

Abstract
The advent of genomic analyses has revolutionized the study of human health. Infectious disease research in particular has experienced an explosion of bacterial genomic, transcriptomic, and proteomic data complementing the phenotypic methods employed in traditional bacteriology. Together, these techniques have revealed novel virulence determinants in numerous pathogens and have provided information for potential chemotherapeutics. The bacterial pathogen, Helicobacter pylori, has been recognized as a class 1 carcinogen and contributes to chronic inflammation within the gastric niche. Genomic analyses have uncovered remarkable coevolution between the human host and H. pylori. Perturbation of this coevolution results in dysregulation of the host-pathogen interaction, leading to oncogenic effects. This review discusses the relationship of H. pylori with the human host and environment and the contribution of each of these factors to disease progression, with an emphasis on features that have been illuminated by genomic tools.
1. Introduction
Helicobacter pylori is a Gram negative, spiral-shaped epsilonproteobacterium that colonizes half of the world's human population [1, 2]. H. pylori is the dominant microorganism within the gastric niche and chronic infection with this pathogen is associated with increased risk for numerous negative disease outcomes including gastritis, peptic and duodenal ulcer, dysplasia, neoplasia, gastric B-cell lymphoma of mucosal-associated lymphoid tissue (MALT lymphoma), and invasive gastric adenocarcinoma [3]. H. pylori persists in the gastric niche despite a robust immune response to infection, indicating that this pathogen has evolved elaborate mechanisms to evade both innate and adaptive arms of the human immune system [4].
H. pylori typically colonizes the human stomach for years or even decades, often without adverse consequences [5]. Recent evidence indicates that there are health benefits associated with H. pylori colonization including protection from allergic airway disease, gastroesophageal reflux disease, Barrett's esophagus, esophageal adenocarcinoma, diarrheal disease, and obesity, implying that the relationship between H. pylori and its human host is complex and dynamic [6–8]. Conversely, numerous factors have been identified that can contribute to the development of negative outcomes with respect to H. pylori infection [9]. Together, these can be clustered into a triad of risk factors including host, pathogen, and environmental features that interact to promote disease progression (Figure 1). In recent years, epidemiologic studies, paired with genomic analyses, have shed light on specific interactions that are associated with increased risk of disease outcomes.
Figure 1.
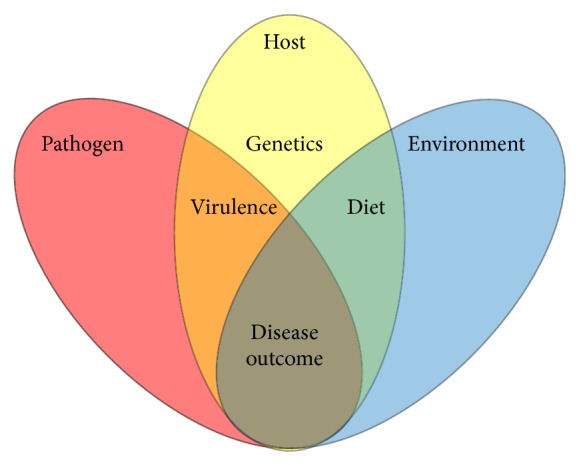
Model of factors influencing H. pylori-related disease outcome. Host genetics, environmental factors, and bacterial strain differences in virulence properties can all contribute to disease progression and increased risk of negative outcomes.
2. Evolution of H. pylori and Geographic Distribution of Strains
H. pylori is an ancient organism that has been prevalent within human populations for over 60,000 years [10]. Certain geographic areas, such as the Latin American Andes Mountain region, have high H. pylori infection rates and very high gastric cancer incidence, characteristics that coincide with low socioeconomic standards [11]. Interestingly, in other regions of the globe with similar socioeconomic conditions, including Africa, India, Thailand, Bangladesh, Pakistan, Iran, Israel, Malaysia, and Saudi Arabia, infection rates are high, but gastric cancer incidence is relatively low [12–14]. These are collectively referred to as “enigmas” within the published literature because the molecular mechanisms behind these differences remain largely obscure.
Genomics tools including whole genome sequencing, restriction fragment length polymorphism (RFLP) genome mapping, and analytical methods, such as maximum likelihood analysis and multilocus sequence typing (MLST), are enhancing the molecular epidemiological methods currently used to study H. pylori pathogenesis [15]. There is an impressive amount of genetic diversity between clinical isolates of H. pylori which is driven by a high mutation rate, frequent recombination events, and random genetic drift as well as positive Darwinian selection and fixation of base substitutions [16]. As human populations migrated across the globe their endemic H. pylori strains diverged alongside them leading to phylogeographic differentiation of this pathogen within human populations that can be classified into European, Amerindian, Asian, and African subgroups [17]. Frequently the phylogeographic origin of an H. pylori strain dictates specific host-adaptive responses through alterations in virulence factor expression. For example, European strains of H. pylori are frequently reported to have elevated virulence when compared to African strains, a characteristic that could explain the “African Enigma” [18]. A better understanding of the phylogenetic relationships between H. pylori strains could reveal novel mechanisms of virulence. Specifically, variations have been analyzed by MLST of housekeeping genes (atpA, efp, ppa, mutY, ureI, trpC, and yphC) to illuminate the genetic origins of H. pylori strains. These techniques have yielded results that have mapped the migration of humans in antiquity out of Africa, across Europe, through Asia, and into the Americas [19]. These analyses also suggest that H. pylori and human coevolution have been perturbed in some geographic areas. For example, H. pylori in India shares common ancestry with European H. pylori strains, indicating a possible acquisition of these strains during colonization by European imperial forces [20]. Conversely, MLST analyses of genomes of H. pylori from native Peruvians suggest that Amerindian strains of H. pylori persisted in these populations in the face of competition from Spanish H. pylori strains. It is likely that the Amerindian strains endemic to native Peruvians acquired Western isotypes of the cag-pathogenicity island, a European-derived virulence factor, resulting in a competitive advantage conferred to the Peruvian strains [21].
