levels, whereas fasted Smn2B/− mice were hyperglycemic. At P21, random fed Smn2B/− mice showed signs of hypoglycemia. Data are mean ± standard deviation (SD); 2-way analysis of variance; **p < 0.01; ***p < 0.001. (D) Intraperitoneal glucose tolerance test (IPGTT) curves of P15 WT (n = 3) and Smn2B/− (n = 5) mice show similar patterns. Data are mean ± SD. (E) Quantification of the IPGTT area under the curve (AUC) shows no difference between P15 WT and Smn2B/− mice. Data are mean ± SD. (F) Individual comparisons of IPGTT curves between P15 WT and Smn2B/− mice show irregularities in the curves of the spinal muscular atrophy (SMA) mice. Smn2B/− #5 displayed a precipitous drop in glucose levels followed by a slow increase, whereas Smn2B/− #2 followed the normal WT curve until the glucose levels began to rise after 60 minutes. (G) Quantification of the change in glucose levels from 60 to 90 minutes and 60 to 120 minutes demonstrates an increase in the Smn2B/− mice and a decrease in the WT mice. Data are mean ± SD; t test, *p = 0.021 (60–90 minutes); p = 0.059 (60–120 minutes). (H) IPGTT curves of P21 WT (n = 5) and Smn2B/− (n = 10) mice show increased glucose levels at 30 minutes and 60 minutes in SMA mice, indicative of glucose intolerance. Data are mean ± SD; t test, **p = 0.002, *p = 0.018. (I) Quantification of the IPGTT AUC of P21 mice shows a significant increase in the AUC of the Smn2B/− mice when compared to WT, suggesting defective glucose clearance. Data are mean ± SD; t test, *p = 0.011.
To determine the ability of fasting Smn2B/− mice to metabolize glucose in an acute setting, we performed an IPGTT.17 At P15, the glucose clearance curve of the Smn2B/− mice was similar to that of WT mice (see Fig 1). Furthermore, the area under the curves (AUCs) of WT and Smn2B/− mice were not significantly different, suggesting that there was no change in the overall glucose tolerance of P15 SMA mice. However, when IPGTT curves were individually recorded, some irregularities became apparent in P15 Smn2B/− mice. For instance, a precipitous drop in glucose levels followed by a slow increase was observed in 1 animal, whereas another followed the normal WT curve until the glucose levels began to rise in the later stages of the IPGTT. When quantifying the change in glucose levels from 60 to 90 minutes and 60 to 120 minutes, we found that Smn2B/− mice displayed an increase, whereas the WTmice showed a decrease. The IPGTT was next repeated in P21 Smn2B/− and control mice. At this time point, the Smn2B/− mice showed signs of glucose intolerance, with glucose levels significantly higher than WT levels at 30 and 60 minutes following the glucose IP injection. Importantly, the AUCs of Smn2B/− mice were significantly greater than those of WT mice. Thus, it appears that at an early age SMA mice have fasting glucose clearance defects, which progressively develop into glucose intolerance.
SMA Mice Display Reduced β Cell and Increased α Cell Numbers
Because the pancreatic islets are among the key players responsible for the maintenance and regulation of blood glucose, we next performed immunohistochemical analysis to assess the cellular profile. Double-labeling of insulin- producing β cells and glucagon-producing α cells revealed a significant decrease in the number of β cells and a significant increase in α cells in the pancreas of Smn2B/− mice relative to WT controls (Fig 2). As early as P7, there was a small shift in pancreatic cell composition, and at P15 the Smn2B/− islets were predominantly composed of α cells. The increase in α cells appeared to plateau at P15, as the percentage of glucagon-positive cells per islet was similar in P21 Smn2B/− islets. The observed decrease in β cells in Smn2B/− islets also corresponded to an overall decrease in β cell mass. The reduced β cell mass was apparent at all 3 time points (P7, P15, and P21), irrespective of β cell composition of the islets. Thus, a loss of β cells and an increase in α cells within Smn2B/− pancreatic islets corresponded to a functional inability to metabolize glucose, at both pre-phenotype and phenotype stages. Of note, Smn2B/− islets appeared smaller than WT islets, which was most likely due to the overall reduction in size of the SMA mice.25
FIGURE 2.

