

Abstract
Mesorhizobium loti strain R7A was isolated in 1993 in Lammermoor, Otago, New Zealand from a Lotus corniculatus root nodule and is a reisolate of the inoculant strain ICMP3153 (NZP2238) used at the site. R7A is an aerobic, Gram-negative, non-spore-forming rod. The symbiotic genes in the strain are carried on a 502-kb integrative and conjugative element known as the symbiosis island or ICEMlSymR7A. M. loti is the microsymbiont of the model legume Lotus japonicus and strain R7A has been used extensively in studies of the plant-microbe interaction. This report reveals that the genome of M. loti strain R7A does not harbor any plasmids and contains a single scaffold of size 6,529,530 bp which encodes 6,323 protein-coding genes and 75 RNA-only encoding genes. This rhizobial genome is one of 100 sequenced as part of the DOE Joint Genome Institute 2010 Genomic Encyclopedia for Bacteria and Archaea-Root Nodule Bacteria (GEBA-RNB) project.
Keywords: Root-nodule bacteria, Nitrogen fixation, Symbiosis, Alphaproteobacteria
Introduction
Mesorhizobium loti strain R7A is a reisolate of strain ICMP3513 (International Culture Collection of Microorganisms from Plants, LandCare Research, Auckland, New Zealand). It was isolated from a root nodule taken from a stand of Lotus corniculatus in Lammermoor, Central Otago, New Zealand, inoculated seven years earlier with strain ICMP3153 [1]. Strain ICMP3153 was a recommended inoculant strain for L. corniculatus in New Zealand and is also known as NZP2238 and Lc265Da. In its guise as NZP2238, it was one of the strains used to define the species Rhizobium loti (now Mesorhizobium loti) [2].
Strain R7A contains a 502-kb symbiosis island, also known as ICEMlSymR7A, that was discovered through its ability to transfer from strain ICMP3153 to indigenous nonsymbiotic mesorhizobia at the Lammermoor field site [1,3]. The symbiosis island encodes 414 genes including all of the genes required for Nod factor synthesis, nitrogen fixation and transfer of the island [4]. Transfer of the island occurs via conjugation involving a rolling-circle process. The transferred island integrates into the chromosome of the recipient cell at the sole phenylalanine tRNA gene. Integration of the island is dependent on a P4-type integrase encoded by intS, located 198 bp downstream of the phe-tRNA gene, which acts on an attachment site (attS) on the circular form of the island and a chromosomal attachment site (attB). Integration of the island reconstructs the entire phe-tRNA gene at one end (arbitrarily termed the left end) and forms a 17-bp repeat of the three-prime end of the phe-tRNA gene at the right end of the integrated island [3-5].
M. loti is the microsymbiont of the model legume Lotus japonicus and strain R7A together with the first M. loti strain sequenced, strain MAFF303099 [6], have been used extensively with L. japonicus in studies of the plant-microbe interaction. Studies using R7A have included characterization of the symbiotic role of the vir Type IV secretion system encoded by the strain [7], determination of the requirements for Nod factor decorations [8] and exopolysaccharides [9] for efficient nodulation of various Lotus species, and characterization of genes required for symbiotic nitrogen fixation [10]. The regulation of symbiosis island transfer in strain R7A has also been extensively characterized [11]. Here we present a summary classification and a set of general features for M. loti strain R7A together with the description of the complete genome sequence and annotation.
Classification and general features
Mesorhizobium loti strain R7A is in the order Rhizobiales of the class Alphaproteobacteria. Cells are described as non-sporulating, Gram-negative, non-encapsulated, rods. The rod-shaped form varies in size with dimensions of 0.25-0.5 μm in width and 1–1.5 μm in length (Figure 1 Left and 1 Center). They are moderately fast growing, forming 2 mm diameter colonies within 4 days and have a mean generation time of approximately 6 h when grown in TY broth at 28°C [1]. Colonies on G/RDM agar [12] and half strength Lupin Agar (½LA) [13] are opaque, slightly domed, mucoid with smooth margins (Figure 1 Right).
Figure 1.

Images of Mesorhizobium loti strain R7A using scanning (Left) and transmission (Center) electron microscopy and the appearance of colony morphology on ½LA (Right).
Strains of this organism are able to tolerate a pH range between 4 and 10. Carbon source utilization and fatty acid profiles of M. loti have been described previously [2,14,15]. Minimum Information about the Genome Sequence (MIGS) is provided in Table 1.
Table 1.