MLST analysis of housekeeping genes can provide insight into phylogeographic differentiation of these loci. Complementary to these techniques, investigations into virulence factors have shown that by evaluating both synonymous and nonsynonymous nucleotide substitutions within the coding region, positive selection for amino acid diversity can be determined. These changes in amino acid sequence can be associated with increased risk for peptic ulcer disease and can also be correlated with variations in geographic origin (Western or Asian) [22].
Besides undergoing phylogeographic differentiation, genomic analyses have revealed that H. pylori has, like other obligate human pathogens including Chlamydia trachomatis and Mycoplasma pneumoniae, undergone reductive evolution by reducing both the number of open reading frames (coding region sequences) and the total size of its genome [23–25]. This likely occurred as a consequence of its coevolution within the human host which provides a specialized niche for bacterial colonization and proliferation and consequently reduces the need to maintain genes involved in vital processes such as macronutrient synthesis and acquisition [26]. These findings underscore the importance of utilizing genomic tools to determine the “core genome” within H. pylori to better understand the basic metabolic requirements for prokaryotic life.
3. H. pylori Genomic Flexibility and Genetic Regulation
H. pylori exhibits unusual genetic flexibility and it is hypothesized that the variability within the genome could potentially account for the organism's ability to adapt to the dynamic environment within the host gastric niche, facilitating chronic colonization. These adaptations include both reversible and irreversible changes to the genome as well as regulatory mechanisms that modulate gene expression [27]. Analyses of numerous whole genome sequences indicate that H. pylori has evolved clusters of genes within genomic islands that harbor distinct areas of variability, termed “plasticity zones.” These plasticity zones are likely involved in horizontal gene transfer, a discovery that is supported by the presence of short conserved integration motifs and coding regions that are orthologous to integrating conjugative elements (ICEs). It is postulated that these ICEs, which are prevalent and widely distributed among all sequenced H. pylori strains, provide a fitness advantage to the bacterium by aiding genetic recombination events, which could ultimately promote immune evasion and increased ability to colonize, as well as other currently unappreciated alterations which would provide a selective advantage for the organism [27]. This hypothesis is supported by recent research which indicates that genetic modifications occur at a rate that is 10 times faster during acute or early infection when H. pylori is initially encountering the host's immune response than during chronic infection. These results also indicate that H. pylori mutation rates are far higher than any other bacteria currently assessed and that many of the mutations occur in genes that encode putative outer membrane proteins [28]. These proteins are implicated in host-pathogen interaction and transmission between hosts and are also likely targets for the adaptive immune response [29]. Thus, modification of these key surface-exposed antigens would likely alter these interactions to promote the establishment of chronic H. pylori infection and therefore the genes encoding these proteins experience the greatest selective pressure.
In addition to mapping genome sequence and structure, next-generation sequencing technology can now profile genome function by determining how and when genes are expressed and the regulatory networks that govern these expression subunits. Global transcriptomic analysis has revealed complexity in the riboregulation of H. pylori gene expression. H. pylori employs approximately 60 small RNAs as well as a surfeit of transcriptional start sites within operons, indicating uncoupling of polycistronic transcriptional regulation. Interestingly, about 5% of the open reading frames encode leaderless messages that lack the canonical translational initiation signals such as the Shine-Dalgarno sequence [30]. Comparison of these features to other Epsilonproteobacteria, such as Campylobacter jejuni, reveals a lack of conservation of operon organization and riboregulatory elements. Together, these results indicate that transcriptional rewiring occurs differently in C. jejuni and H. pylori to compensate for the genetic variations that occurred after these two species diverged from a common ancestor [31].
Epigenetics has emerged as an important area of study to better understand the subtle and complex nature of gene regulation. Epigenetic modifications such as DNA methylation carried out by DNA methyltransferases can have drastic effects on both genome architecture and gene expression. H. pylori encodes numerous DNA methyltransferases and single-molecule real-time sequence analyses of the methylome of closely related strains revealed great diversity in the methylation of target sequences. This result is attributed to variation in the specificity of the methyltransferase domain as well as variation within the methylation target sequence [32, 33]. Together, these features contribute to changes in gene regulation including modulating expression of flgE (encoding a flagellar component), cagY (encoding a type IV secretion component), and ureC (encoding a subunit of the urease complex) [32]. These results underscore the importance of expression dynamics and the necessity to identify the numerous regulators responsible for mediating complex interactions between the host and pathogen to further our understanding of chronic infectious processes.