Pancreatic defects in Smn2B/− mice. (A) Representative cross-sections of P7, P15, and P21 pancreatic islets of wild-type (WT) and Smn2B/− mice. Islets were colabeled for insulin (red; β cells), glucagon (green; α cells), and Hoechst (blue; nuclei). Arrowhead shows a yellow cell in an Smn2B/− islet, positive for both insulin and glucagon. Scale bar = 50μm. (B) Quantification of the percentage of insulin-positive cells per islet shows a significant decrease of β cells in Smn2B/− islets (n = 3 mice) at all time points when compared to WT islets (n = 3 mice). Data are mean ± standard deviation (SD); 2-way analysis of variance (ANOVA), ***p < 0.001, ****p < 0.0001. (C) Quantification of the percentage of glucagon-positive cells per islet shows a significant increase of α cells in Smn2B/− islets (n = 3 mice) at all time points relative to WT (n = 3 mice). Data are mean ± SD; 2-way ANOVA, ***p < 0.001, ****p < 0.0001. (D) Representative images of P7, P15, and P21 pancreatic cross-sections of WT and Smn2B/− mice. Sections were stained for insulin (red; β cells) and Hoechst (blue; nuclei). Scale bar = 100μm. (E) Quantification of the β cell mass (β cell area/whole pancreatic section area). The Smn2B/− mice (n = 3 mice per time point) display a significant reduction in β cell mass compared to WT mice (n = 3 mice per time point). Data are mean ± SD; t test; *p = 0.021.
SMA Mice Have Hyperglucagonemia and Increased Insulin Sensitivity
We next assessed whether the loss of β cells and the gain of α cells in Smn2B/− mice was reflected in the overall blood insulin and glucagon levels. Serum was collected from random fed P7, P15, and P21 WT and Smn2B/− mice. In addition, sera from P21 mice were analyzed 30 minutes following a glucose injection, as this time point corresponds to the greatest observed difference in glucose levels between WT and Smn2B/− mice in the IPGTT.
Serum glucagon levels from Smn2B/− mice were significantly higher than those observed in WT mice at all time points (Fig 3A), suggesting that both pre-phenotype and phenotypic SMA mice were hyperglucagonemic. Interestingly, insulin levels were similar at all time points between Smn2B/− and WT mice (see Fig 3B), indicating the functional competence of the remaining insulin-producing β cells in Smn2B/− islets.
FIGURE 3.

Smn2B/− mice have normoinsulinemia, hyperglucagonemia, and increased insulin sensitivity. (A, B) Serum glucagon (A) and insulin (B) levels in random fed P7, P15, P21, and P21 30 minutes post–intraperitoneal glucose tolerance test (IPGTT) wild-type (WT; n = 3 [P7], 4 [P15], 7 [P21], and 4 [P21, 30 minutes]) and Smn2B/− (n = 4 [P7], 3 [P15], 3 [P21], and 6 [P21, 30 minutes]) mice. Whereas insulin levels were similar between both groups (B), glucagon levels were significantly higher in the Smn2B/− mice when compared to WT (A). Data are mean ± standard deviation; t test, **p = 0.0042. (C) Representative immunoblot of phospho-AKT (p-AKT) and total AKT in extracts of random fed livers of P21 WT and Smn2B/− mice. (D) Quantification of the p-AKT/total AKT ratios shows a trend (p = 0.0536) toward increased p-AKT in Smn2B/− livers (n = 4 mice) compared to WT (n = 4 mice), indicative of increased insulin sensitivity.
Because P21 Smn2B/− mice are glucose intolerant, we next addressed whether they displayed signs of insulin resistance. To do this, we measured levels of p-AKT in the liver, as reduced levels are indicative of insulin resistance (reviewed in Taniguchi et al26). Livers from P21 random fed Smn2B/− mice displayed higher levels of p- AKT when compared to WT livers (see Fig 3C, D), suggesting instead a hypersensitivity to insulin. The absence of insulin resistance in the livers of Smn2B/− mice was consistent with their normal levels of insulin, as the pancreas typically responds to insulin resistance by increasing its insulin production (reviewed in Shanik et al27).
Thus, the above results suggest that a critical consequence of the increased number of α cells within Smn2B/− islets is hyperglucagonemia, whereas loss of β cells within Smn2B/− islets does not significantly impact upon insulin levels.