Classification and general features of Mesorhizobium loti strain R7A according to the MIGS recommendations [16,17]
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| |
Current classification |
Domain Bacteria |
TAS [17] |
| Phylum Proteobacteria |
TAS [18] |
||
| Class Alphaproteobacteria |
TAS [19] |
||
| Order Rhizobiales |
TAS [20,21] |
||
| Family Phyllobacteriaceae |
TAS [21,22] |
||
| Genus Mesorhizobium |
TAS [14] |
||
| Species Mesorhizobium loti |
TAS [14] |
||
| Strain R7A |
TAS [1] |
||
| |
Gram stain |
Negative |
IDA |
| |
Cell shape |
Rod |
IDA |
| |
Motility |
Motile |
IDA |
| |
Sporulation |
Non-sporulating |
NAS |
| |
Temperature range |
Mesophile |
NAS |
| |
Optimum temperature |
28°C |
NAS |
| |
Salinity |
Unknown |
NAS |
| MIGS-22 |
Oxygen requirement |
Aerobic |
TAS [2] |
| |
Carbon source |
Various |
TAS [23] |
| |
Energy source |
Chemoorganotroph |
TAS [23] |
| MIGS-6 |
Habitat |
Soil, root nodule, host |
TAS [2] |
| MIGS-15 |
Biotic relationship |
Free living, Symbiotic |
TAS [2] |
| MIGS-14 |
Pathogenicity |
None |
NAS |
| |
Biosafety level |
1 |
TAS [24] |
| |
Isolation |
Root nodule of Lotus corniculatus |
TAS [1] |
| MIGS-4 |
Geographic location |
Lammermoor, Otago, NZ |
TAS [1] |
| MIGS-5 |
Nodule collection date |
1993 |
TAS [1] |
| MIGS-4.1 |
Latitude |
-45.53 |
TAS [1] |
| MIGS-4.2 |
Longitude |
169.9415 |
TAS [1] |
| MIGS-4.3 |
Depth |
5 cm |
IDA |
| MIGS-4.4 | Altitude | 885 meters | IDA |
Evidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [25].
Figure 2 Phylogenetic tree showing the relationships of Mesorhizobium loti R7A with other root nodule bacteria based on aligned sequences of the 16S rRNA gene (1,290 bp internal region). All sites were informative and there were no gap-containing sites. Phylogenetic analyses were performed using MEGA [26], version 5. The tree was built using the Maximum-Likelihood method with the General Time Reversible model [27]. Bootstrap analysis [28] with 500 replicates was performed to assess the support of the clusters. Type strains are indicated with a superscript T. Brackets after the strain name contain a DNA database accession number and/or a GOLD ID (beginning with the prefix G) for a sequencing project registered in GOLD [29]. Published genomes are indicated with an asterisk.
Figure 2.

Shows the phylogenetic neighborhood of M. loti strain R7A in a 16S rRNA gene sequence based tree. This strain has 100% (1,367/1,367 bp) 16S rRNA gene sequence identity to MAFF303099 (GOLD ID: Gc00040) and 99.8% sequence identity (1,364/1,397 bp) to M. opportunistum WSM2075 (GOLD ID: Gc01853).
Symbiotaxonomy
M. loti strain R7A is a field reisolate of strain ICMP3153 that was originally isolated from a Lotus corniculatus nodule in Ireland. It forms effective symbioses with L. tenuis, L. corniculatus, L. japonicus (including ecotypes Gifu and MG-20), L. filicaulis and L. burttii. It also induces but does not infect nodule primordia on L. pedunculatus and Leucaena leucocephala[7,8]. Mutants of strain R7A defective in the vir Type IV secretion system encoded on the symbiosis island are able to form effective nodules on Leucaena leucocephala but not L. pedunculatus[7]. A nonsymbiotic derivative of R7A cured of the symbiosis island and therefore unable to form root nodules has also been isolated and is called R7ANS [5].
Genome sequencing and annotation information
Genome project history
This organism was selected for sequencing on the basis of its environmental and agricultural relevance to issues in global carbon cycling, alternative energy production, and biogeochemical importance, and is part of the Community Sequencing Program at the U.S. Department of Energy, Joint Genome Institute (JGI) for projects of relevance to agency missions. The genome project is deposited in the Genomes OnLine Database [29] and an improved-high-quality-draft genome sequence in IMG. Sequencing, finishing and annotation were performed by the JGI. A summary of the project information is shown in Table 2.
Table 2.