4. Toxins Encoded in the H. pylori Genome
H. pylori exerts an immunomodulatory effect within this host tissue as a strategy to circumnavigate both innate and adaptive immune systems. Two toxins, the vacuolating cytotoxin (VacA) and the cancer-associated gene toxin (CagA), have been implicated in perturbing host immunological responses [34–36]. VacA is a pore-forming toxin secreted by H. pylori that causes a wide variety of alterations in host cell biology including cell vacuolation, autophagy, inhibition of T-cell proliferation, and induction of programmed necrosis [36–38]. The gene that encodes VacA has been shown to have variation between strains and the s1m1 variant is associated with the greatest risk for development of diseases including precancerous lesions and intestinal metaplasia [39, 40]. Sequencing data paired with epidemiological studies have revealed that polymorphisms within the intermediate region (i1-type) of VacA are associated with increased risk of peptic ulcer disease [40]. Additionally, evaluation of the molecular evolution of VacA reveals that positive selection has modified the sequence encoding VacA in a process that is independent of the evolution of the core genome [41]. This type of separate positive selection is also observed in the gene encoding the major surface antigen, CagA [41]. CagA is a cytotoxin that is translocated into host cells by the cag-type IV secretion system (cag-T4SS), a macromolecular nanomachine encoded by several genes within the cag pathogenicity island (cag-PAI) [42]. This T4SS is assembled at the host-pathogen interface (Figure 2) and is implicated in secretion of peptidoglycan and the aforementioned cytotoxin, CagA which results in numerous changes to host cell biology including upregulation of proinflammatory cytokines, alteration of actin cytoskeleton, disruption of metal homeostasis, and aberrant cell signaling [42–46]. Evaluation of cagA sequences has revealed that amino acid polymorphisms within the Glu-Pro-Ile-Tyr-Ala (EPIYA) segments contribute significantly to carcinogenesis [47]. This is interesting, because these regions are phosphorylated by host tyrosine kinases and are involved in the modulation of host signal transduction events. Together, these studies have revealed that polymorphisms within the coding regions for the cytotoxins VacA and CagA, specifically the i1 intermediate region and the EPIYA motif, respectively, can contribute to increased risk for development of gastric diseases [48, 49].
Figure 2.
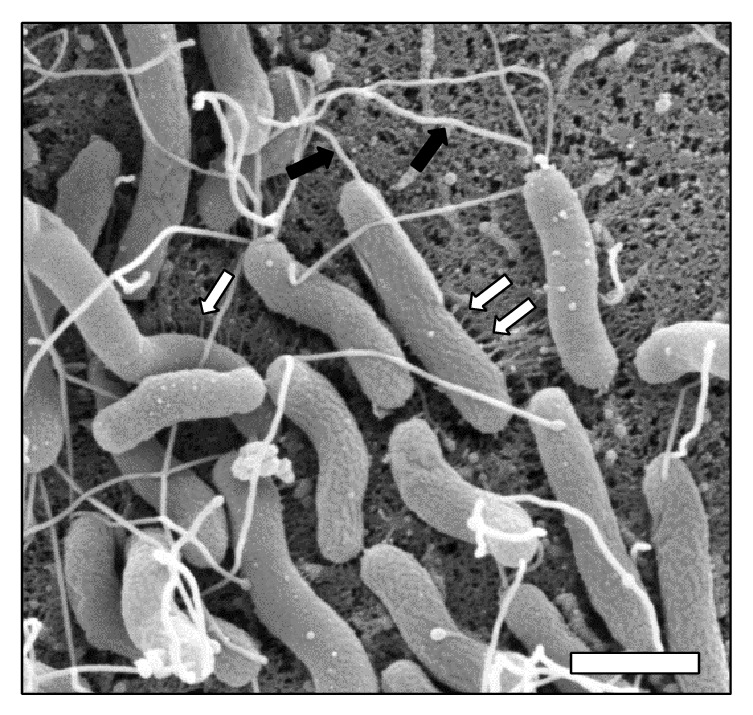
High resolution scanning electron micrograph of H. pylori in contact with human gastric epithelial cells. H. pylori virulence factors including flagella (black arrows) and cag-T4SS pili (white arrows) are present on the bacterial cell surface during host-pathogen interaction. Flagella aid in cell motility through the mucus layer to penetrate host tissues. The cag-T4SS pili induce proinflammatory and oncogenic cellular responses. Magnification bar indicates 1 μm.
5. Expression of H. pylori Virulence Factors
Successful chronic infection of a vertebrate host is a delicate balance between host and pathogen. The pathogen expresses virulence factors to (1) elicit an immune response which eliminates resident microbiota, (2) acquire nutrients, (3) permit bacteria to penetrate host tissues, and (4) allow bacteria to turn the host tissue into a replicative niche. Bacteria have evolved to tightly control the expression of virulence factors to promote successful colonization. H. pylori has evolved to respond to environmental stimuli, such as gastric acid, with a repertoire of regulatory elements such as two-component systems comprised of a sensor kinase (ArsS) and a response regulator (ArsR), which ultimately modulates the expression of virulence genes involved in motility and cag-T4SS function [50]. Motility of H. pylori cells through the gastric mucosa is accomplished by utilization of numerous lophotrichous flagella (Figure 2). Once H. pylori penetrates the gastric mucosa, it interacts with host epithelial cells and elaborates cag-T4SS pili (Figure 2). The cag-T4SS encoded within the cag-PAI is organized into multiple overlapping operons that are likely coregulated as well as divergently regulated by numerous types of stimuli found within the gastric niche [51]. Besides pH, H. pylori senses diverse environmental cues including iron, nickel, cobalt, and zinc and responds to these cues by altering virulence expression [52]. For example, in conditions of low iron availability, H. pylori increases cag-T4SS activity and pilus biogenesis as well as expression of numerous flagellar components [53]. Conversely, in conditions of low zinc availability, H. pylori represses cag-T4SS machinery [54]. These data indicate that environmental cues present in the host can alter the carcinogenic potential of H. pylori and increase the risk of negative disease outcomes.