Pancreatic Islets in SMA Mice Do Not Have Increased Apoptotic or Proliferative Cells
To determine whether the loss of β cells in pancreata from Smn2B/− mice was due to increased apoptosis, we performed a TUNEL assay on P10 tissue. This time point was chosen because it bridges the minimal loss in β cells observed at P7 and the maximal loss observed at P15. Very few apoptotic cells were observed in either WT or Smn2B/− pancreatic islets (Fig 4). Indeed, both the number of TUNEL-positive islets and the number of TUNEL-positive cells per islet were similarly low in both groups. We next assessed whether the increase in α cells in Smn2B/− islets was due to an increase in proliferation. Double-labeling of glucagon and Ki67, a proliferation marker, was performed on sections from pancreata of P10 WT and Smn2B/− mice. The number of islets containing Ki67-positive glucagon-positive cells was comparable between WT and Smn2B/− mice. Therefore, the decrease in β cells and increase in α cells in Smn2B/− pancreatic islets were neither a result of increased β cell apoptosis nor an increase in α cell proliferation.
FIGURE 4.

Smn2B/− islets do not show increased β cell apoptosis or α cell proliferation. (A) Representative images of P10 pancreatic islets of wild-type (WT) and Smn2B/− mice colabeled for insulin (Ins; red; β cells), TUNEL (green), and Hoechst (blue; nuclei). Scale bar = 100μm. (B, C) Quantification of the percentage of TUNEL-positive islets (B) and the percentage of tunnelpositive insulin cells per islet (C) shows no difference between WT (n = 3) and Smn2B/− (n = 3) mice. Data are mean ± standard deviation (SD). (D) Representative images of P10 pancreatic islets of WT and Smn2B/− mice colabeled with Ki67 (red), glucagon (Gluc; green; α cells), and Hoechst (blue; nuclei). Scale bar = 50μm. (E) Quantification of the percentage of islets with Ki67-positive/ glucagon-positive cells shows no difference between WT (n = 3) and Smn2B/− (n = 3) mice. Data are mean ± SD.
β Cell Identity Transcription Factors Are Unchanged in Smn2B/− Islets
The results from the apoptosis and proliferation analysis suggest that there is another mechanism responsible for the gradual change in composition of Smn2B/− pancreatic islets. At P15, we observed cells co-labeling for insulin and glucagon (see Fig 2A, arrowhead), hinting at a temporal and progressive fate switch from β to α cell. To investigate this possibility, we examined the expression of various transcription factors responsible for the determination and maintenance of β cell identity.28 RT-PCR was performed on RNA extracted from isolated pancreatic islets of P15 mice, an early phenotypic time point for the Smn2B/− mice, in terms of both SMA and glucose tolerance and pancreatic defects. We did not observe any changes in mRNA expression of transcription factors responsible for the regulation of pancreatic progenitors (Pdx1), general growth and organization of pancreatic islets (Pax6), maturation and identity of β cells (NeuroD), and function of differentiated β cells (Pdx1 and Islet1; Fig 5).28 Insulin mRNA was also similar between WT and Smn2B/− islets (see Fig 5), consistent with the normoinsulinemic profile of the serum of P15 Smn2B/− mice (see Fig 3B). Glucagon mRNA was slightly higher in Smn2B/− islets (see Fig 5), also consistent with the hyperglucagonemia observed in P15 Smn2B/− serum (see Fig 3A).
FIGURE 5.
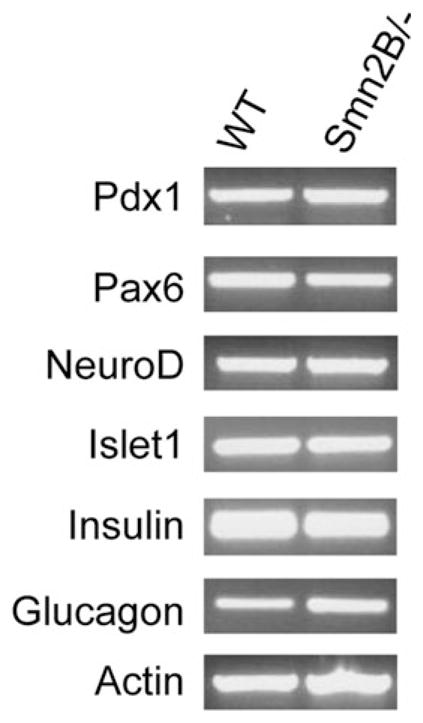
The expression profile of β cell identity transcription factors is unchanged in Smn2B/− islets. Representative reverse transcriptase polymerase chain reaction results from RNA isolated from P15 pancreatic islets of wild-type (WT; n = 3, pooled) and Smn2B/− (n = 2, pooled) mice. Pdx1, Pax6, NeuroD, and Islet1 display similar mRNA levels in both WT and Smn2B/− islets. The mRNA levels for insulin were normal, whereas levels for glucagon were increased. Actin transcripts were used as a loading control.