Genome sequencing project information for Mesorhizobium loti R7A
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 |
Finishing quality |
Improved-high-quality-draft |
| MIGS-28 |
Libraries used |
Illumina Standard (short PE) and CLIP (long PE) libraries |
| MIGS-29 |
Sequencing platforms |
Illumina HiSeq2000 technology |
| MIGS-31.2 |
Sequencing coverage |
Illumina: 563× |
| MIGS-30 |
Assemblers |
Velvet version 1.1.05; Allpaths-LG version r38445 phrap, version 4.24 |
| MIGS-32 |
Gene calling method |
Prodigal 1.4, GenePRIMP |
| |
Genbank accession |
AZAM00000000 |
| |
Genbank Registration Date |
07-FEB-2014 |
| |
GOLD ID |
Gi08825 |
| |
NCBI project ID |
74389 |
| |
Database: IMG |
2512875016 |
| Project relevance | Symbiotic nitrogen fixation, agriculture |
Growth conditions and DNA isolation
M. loti strain R7A was grown to mid logarithmic phase in TY rich medium [30] on a gyratory shaker at 28°C at 250 rpm. DNA was isolated from 60 mL of cells using a CTAB (Cetyl trimethyl ammonium bromide) bacterial genomic DNA isolation method [31].
Genome sequencing and assembly
The draft genome of M. loti R7A was generated at the DOE Joint Genome Institute (JGI) using Illumina data [32]. For this genome, we constructed and sequenced an Illumina short-insert paired-end library with an average insert size of 270 bp which generated 21,315,208 reads and an Illumina long-insert paired-end library with an average insert size of 10487.44 +/- 2154.53 bp which generated 3,077,470 reads totaling 3,659 Mbp of Illumina data (unpublished, Feng Chen). All general aspects of library construction and sequencing performed at the JGI can be found at the DOE Joint Genome Institute website [33].
The initial draft assembly contained 12 contigs in 1 scaffold. The initial draft data was assembled with Allpaths, version 38445, and the consensus was computationally shredded into 10 Kbp overlapping fake reads (shreds). The Illumina draft data were also assembled with Velvet, version 1.1.05 [34], and the consensus sequences were computationally shredded into 1.5 Kbp overlapping fake reads (shreds). The Illumina draft data was assembled again with Velvet using the shreds from the first Velvet assembly to guide the next assembly. The consensus from the second VELVET assembly was shredded into 1.5 Kbp overlapping fake reads. The fake reads from the Allpaths assembly and both Velvet assemblies and a subset of the Illumina CLIP paired-end reads were assembled using parallel phrap, version SPS 4.24 (High Performance Software, LLC). Possible mis-assemblies were corrected with manual editing in Consed [35-37]. Gap closure was accomplished using repeat resolution software (Wei Gu, unpublished), and sequencing of bridging PCR fragments with Sanger technology. A total of 40 additional sequencing reactions were completed to close gaps and to raise the quality of the final sequence. There are 3 contigs and 1 scaffold in the current assembly. The estimated size of the genome is 6.5 Mbp and the final assembly is based on 3,659 Mb of Illumina draft data, which provides an average 563× coverage of the genome.
Genome annotation
Genes were identified using Prodigal [38] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePrimp pipeline [39]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Non-coding genes and miscellaneous features were predicted using tRNAscan-SE [40], RNAMMer [41], Rfam [42], TMHMM [43], and SignalP [44]. Additional gene prediction analyses and functional annotation were performed within the Integrated Microbial Genomes (IMG-ER) platform [45].
Genome properties
The genome is 6,529,530 nucleotides with 62.93% GC content (Table 3 and Figure 3) and is comprised of a single scaffold and no plasmids. From a total of 6,398 genes, 6,323 were protein encoding and 75 RNA-only encoding genes. Within the genome, 203 pseudogenes were also identified. The majority of genes (80.10%) were assigned a putative function whilst the remaining genes were annotated as hypothetical. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3.
Genome statistics for Mesorhizobium loti R7A
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) |
6,529,530 |
100.00 |
| DNA coding region (bp) |
5697197 |
87.25 |
| DNA G + C content (bp) |
4108774 |
62.93 |
| Number of scaffolds |
1 |
|
| Number of contigs |
3 |
|
| Total genes |
6,398 |
100.00 |
| RNA genes |
75 |
1.17 |
| rRNA operons |
2* |
|
| Protein-coding genes |
6,323 |
98.83 |
| Genes with function prediction |
5,125 |
80.10 |
| Genes assigned to COGs |
5,127 |
80.13 |
| Genes assigned Pfam domains |
5,333 |
83.35 |
| Genes with signal peptides |
565 |
8.83 |
| Genes coding transmembrane proteins | 1,518 | 23.73 |
*3 copies of 5S, 2 copies of 16S and 3 copies of 23S rRNA genes.
Figure 3.