6. Dietary Contribution
Bacteria respond to their environment and alter virulence factor expression accordingly as described above. One of the numerous environmental stimuli that H. pylori encounters in the gastric niche are molecules derived from the host diet. Numerous dietary habits such as iron deficiency, salt preference, nitrite, protein, and fat intake have been epidemiologically linked with increased risk of H. pylori-related disease [55]. However, precious few of these dietary factors have been recapitulated in an animal model of H. pylori infection with some exceptions. Dietary iron deficiency has been correlated with increased risk of H. pylori-related disease progression [56]. In a rodent model of H. pylori infection, animals fed a low-iron diet exhibited higher incidences of gastric cancer compared to animals fed an iron-replete diet. Proteomics analyses of strains of H. pylori derived from these animals revealed that low-iron conditions induced expression of numerous virulence factors including flagellar proteins, a VacA paralog, CagA, HopQ, and urease [53]. Concomitantly, conditions of low iron availability increased H. pylori CagA T4SS induction of host proinflammatory IL-8 secretion, a result that correlated with the increase in gastric inflammation in animals fed a low-iron diet. Similarly, dietary salt intake has been associated with increased risk of gastric disease. In a rodent model of H. pylori infection, animals fed a high-salt diet exhibited higher incidences of gastric cancer and inflammation compared to animals fed a regular salt diet. Analysis of bacterial and host transcripts revealed that cagA and IL-1β, respectively, were highly upregulated in H. pylori-infected animals in response to dietary salt intake, a result that correlated with disease phenotypes [57]. Thus, variations in dietary ion consumption could lead to changes in bacterial virulence factor expression that ultimately alter disease progression.
7. Host Factors Associated with Disease
In addition to the numerous bacterial factors that have been demonstrated to affect disease outcome, genomic approaches have revealed host factors that are correlated with H. pylori-associated disease manifestation. MALT lymphoma has been characterized by microarray analyses which revealed pronounced infiltration of gastric tissue with CD4(+) T cells expressing CD28 and CD69 as well as an increased expression of calprotectin [58]. These results indicate that MALT lymphoma tumor cell proliferation is driven by Th2-polarized activated T cells and innate immune cells. Complementing this study, numerous subsequent studies have implicated Th1, Th17, and the host neutrophil-associated protein calprotectin as host molecules that are induced in response to H. pylori infection and associated with gastric inflammation [59]. Interestingly, whole genome expression profiles and sequencing revealed that polymorphisms in IL-1β and the IL-8 promoter region can increase the risk of H. pylori-related diseases such as gastric cancer [62, 63]. Both IL-1β and IL-8 are powerful proinflammatory cytokines implicated in gastric inflammation and carcinogenesis in response to H. pylori. There are numerous reports supporting that the dysregulation of the host-pathogen interaction, initiated through promotion of inflammation, demolishes resident microbiota to tip the balance in favor of a pathogen, ultimately resulting in disease progression [64, 65]. Aligned with these, genomic analyses have revealed that H. pylori has coevolved with its human host to promote less severe gastric lesions, and that disruption of the coevolution (introduction of nonnative strains of H. pylori into a human host) contributes to differences in disease severity [66, 67]. Together, these host-specific factors should be taken into account when crafting models of infectious disease risk.
8. Antibiotic Resistance Mechanisms
Once chronic infection is established, clearance of the colonizing bacteria requires administration of antimicrobial chemotherapeutics. The standard treatment for H. pylori infection is triple therapy with a proton pump inhibitor (PPI), amoxicillin, and clarithromycin, but current treatment paradigms in populations predominantly colonized with resistant strains favor quadruple therapy with the addition of metronidazole [68]. Antimicrobial resistance is a widespread problem among bacterial pathogens and recent evidence indicates the emergence of antibiotic-resistant strains of H. pylori in clinical samples. In an effort to better understand the molecular mechanisms that govern antimicrobial resistance, whole genome sequencing has been performed to characterize patterns of genotypic changes that can be correlated with phenotypic changes in antibiotic susceptibility [69]. The results indicate that resistance to clarithromycin can be conferred in H. pylori via mutations in 23S rRNA genes as well as modifications within genes that encode the TolC-family of efflux pumps [70]. Additionally, resistance to amoxicillin was conferred through mutations within outer membrane protein coding regions (hopC, hofH, and hefC) and penicillin-binding proteins (pbp1 and pbp2) [71]. Metronidazole resistance has also emerged through frameshift and nonsense mutations in the rdxA or frxA coding regions or alternately in the ferric uptake regulator (fur) locus, rendering quadruple therapy ineffective in many cases [72]. In these instances where canonical triple or quadruple therapies fail, second line drugs such as tetracycline and fluoroquinolones are often employed. Unfortunately, both tetracycline and fluoroquinolone resistant isolates of H. pylori have emerged. Sequencing reveals that 16S rRNA substitutions confer tetracycline resistance, while gyrA mutation at codon 87 or 91 results in fluoroquinolone resistance [72, 73]. Collectively, these data demonstrate that H. pylori has the capacity to modify its genome to circumnavigate the selective pressure of antimicrobial chemotherapeutics. Understanding the genetic elements present in antibiotic-resistant strains of H. pylori could provide novel targets for new antimicrobial strategies that will restore the utility of existing therapies.