Metabolic Abnormalities in Human SMA Subjects
All 6 infants from whom pancreas samples were collected at autopsy had been followed prospectively in our clinical outcomes study “Clinical and Genetic Studies in SMA.” The type I SMA patients were genetically confirmed by a homozygous deletion of SMN1, and all of them possessed 2 SMN2 copies. Clinical and laboratory records were examined for evidence of abnormal glucose levels, but no formal evaluation of glucose metabolism in the form of glucose tolerance testing was performed, nor were insulin or glucagon levels obtained. In 2 of 6 subjects (UT-SMA206 and UT-SMA217), point of care testing was performed on several occasions in the home, via glucometer, to assess for hypoglycemia that is often associated with an intolerance of gastric tube feeds. In this setting, glucose levels ranged from low (45mg/dl; normal range, >60mg/dl) to mild hyperglycemia (128mg/dl; normal range, <108mg/dl). We further reviewed all glucose levels obtained from all subjects during hospitalizations for illness or elective admissions, and during elective outpatient evaluations. One subject, UTSMA177, underwent an elective surgical repair for coarctation of the aorta at 9 days of age, prior to her diagnosis of SMA at 3 months of age. A glucose reading obtained in the perioperative period was noted to be significantly elevated at 209mg/dl (normal range, 60–108mg/dl). Of note, she was catabolic and had not received intravenous glucose or parenteral nutrition before or during the procedure. This same subject had at least 2 other mildly elevated glucose levels (113 and 111mg/dl) during subsequent hospitalizations. At least 2 other subjects had at least 1 documented elevated glucose level. However, in the other 3 subjects, no abnormal glucose levels were documented, and in none of the 6 cases was glucose tolerance formally assessed.
Pancreas from Human SMA Displays Abnormal Islet Composition
We next investigated whether SMA patients had similar pancreatic defects as those observed in the SMA mice. Pancreatic samples were not collected for any given observed abnormality or suggestive clinical history, and were grossly normal pathologically; rather, the tissue was obtained as part of a comprehensive collection of tissues in willing study participants, which included all major organs in addition to skin, muscle, nerve, spinal cord, and brain. To this end, we performed immunohistochemistry on 6 type I SMA pancreatic samples, co-labeling for insulin-producing β cells and glucagon-producing α cells (Fig 6). Astonishingly, every SMA pancreas contained islets predominantly composed of α cells and/or displaying an overall disorganized appearance. Quantification shows that pancreatic islets from type I SMA patients were composed of significantly more α cells compared to control islets. We further compared SMA patients individually and found that they displayed similar pancreatic islet compositions, suggesting, at least in the 6 samples analyzed herein, that the age at which type I SMA patients succumbed to the disease did not influence islet pathology. It is important to note that human islets differ from rodent islets in that they do not display a core-mantle segregation of β and α cells.29 Furthermore, human islets also contain fewer β cells, because α cells localize along the mantle and internal vascular channels. 29,30 Nevertheless, the core of normal human islets is typically composed of more β cells than α cells.29 Both SMA mice and human patients displayed pancreatic islets with an abnormal increase in α cell number, suggesting a conserved role for the SMN protein in normal pancreatic development.
FIGURE 6.

Human type I spinal muscular atrophy (SMA) pancreatic islets display an abnormal composition of insulin-secreting β cells and glucagon-secreting α cells. (A) Representative pancreatic cross-sections from pediatric controls and type I SMA patients colabeled for insulin (red; β cells), glucagon (green; α cells), and Hoechst (blue; nuclei). Scale bar = 50μm. (B) Quantification of islet composition shows that type I SMA islets display significantly more α cells and significantly fewer β cells compared to control islets. Data are mean ± standard deviation (SD); t test, ***p = 0.0004. (C) Comparison of the percentage of glucagon-positive cells per islet between type I SMA patients shows that their pancreatic islet compositions are not significantly different. Data are mean ± SD; NS = not significant.