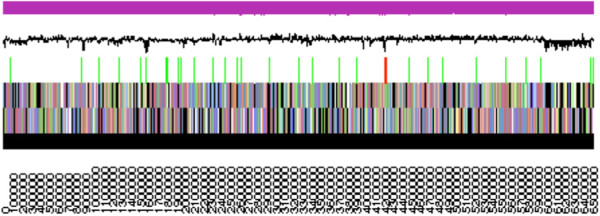
Graphical map of the single scaffold of Mesorhizobium loti R7A. From bottem to the top: Genes on forward strand (color by COG categories as denoted by the IMG platform), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, sRNAs red, other RNAs black), GC content, GC skew.
Table 4.
Number of protein coding genes ofMesorhizobium lotiR7A associated with the general COG functional categories
| Code | Value | % age | COG category |
|---|---|---|---|
| J |
199 |
3.49 |
Translation, ribosomal structure and biogenesis |
| A |
0 |
0.00 |
RNA processing and modification |
| K |
521 |
9.13 |
Transcription |
| L |
172 |
3.01 |
Replication, recombination and repair |
| B |
6 |
0.11 |
Chromatin structure and dynamics |
| D |
30 |
0.53 |
Cell cycle control, mitosis and meiosis |
| Y |
0 |
0.00 |
Nuclear structure |
| V |
65 |
1.14 |
Defense mechanisms |
| T |
217 |
3.80 |
Signal transduction mechanisms |
| M |
296 |
5.19 |
Cell wall/membrane biogenesis |
| N |
53 |
0.93 |
Cell motility |
| Z |
0 |
0.00 |
Cytoskeleton |
| W |
1 |
0.02 |
Extracellular structures |
| U |
124 |
2.17 |
Intracellular trafficking and secretion |
| O |
195 |
3.42 |
Posttranslational modification, protein turnover, chaperones |
| C |
304 |
5.33 |
Energy production conversion |
| G |
511 |
8.95 |
Carbohydrate transport and metabolism |
| E |
675 |
11.83 |
Amino acid transport metabolism |
| F |
89 |
1.56 |
Nucleotide transport and metabolism |
| H |
216 |
3.78 |
Coenzyme transport and metabolism |
| I |
242 |
4.24 |
Lipid transport and metabolism |
| P |
249 |
4.36 |
Inorganic ion transport and metabolism |
| Q |
181 |
3.17 |
Secondary metabolite biosynthesis, transport and catabolism |
| R |
750 |
13.14 |
General function prediction only |
| S |
612 |
10.72 |
Function unknown |
| - | 1,271 | 19.87 | Not in COGS |
Conclusions
The M. loti R7A genome consists of a single 6.5-Mb chromosome which encodes 6,398 genes. The sequencing was completed to the stage where a single scaffold comprising 3 contigs was obtained. M. loti strain R7A and M. loti strain MAFF303099 are currently the two most widely studied M. loti strains. Strain R7A differs from MAFF303099 in that the genome lacks plasmids whereas the genome of MAFF303099 includes two plasmids pMLa and pMLb [6]. The R7A symbiosis island remains mobile whereas the MAFF303099 symbiosis island is likely immobile due at least in part to a transposon insertion within the origin of transfer (oriT) [3,5]. M. loti strain R7A represents an important resource for the study of the mechanism and regulation of transfer of large mobile integrative and conjugative elements (ICEs). It is also widely used in conjunction with the model legume Lotus japonicus for ongoing molecular analyses of the plant-microbe interactions required for the establishment of a nitrogen-fixing symbiosis.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JS and CR supplied the strain and background information for this project and helped WR write the paper, TR supplied DNA to JGI and performed all imaging, WR coordinated the project and all other authors were involved in either sequencing the genome and/or editing the paper. All authors read and approved the final manuscript.
Contributor Information
Simon Kelly, Email: kelly@mb.au.dk.
John Sullivan, Email: john.sullivan@otago.ac.nz.
Clive Ronson, Email: clive.ronson@otago.ac.nz.
Rui Tian, Email: protein17@yahoo.com.sg.
Lambert Bräu, Email: lambert.brau@deakin.edu.au.
Christine Munk, Email: christinem@lanl.gov.
Lynne Goodwin, Email: lynneg@lanl.gov.
Cliff Han, Email: han_cliff@lanl.gov.
Tanja Woyke, Email: twoyke@lbl.gov.
Tatiparthi Reddy, Email: TBReddy@lbl.gov.
Marcel Huntemann, Email: mhuntemann@lbl.gov.
Amrita Pati, Email: APati@lbl.gov.
Konstantinos Mavromatis, Email: kmavrommatis@lbl.gov.
Victor Markowitz, Email: VMMarkowitz@lbl.gov.