9. Conclusions
H. pylori is an ancient pathogen that has evolved to persistently colonize the human gastric niche. Infection with this pathogen leads to diverse outcomes ranging from asymptomatic gastritis to invasive adenocarcinoma. Factors that modulate risk fall into three major categories including pathogen attributes, host genetics, and environmental stimuli. Genomic tools have revealed numerous aspects of the host-pathogen interaction, including dietary ion consumption, expression of virulence factors such as toxins, secretion systems, and flagella, induction of proinflammatory signaling, and cytokine secretion (IL-1 and IL-8) which ultimately leads to adaptive immune responses including Th1 and Th17 expansion (Figure 3). These events at the host-pathogen interface could be exploited to influence the outcome of H. pylori-related diseases. Future work is required to develop chemotherapeutic strategies tailored to manipulate these interactions and tip the balance in favor of the health of the human host.
Figure 3.

Model of H. pylori-host interaction in vivo. (A) H. pylori encounters numerous ions in the gastric niche and utilizes flagella to penetrate the mucus layer and reach the gastric epithelia. (B) H. pylori secretes VacA and CagA cytotoxins, causing changes in host cell biology. (C) The adaptive immune response as a consequence of H. pylori infection is skewed to expand Th1 and Th17 populations.
Acknowledgments
This work has been funded primarily by a Career Development Award IK2BX001701 (to Jennifer A. Gaddy) from the Office of Medical Research, Department of Veterans Affairs. Core Services including use of the Cell Imaging Shared Resource were performed through both Vanderbilt University Medical Center's Digestive Disease Research Center supported by NIH Grant P30DK058404 Core Scholarship and Vanderbilt Institute for Clinical and Translational Research Program supported by the National Center for Research Resources, Grant UL1 RR024975-01, and the National Center for Advancing Translational Sciences, Grant 2 UL1 TR000445-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interests
The authors declare that this work was conducted in the absence of any commercial or financial relationships that could create a potential conflict of interests.
Authors' Contribution
Kathryn P. Haley and Jennifer A. Gaddy reviewed the literature and wrote and revised the paper.
References
- 1.Stolte M. Helicobacter pylori gastritis and gastric MALT-lymphoma. The Lancet. 1992;339(8795):745–746. doi: 10.1016/0140-6736(92)90645-j. [DOI] [PubMed] [Google Scholar]
- 2.Pernitzsch S. R., Sharma C. M. Transcriptome complexity and riboregulation in the human pathogen Helicobacter pylori . Frontiers in Cellular and Infection Microbiology. 2012;2, article 14 doi: 10.3389/fcimb.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makola D., Peura D. A., Crowe S. E. Helicobacter pylori infection and related gastrointestinal diseases. Journal of Clinical Gastroenterology. 2007;41(6):548–558. doi: 10.1097/mcg.0b013e318030e3c3. [DOI] [PubMed] [Google Scholar]
- 4.Lewis N. D., Asim M., Barry D. P., et al. Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. The Journal of Immunology. 2011;186(6):3632–3641. doi: 10.4049/jimmunol.1003431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pacifico L., Anania C., Osborn J. F., Ferraro F., Chiesa C. Consequences of Helicobacter pylori infection in children. World Journal of Gastroenterology. 2010;16(41):5181–5194. doi: 10.3748/wjg.v16.i41.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnold I. C., Dehzad N., Reuter S., et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. Journal of Clinical Investigation. 2011;121(8):3088–3093. doi: 10.1172/jci45041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kandulski A., Malfertheiner P. Helicobacter pylori and gastroesophageal reflux disease. Current Opinion in Gastroenterology. 2014;30(4):402–407. doi: 10.1097/mog.0000000000000085. [DOI] [PubMed] [Google Scholar]
- 8.Cover T. L., Blaser M. J. Helicobacter pylori in health and disease. Gastroenterology. 2009;136(6):1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atherton J. C. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annual Review of Pathology. 2006;1:63–96. doi: 10.1146/annurev.pathol.1.110304.100125. [DOI] [PubMed] [Google Scholar]
- 10.Moodley Y., Linz B., Bond R. P., et al. Age of the association between Helicobacter pylori and man. PLoS Pathogens. 2012;8(5) doi: 10.1371/journal.ppat.1002693.e1002693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Correa P., Piazuelo M. B. Natural history of Helicobacter pylori infection. Digestive and Liver Disease. 2008;40(7):490–496. doi: 10.1016/j.dld.2008.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellmig S., Hampe J., Schreiber S. Helicobacter pylori infection in Africa and Europe: enigma of host genetics. Gut. 2003;52(12):p. 1799. doi: 10.1136/gut.52.12.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misra V., Pandey R., Misra S. P., Dwivedi M. Helicobacter pylori and gastric cancer: Indian enigma. World Journal of Gastroenterology. 