Discussion
Previous clinical reports have identified abnormal fatty acid metabolism19,22 and hyperlipoproteinemia20,21 in SMA patients. Interestingly, all of these defects can originate from an inappropriate regulation of glucose metabolism. However, until now a specific defect in glucose metabolism or pancreatic islet cell function in mice or humans with SMA has not been recognized. In the present study, we demonstrate that pancreatic defects and as a consequence, alterations in glucose metabolism are associated with SMA. Indeed, SMA mice have fasting hyperglycemia, glucose intolerance, hypersensitivity to insulin, and hyperglucagonemia. These metabolic defects are accompanied by a temporal shift in pancreatic islet composition, from the typical β cell core to an abnormal increase in α cell number. In addition, we demonstrate similar histopathologic abnormalities in autopsy specimens from SMA type I infants and children, with pancreatic islets that predominantly contain glucagon-producing α cells. Interestingly, although the age of death of the SMA patients ranged from 7 to 35 months, they displayed comparable pancreatic islet composition. A thorough examination of pancreas samples from type I, II, and III SMA patients would most likely reveal a progressive increase in the severity of islet abnormalities.
Although we did not perform systematic studies of glucose metabolism in these children prior to their deaths, review of clinical records provides some evidence of potential abnormalities in glucose metabolism in at least 3 of the 6 subjects for whom we had pancreas samples from autopsy available for study. However, the relevance of such abnormalities, if any, to the clinical outcomes in these patients remains unclear at this time. In several other SMA type I children enrolled in our studies, we have observed hyperglycemia in the setting of a catabolic state, particularly if the only source of calories provided is intravenous glucose. However, such abnormalities were attributed to their illness and severely reduced lean body mass, and neither formal glucose tolerance testing nor insulin or glucagon levels were examined prior to their deaths, which were invariably related to acute and/or chronic respiratory failure in the setting of severe muscle weakness. We have never clearly documented fasting hyperglycemia in SMA type I subjects. In general, we make every effort to limit fasting in this population, due to their severely diminished lean body mass and energy reserves, and prior anecdotal reports of hypoglycemia in this setting. However, several type II and type III SMA subjects enrolled in our research studies have been formally diagnosed with diabetes or glucose intolerance requiring medical treatment. Clearly, further systematic studies in human subjects are needed to clarify the relevance of these observations to SMA patients.
Other neurodegenerative diseases are also characterized by symptoms of metabolic disorders. Indeed, numerous studies suggest a role for impaired glucose uptake and insulin resistance in the pathology and progression of Alzheimer disease.31,32 Recently, an analysis of a mouse model of Prader Willi syndrome, a rare genetic neurodevelopmental disorder,33 revealed hypoinsulinemia and hypoglucagonemia due to increased apoptosis of α and β pancreatic cells.34 Although the mechanisms that lead to the glucose homeostasis perturbations vary significantly between these various pathologies, their occurrences support the idea that the pathological hallmarks of a specific disease may involve >1 organ and/or compartment, albeit with different severities and functional consequences.
The phenotypic picture painted herein, at least in the SMA mouse model, is of a progressive pathology. In pre-phenotype (P7) and early phenotype (P15) stages, random fed SMA mice were normoglycemic, whereas fasting SMA mice were hyperglycemic. At a later phenotypic stage (P21), random fed Smn2B/− mice were hypoglycemic and continued to display fasting hyperglycemia. Furthermore, whereas P15 Smn2B/− mice had normal IPGTT curves, P21 Smn2B/− mice had become glucose intolerant. Interestingly, insulin levels in SMA mice remained normal, and P21 Smn2B/− livers showed signs of increased insulin sensitivity, suggesting that the defects in glucose metabolism were not due to hypoinsulinemia or insulin resistance. Rather, the observed metabolic abnormalities most likely stemmed from elevated glucagon levels, which subsequently enhanced hepatic glucose output. In all probability, the increased glucagon release was a direct consequence of the increased number of α cells within Smn2B/− islets. It will therefore be of future interest to determine whether preventing glucagon release, through the administration of glucagon receptor antagonist II,35,36 can benefit the glucose and pancreatic defects identified in the Smn2B/− mice.