2014;20(6):1503–1509. doi: 10.3748/wjg.v20.i6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hussein N. R. Helicobacter pylori and gastric cancer in the Middle East: a new enigma? World Journal of Gastroenterology. 2010;16(26):3226–3234. doi: 10.3748/wjg.v16.i26.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki R., Shiota S., Yamaoka Y. Molecular epidemiology, population genetics, and pathogenic role of Helicobacter pylori . Infection, Genetics and Evolution. 2012;12(2):203–213. doi: 10.1016/j.meegid.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres-Morquecho A., Giono-Cerezo S., Camorlinga-Ponce M., Vargas-Mendoza C. F., Torres J. Evolution of bacterial genes: evidences of positive Darwinian selection and fixation of base substitutions in virulence genes of Helicobacter pylori . Infection, Genetics and Evolution. 2010;10(6):764–776. doi: 10.1016/j.meegid.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Camorlinga-Ponce M., Perez-Perez G., Gonzalez-Valencia G., et al. Helicobacter pylori genotyping from american indigenous groups shows novel amerindian vacA and cagA alleles and Asian, African and European admixture. PLoS ONE. 2011;6(11) doi: 10.1371/journal.pone.0027212.e27212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheh A., Chaturvedi R., Merrell D. S., Correa P., Wilson K. T., Fox J. G. Phylogeographic origin of Helicobacter pylori determines host-adaptive responses upon coculture with gastric epithelial cells. Infection and Immunity. 2013;81(7):2468–2477. doi: 10.1128/iai.01182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mane S. P., Dominguez-Bello M. G., Blaser M. J., et al. Host-interactive genes in Amerindian Helicobacter pylori diverge from their old world homologs and mediate inflammatory responses. Journal of Bacteriology. 2010;192(12):3078–3092. doi: 10.1128/jb.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devi S. M., Ahmed I., Francalacci P., et al. Ancestral European roots of Helicobacter pylori in India. BMC Genomics. 2007;8, article 184 doi: 10.1186/1471-2164-8-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devi S. M., Ahmed I., Khan A. A., et al. Genomes of Helicobacter pylori from native Peruvians suggest admixture of ancestral and modern lineages and reveal a western type cag-pathogenicity island. BMC Genomics. 2006;7, article 191 doi: 10.1186/1471-2164-7-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao P., Kerry J. L., Blaser M. J., Cover T. L. Analysis of hopQ alleles in East Asian and Western strains of Helicobacter pylori . FEMS Microbiology Letters. 2005;251(1):37–43. doi: 10.1016/j.femsle.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 23.Dong Q.-J., Wang L.-L., Tian Z.-B., Yu X.-J., Jia S.-J., Xuan S.-Y. Reduced genome size of Helicobacter pylori originating from East Asia. World Journal of Gastroenterology. 2014;20(19):5666–5671. doi: 10.3748/wjg.v20.i19.5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie G., Bonner C. A., Jensen R. A. Dynamic diversity of the tryptophan pathway in chlamydiae: reductive evolution and a novel operon for tryptophan recapture. Genome Biology. 2002;3(9) doi: 10.1186/gb-2002-3-9-research0051.research0051.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meseguer M. A., Álvarez A., Rejas M. T., Sánchez C., Pérez-Díaz J. C., Baquero F. Mycoplasma pneumoniae: a reduced-genome intracellular bacterial pathogen. Infection, Genetics and Evolution. 2003;3(1):47–55. doi: 10.1016/s1567-1348(02)00151-x. [DOI] [PubMed] [Google Scholar]
- 26.Toft C., Andersson S. G. E. Evolutionary microbial genomics: insights into bacterial host adaptation. Nature Reviews Genetics. 2010;11(7):465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- 27.Fischer W., Breithaupt U., Kern B. I., Smith S. I., Spicher C., Haas R. A comprehensive analysis of Helicobacter pylori plasticity zones reveals that they are integrating conjugative elements with intermediate integration specificity. BMC Genomics. 2014;15, article 310 doi: 10.1186/1471-2164-15-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linz B., Windsor H. M., McGraw J. J., et al. A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nature Communications. 2014;5(article 4165) doi: 10.1038/ncomms5165. [DOI] [PubMed] [Google Scholar]
- 29.Linz B., Windsor H. M., Gajewski J. P., et al. Helicobacter pylori genomic microevolution during naturally occurring transmission between adults. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0082187.e82187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma C. M., Hoffmann S., Darfeuille F., et al. The primary transcriptome of the major human pathogen Helicobacter pylori . Nature. 2010;464(7286):250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 31.Porcelli I., Reuter M., Pearson B. M., Wilhelm T., van Vliet A. H. M. Parallel evolution of genome structure and transcriptional landscape in the Epsilonproteobacteria. BMC Genomics. 2013;14(1, article 616) doi: 10.1186/1471-2164-14-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furuta Y., Namba-Fukuyo H., Shibata T. F., et al. Methylome diversification through changes in DNA methyltransferase sequence specificity. PLoS Genetics. 2014;10(4) doi: 10.1371/journal.pgen.1004272.e1004272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krebes J., Morgan R. D., Bunk B., et al. The complex methylome of the human gastric pathogen Helicobacter pylori . Nucleic Acids Research. 2014;42(4):2415–2432. doi: 10.1093/nar/gkt1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cover T. L., Blaser M. J. Purification and characterization of the vacuolating toxin from Helicobacter pylori . The Journal of Biological Chemistry. 1992;267(15):10570–10575. [PubMed] [Google Scholar]