Interestingly, the observed shift in islet composition in SMA mice did not appear to be a consequence of increased β cell apoptosis or α cell proliferation, which is what typically occurs in type 2 diabetes mellitus models. 36,37 This finding suggests that the dramatic increase in the number of α cells within SMA islets may have been due to a time-dependent fate switch. Future experiments should therefore focus on the identification of β and α cell lineage determinants modulated by the Smn protein. As the RT-PCR experiment did not reveal a general change in the transcription factor profiles, it is most likely that the developmental pancreatic defects in SMA stem specifically from 1 or a few essential components.
It is important to note that the cellular composition of pancreatic islets is species and location dependent.29,38 Taking into account these factors, quantitative analyses were performed solely on intraspecies comparisons from islets that were localized throughout the entire pancreatic sections (tail, body, and head). Furthermore, the increase in α cells in the islets of SMA mice is accompanied by hyperglucagonemia, highlighting a functional consequence of the observed change in cellular makeup. Thus, the progressive shift in SMA islet composition described in our study can be attributed to SMN loss and not pattern variability.
Our report identifies a novel role for the SMN protein in the pancreas, the loss of which results in pancreatic development and glucose metabolism defects in an SMA mouse model and human type I SMA patients. The ensuing question is whether this pathology is SMA dependent or independent. The pathological hallmarks in SMA are motor neuron loss and muscular atrophy.2 Although not the primary targets, motor neuron innervation and degeneration occur in late stage diabetic models. 39,40 As for muscle, it is undeniably a central player in glucose metabolism via its role in insulin-dependent glucose uptake.41 Furthermore, various neurodegenerative and neuromuscular disorders have been associated with pancreatic and glucose metabolism defects.42–44 Thus, it is possible that the glucose and pancreas pathologies observed in the Smn2B/− mice were a direct consequence of the loss of functional motor neurons and compromised skeletal muscle. However, P7 Smn2B/− mice, which represent an SMA pre-phenotype stage, displayed fasting hyperglycemia and an increased number of pancreatic islet α cells. This suggests that glucose metabolism and pancreatic defects in the Smn2B/− mice are SMA independent, because they arise before disease onset, and therefore occur as a direct consequence of SMN depletion and loss of its unique role in the pancreas.
Although the defects in glucose metabolism and pancreatic development in the Smn2B/− mice are most likely independent of disease onset, their impact on the progression of SMA should not be underestimated. Indeed, various therapeutic strategies such as administration of insulin-like growth factor 1 to muscle, neuronal depletion of phosphatase and tensin homolog, and systemic trichostatin A administration, have shown beneficial effects in SMA mouse models and also act on glucose metabolism. 45–51 Thus, although the SMA therapeutic strategies described above were aimed at motor neuron and muscle pathology, it is possible that the positive outcomes were due in part to improvements in glucose metabolism and pancreatic development. Furthermore, it has previously been reported that specific maternal rodent diets as well as neonatal nutritional support can increase the lifespan of SMA mice.24,52 Because diet has a major influence on glucose metabolism,53 these studies support the hypothesis that metabolism defects may indeed contribute to SMA pathology. Future development and assessment of SMA therapies should therefore also evaluate their effect on glucose metabolism and pancreatic abnormalities.
Finally, it would be of interest to perform a more in-depth characterization of the glucose defects in SMA patients and assess whether they are linked to disease severity. A key component issuing from our work is that as SMA patients are surviving longer, either through mechanical or therapeutic strategies, additional tissue-specific pathologies are becoming apparent and impact the well-being of these patients. Merging assessment and therapeutic intervention for metabolic impairments with existing clinical care management of SMA patients is therefore of paramount importance.
Acknowledgments
This project was funded by grants from the Canadian Institutes of Health Research (CIHR grant number MOP 38040) and Muscular Dystrophy Association to R.K. M.B. and J.-P.M are recipients of a Frederick Banting and Charles Best CIHR Doctoral Research Award. R.K. is a recipient of a University Health Research Chair from the University of Ottawa. R.A.S. holds the Canada Research Chair in Apoptotic Signaling. K.J.S. received a grant and travel support from the NIH National Institute of Child Health and Human Development. (Therapeutic Opportunity in Spinal Muscular Atrophy, 5R01HD054599-05).