- 35.Tummuru M. K. R., Cover T. L., Blaser M. J. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infection and Immunity. 1993;61(5):1799–1809. doi: 10.1128/iai.61.5.1799-1809.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundrud M. S., Torres V. J., Unutmaz D., Cover T. L. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(20):7727–7732. doi: 10.1073/pnas.0401528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radin J. N., González-Rivera C., Ivie S. E., McClain M. S., Cover T. L. Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infection and Immunity. 2011;79(7):2535–2543. doi: 10.1128/iai.01370-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yahiro K., Satoh M., Nakano M., et al. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. Journal of Biological Chemistry. 2012;287(37):31104–31115. doi: 10.1074/jbc.m112.387498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Höcker M., Hohenberger P. Helicobacter pylori virulence factors—one part of a big picture. The Lancet. 2003;362(9391):1231–1233. doi: 10.1016/s0140-6736(03)14547-3. [DOI] [PubMed] [Google Scholar]
- 40.Jones K. R., Jang S., Chang J. Y., et al. Polymorphisms in the intermediate region of VacA impact Helicobacter pylori-induced disease development. Journal of Clinical Microbiology. 2011;49(1):101–110. doi: 10.1128/jcm.01782-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gangwer K. A., Shaffer C. L., Suerbaum S., Lacy D. B., Cover T. L., Bordenstein S. R. Molecular evolution of the Helicobacter pylori vacuolating toxin gene vacA . Journal of Bacteriology. 2010;192(23):6126–6135. doi: 10.1128/jb.01081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson E. M., Gaddy J. A., Voss B. J., Hennig E. E., Cover T. L. Genes required for assembly of pili associated with the Helicobacter pylori cag type IV secretion system. Infection and Immunity. 2014;82(8):3457–3470. doi: 10.1128/iai.01640-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutton M. L., Kaparakis-Liaskos M., Turner L., Cardona A., Kwok T., Ferrero R. L. Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-κB-dependent responses and peptidoglycan delivery in epithelial cells. Infection and Immunity. 2010;78(11):4523–4531. doi: 10.1128/iai.00439-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guillemin K., Salama N. R., Tompkins L. S., Falkow S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(23):15136–15141. doi: 10.1073/pnas.182558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tegtmeyer N., Wessler S., Backert S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS Journal. 2011;278(8):1190–1202. doi: 10.1111/j.1742-4658.2011.08035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan S., Noto J. M., Romero-Gallo J., Peek R. M., Jr., Amieva M. R. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathogens. 2011;7(5) doi: 10.1371/journal.ppat.1002050.e1002050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen C. Y., Wang F. Y., Wan H. J., et al. Amino acid polymorphisms flanking the EPIYA-A motif of Helicobacter pylori CagA C-terminal region is associated with gastric cancer in East China: Experience from a single center. Journal of Digestive Diseases. 2013;14(7):358–365. doi: 10.1111/1751-2980.12056. [DOI] [PubMed] [Google Scholar]
- 48.Basso D., Zambon C.-F., Letley D. P., et al. Clinical relevance of Helicobacter pylori cagA and vacA Gene Polymorphisms. Gastroenterology. 2008;135(1):91–99. doi: 10.1053/j.gastro.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 49.McClain M. S., Shaffer C. L., Israel D. A., Peek R. M., Jr., Cover T. L. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics. 2009;10, article 13 doi: 10.1186/1471-2164-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scott D. R., Marcus E. A., Wen Y., Oh J., Sachs G. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(17):7235–7240. doi: 10.1073/pnas.0702300104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ta L. H., Hansen L. M., Sause W. E., et al. Conserved transcriptional unit organization of the cag pathogenicity island among Helicobacter pylori strains. Frontiers in Cellular and Infection Microbiology. 2012;2, article 46 doi: 10.3389/fcimb.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Contreras M., Thiberge J. M., Mandrand-Berthelot M. A., Labigne A. Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori . Molecular Microbiology. 2003;49(4):947–963. doi: 10.1046/j.1365-2958.2003.03621.x. [DOI] [PubMed] [Google Scholar]