We thank the Kothary Laboratory for helpful discussions.
Footnotes
Potential Conflicts of Interest
K.J.S.: grants/grants pending, ISIS Pharmaceuticals, National Institute of Child Health and Human Development, Alternating Hemiplegia of Childhood Foundation, Biomarin Pharmaceutical, Muscular Dystrophy Association, Orphamed Pharmaceuticals.
References
- 1.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;15:409–413. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 4.Jablonka S, Schrank B, Kralewski M, et al. Reduced survival motor neuron (Smn) gene dose in mice leads to motor neuron degeneration: an animal model for spinal muscular atrophy type III. Hum Mol Genet. 2000;9:341–346. doi: 10.1093/hmg/9.3.341. [DOI] [PubMed] [Google Scholar]
- 5.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 7.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 8.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 9.Walker MP, Rajendra TK, Saieva L, et al. SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum Mol Genet. 2008;17:3399–3410. doi: 10.1093/hmg/ddn234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murray LM, Comley LH, Thomson D, et al. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 11.Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19:3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shababi M, Habibi J, Yang HT, et al. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19:4059–4071. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- 13.Bevan AK, Hutchinson KR, Foust KD, et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowerman M, Anderson CL, Beauvais A, et al. SMN, profilin IIa and plastin 3: a link between the deregulation of actin dynamics and SMA pathogenesis. Mol Cell Neurosci. 2009;42:66–74. doi: 10.1016/j.mcn.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 15.DiDonato CJ, Lorson CL, De Repentigny Y, et al. Regulation of murine survival motor neuron (Smn) protein levels by modifying Smn exon 7 splicing. Hum Mol Genet. 2001;10:2727–2736. doi: 10.1093/hmg/10.23.2727. [DOI] [PubMed] [Google Scholar]
- 16.Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heikkinen S, Argmann CA, Champy MF, Auwerx J. Evaluation of glucose homeostasis. Curr Protoc Mol Biol. 2007;Chapter 29(Unit 29B.3) doi: 10.1002/0471142727.mb29b03s77. [DOI] [PubMed] [Google Scholar]
- 18.Fu A, Ng AC, Depatie C, et al. Loss of Lkb1 in adult beta cells increases beta cell mass and enhances glucose tolerance in mice. Cell Metab. 2009;10:285–295. doi: 10.1016/j.cmet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Crawford TO, Sladky JT, Hurko O, et al. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann Neurol. 1999;45:337–343. doi: 10.1002/1531-8249(199903)45:3<337::aid-ana9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 20.Quarfordt SH, DeVivo DC, Engel WK, et al. Familial adult-onset proximal spinal muscular atrophy. Report of a family with type II hyperlipoproteinemia. Arch Neurol. 1970;22:541–549. doi: 10.1001/archneur.1970.00480240061009. [DOI] [PubMed] [Google Scholar]
- 21.Dahl DS, Peters HA. Lipid disturbances associated with spiral muscular atrophy. Clinical, electromyographic, histochemical, and lipid studies. Arch Neurol. 1975;32:195–203. doi: 10.1001/archneur.1975.00490450075011. [DOI] [PubMed] [Google Scholar]
- 22.Tein I, Sloane AE, Donner EJ, et al. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: primary or secondary defect(s)? Pediatr Neurol. 1995;12:21–30. doi: 10.1016/0887-8994(94)00100-g. [DOI] [PubMed] [Google Scholar]
- 23.Bowerman M, Beauvais A, Anderson CL, Kothary R. Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum Mol Genet. 2010;19:1468–1478. doi: 10.1093/hmg/ddq021. [DOI] [PubMed] [Google Scholar]
- 24.Butchbach ME, Rose FF, Jr, Rhoades S, et al. Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2010;391:835–840. doi: 10.1016/j.bbrc.2009.11.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowerman M, Murray LM, Beauvais A, et al. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul Disord. 2012;22:263–276. doi: 10.1016/j.nmd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 27.Shanik MH, Xu Y, Skrha J, et al. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care. 2008;31(suppl 2):S262–S268. doi: 10.2337/dc08-s264. [DOI] [PubMed] [Google Scholar]
- 28.Rojas A, Khoo A, Tejedo JR, et al. Islet cell development. Adv Exp Med Biol. 2010;654:59–75. doi: 10.1007/978-90-481-3271-3_4. [DOI] [PubMed] [Google Scholar]
- 29.Bosco D, Armanet M, Morel P, et al. Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes. 2010;59:1202–1210. doi: 10.2337/db09-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabrera O, Berman DM, Kenyon NS, et al. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A. 2006;103:2334–2339. doi: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dodart JC, Mathis C, Bales KR, et al. Early regional cerebral glucose hypometabolism in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Neurosci Lett. 1999;277:49–52. doi: 10.1016/s0304-3940(99)00847-2. [DOI] [PubMed] [Google Scholar]
- 32.de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes— evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 34.Stefan M, Simmons RA, Bertera S, et al. Global deficits in development, function, and gene expression in the endocrine pancreas in a deletion mouse model of Prader-Willi syndrome. Am J Physiol Endocrinol Metab. 2011;300:E909–E922. doi: 10.1152/ajpendo.00185.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan H, Gu W, Yang J, et al. Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J Pharmacol Exp Ther. 2009;329:102–111. doi: 10.1124/jpet.108.147009. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z, Kim W, Chen Z, et al. Insulin and glucagon regulate pancreatic alpha-cell proliferation. PLoS One. 2011;6:e16096. doi: 10.1371/journal.pone.0016096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pick A, Clark J, Kubstrup C, et al. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998;47:358–364. doi: 10.2337/diabetes.47.3.358. [DOI] [PubMed] [Google Scholar]
- 38.Kim A, Miller K, Jo J, et al. Islet architecture: a comparative study. Islets. 2009;1:129–136. doi: 10.4161/isl.1.2.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zochodne DW, Ramji N, Toth C. Neuronal targeting in diabetes mellitus: a story of sensory neurons and motor neurons. Neuroscientist. 2008;14:311–318. doi: 10.1177/1073858408316175. [DOI] [PubMed] [Google Scholar]
- 40.Ramji N, Toth C, Kennedy J, Zochodne DW. Does diabetes mellitus target motor neurons? Neurobiol Dis. 2007;26:301–311. doi: 10.1016/j.nbd.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Sinnreich M, Sorenson EJ, Klein CJ. Neurologic course, endocrine dysfunction and triplet repeat size in spinal bulbar muscular atrophy. Can J Neurol Sci. 2004;31:378–382. doi: 10.1017/s0317167100003486. [DOI] [PubMed] [Google Scholar]
- 43.Barris RW. Hyperinsulinism and neuromuscular disorders; a consideration of the association of pancreatic adenoma with wasting states. Calif Med. 1953;78:224–226. [PMC free article] [PubMed] [Google Scholar]
- 44.Takeda S, Sato N, Uchio-Yamada K, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Cola G, Cool MH, Accili D. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J Clin Invest. 1997;99:2538–2544. doi: 10.1172/JCI119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ranke MB. Insulin-like growth factor-I treatment of growth disorders, diabetes mellitus and insulin resistance. Trends Endocrinol Metab. 2005;16:190–197. doi: 10.1016/j.tem.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 47.Bosch-Marce M, Wee CD, Martinez TL, et al. Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum Mol Genet. 2011;20:1844–1853. doi: 10.1093/hmg/ddr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stiles BL, Kuralwalla-Martinez C, Guo W, et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol. 2006;26:2772–2781. doi: 10.1128/MCB.26.7.2772-2781.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ning K, Drepper C, Valori CF, et al. PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum Mol Genet. 2010;19:3159–3168. doi: 10.1093/hmg/ddq226. [DOI] [PubMed] [Google Scholar]
- 50.Avila AM, Burnett BG, Taye AA, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun C, Zhou J. Trichostatin A improves insulin stimulated glucose utilization and insulin signaling transduction through the repression of HDAC2. Biochem Pharmacol. 2008;76:120–127. doi: 10.1016/j.bcp.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 52.Narver HL, Kong L, Burnett BG, et al. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64:465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rivellese AA, De Natale C, Lilli S. Type of dietary fat and insulin resistance. Ann N Y Acad Sci. 2002;967:329–335. doi: 10.1111/j.1749-6632.2002.tb04288.x. [DOI] [PubMed] [Google Scholar]