- 53.Noto J. M., Gaddy J. A., Lee J. Y., et al. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. The Journal of Clinical Investigation. 2013;123(1):479–492. doi: 10.1172/jci64373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaddy J. A., Radin J. N., Loh J. T., et al. The host protein calprotectin modulates the Helicobacter pylori cag type IV secretion system via zinc sequestration. PLoS Pathogens. 2014;10(10) doi: 10.1371/journal.ppat.1004450.e1004450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y.-W., Eom S.-Y., Yim D.-H., et al. Evaluation of the relationship between dietary factors, CagA-positive Helicobacter pylori infection, and RUNX3 promoter hypermethylation in gastric cancer tissue. World Journal of Gastroenterology. 2013;19(11):1778–1787. doi: 10.3748/wjg.v19.i11.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pra D., Franke S. I. R., Henriques J. A. P., Fenech M. A possible link between iron deficiency and gastrointestinal carcinogenesis. Nutrition and Cancer. 2009;61(4):415–426. doi: 10.1080/01635580902803701. [DOI] [PubMed] [Google Scholar]
- 57.Gaddy J. A., Radin J. N., Loh J. T., et al. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infection and Immunity. 2013;81(6):2258–2267. doi: 10.1128/iai.01271-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mueller A., O'Rourke J., Chu P., et al. The role of antigenic drive and tumor-infiltrating accessory cells in the pathogenesis of helicobacter-induced mucosa-associated lymphoid tissue lymphoma. The American Journal of Pathology. 2005;167(3):797–812. doi: 10.1016/s0002-9440(10)62052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scott Algood H. M., Gallo-Romero J., Wilson K. T., Peek R. M., Jr., Cover T. L. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunology and Medical Microbiology. 2007;51(3):577–586. doi: 10.1111/j.1574-695x.2007.00338.x. [DOI] [PubMed] [Google Scholar]
- 60.Algood H. M., Allen S. S., Washington M. K., Peek R. M., Jr., Miller G. G., Cover T. L. Regulation of gastric B cell recruitment is dependent on IL-17 receptor A signaling in a model of chronic bacterial infection. The Journal of Immunology. 2009;183(9):5837–5846. doi: 10.4049/jimmunol.0901206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leach S. T., Mitchell H. M., Geczy C. L., Sherman P. M., Day A. S. S100 calgranulin proteins S100A8, S100A9 and S100A12 are expressed in the inflamed gastric mucosa of Helicobacter pylori-infected children. Canadian Journal of Gastroenterology. 2008;22(5):461–464. doi: 10.1155/2008/308942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang S. Y., Shen X. Y., Wu C. Y., et al. Analysis of whole genomic expression profiles of Helicobacter pylori related chronic atrophic gastritis with IL-1B-31CC/-511TT genotypes. Journal of Digestive Diseases. 2009;10(2):99–106. doi: 10.1111/j.1751-2980.2009.00367.x. [DOI] [PubMed] [Google Scholar]
- 63.Xue H., Liu J., Lin B., Wang Z., Sun J., Huang G. A meta-analysis of interleukin-8-251 promoter polymorphism associated with gastric cancer risk. PLoS ONE. 2012;7(1) doi: 10.1371/journal.pone.0028083.e28083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Behnsen J., Jellbauer S., Wong C. P., et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40(2):262–273. doi: 10.1016/j.immuni.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sassone-Corsi M., Raffatellu M. A hydrogen boost for Salmonella . Cell Host & Microbe. 2013;14(6):603–604. doi: 10.1016/j.chom.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 66.Kodaman N., Pazos A., Schneider B. G., et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(4):1455–1460. doi: 10.1073/pnas.1318093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kodaman N., Sobota R. S., Mera R., Schneider B. G., Williams S. M. Disrupted human—pathogen co-evolution: a model for disease. Frontiers in Genetics. 2014;5, article 290 doi: 10.3389/fgene.2014.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferreira J., Moss S. F. Current paradigm and future directions for treatment of Helicobacter pylori infection. Current Treatment Options in Gastroenterology. 2014;12(4):373–384. doi: 10.1007/s11938-014-0027-6. [DOI] [PubMed] [Google Scholar]
- 69.Slinger R., Yan L., Chan F., et al. Pyrosequencing assay to rapidly detect clarithromycin resistance mutations in Canadian Helicobacter pylori isolates. Canadian Journal of Gastroenterology. 2009;23(9):609–612. doi: 10.1155/2009/695349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iwamoto A., Tanahashi T., Okada R., et al. Whole-genome sequencing of clarithromycin resistant Helicobacter pylori characterizes unidentified variants of multidrug resistant efflux pump genes. Gut Pathogens. 2014;6(1, article 27) doi: 10.1186/1757-4749-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qureshi N. N., Gallaher B., Schiller N. L. Evolution of amoxicillin resistance of Helicobacter pylori in vitro : characterization of resistance mechanisms. Microbial Drug Resistance. 2014;20(6):509–516. doi: 10.1089/mdr.2014.0019. [DOI] [PubMed] [Google Scholar]
- 72.Alfizah H., Norazah A., Hamizah R., Ramelah M. Resistotype of Helicobacter pylori isolates: the impact on eradication outcome. Journal of Medical Microbiology. 2014;63(5):703–709. doi: 10.1099/jmm.0.069781-0. [DOI] [PubMed] [Google Scholar]
- 73.Choi S. S., Chivers P. T., Berg D. E. Point mutations in Helicobacter pylori's fur regulatory gene that alter resistance to metronidazole, a prodrug activated by chemical reduction. PLoS ONE. 2011;6(3) doi: 10.1371/journal.pone.0018236.e18236 [DOI] [PMC free article] [PubMed] [Google Scholar]