Natalia Ivanova, Email: NNIvanova@lbl.gov.
Nikos Kyrpides, Email: nckyrpides@lbl.gov.
Wayne Reeve, Email: W.Reeve@murdoch.edu.au.
Acknowledgements
This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396.
References
- Sullivan JT, Patrick HN, Lowther WL, Scott DB, Ronson CW. Nodulating strains of Rhizobium loti arise through chromosomal symbiotic gene transfer in the environment. Proc Natl Acad Sci U S A. 1995;92:8985–9. doi: 10.1073/pnas.92.19.8985. PubMed http://dx.doi.org/10.1073/pnas.92.19.8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis BDW, Pankhurst CE, Patel JJ. Rhizobium loti, a new species of legume root nodule bacteria. Int J Syst Bacteriol. 1982;32:378–80. doi: 10.1099/00207713-32-3-378. http://dx.doi.org/10.1099/00207713-32-3-378. [DOI] [Google Scholar]
- Sullivan JT, Ronson CW. Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc Natl Acad Sci U S A. 1998;95:5145–9. doi: 10.1073/pnas.95.9.5145. PubMed http://dx.doi.org/10.1073/pnas.95.9.5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JT, Trzebiatowski JR, Cruickshank RW, Gouzy J, Brown SD, Elliot RM, Fleetwood DJ, McCallum NG, Rossbach U, Stuart GS, Weaver JE, Webby RJ, de Bruijn FJ, Ronson CW. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A. J Bacteriol. 2002;184:3086–95. doi: 10.1128/JB.184.11.3086-3095.2002. PubMed http://dx.doi.org/10.1128/JB.184.11.3086-3095.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay JP, Sullivan JT, Stuart GS, Lamont IL, Ronson CW. Excision and transfer of the Mesorhizobium loti R7A symbiosis island requires an integrase IntS, a novel recombination directionality factor RdfS, and a putative relaxase RlxS. Mol Microbiol. 2006;62:723–34. doi: 10.1111/j.1365-2958.2006.05396.x. PubMed http://dx.doi.org/10.1111/j.1365-2958.2006.05396.x. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000;7:331–8. doi: 10.1093/dnares/7.6.331. PubMed http://dx.doi.org/10.1093/dnares/7.6.331. [DOI] [PubMed] [Google Scholar]
- Hubber A, Vergunst AC, Sullivan JT, Hooykaas PJJ, Ronson CW. Symbiotic phenotypes and translocated effector proteins of the Mesorhizobium loti strain R7A VirB/D4 type IV secretion system. Mol Microbiol. 2004;54:561–74. doi: 10.1111/j.1365-2958.2004.04292.x. [DOI] [PubMed] [Google Scholar]
- Rodpothong P, Sullivan JT, Songsrirote K, Sumpton D, Cheung KWJT, Thomas-Oates J, Radutoiu S, Stougaard J, Ronson CW. Nodulation gene mutants of Mesorhizobium loti R7A - nodZ and nolL mutants have host-specific phenotypes on Lotus spp. Mol Plant Microbe Interact. 2009;22:1546–54. doi: 10.1094/MPMI-22-12-1546. PubMed http://dx.doi.org/10.1094/MPMI-22-12-1546. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Muszynski A, Kawaharada Y, Hubber AM, Sullivan JT, Sandal N, Carlson RW, Stougaard J, Ronson CW. Conditional requirement for exopolysaccharide in the Mesorhizobium-Lotus symbiosis. Mol Plant Microbe Interact. 2013;26:319–29. doi: 10.1094/MPMI-09-12-0227-R. PubMed http://dx.doi.org/10.1094/MPMI-09-12-0227-R. [DOI] [PubMed] [Google Scholar]
- Sullivan JT, Brown SD, Ronson CW. The NifA-RpoN regulon of Mesorhizobium loti strain R7A and its symbiotic activation by a novel LacI/GalR-family regulator. PLoS One. 2013;8(1):e53762. doi: 10.1371/journal.pone.0053762. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay JP, Major AS, Komarovsky VM, Sullivan JT, Dy RL, Hynes MF, Salmond GPC, Ronson CW. A widely conserved molecular switch controls quorum sensing and symbiosis island transfer in Mesorhizobium loti through expression of a novel antiactivator. Mol Microbiol. 2013;87:1–13. doi: 10.1111/mmi.12079. PubMed http://dx.doi.org/10.1111/mmi.12079. [DOI] [PubMed] [Google Scholar]
- Ronson CW, Nixon BT, Albright LM, Ausubel FM. Rhizobium meliloti ntrA (rpoN) gene is required for diverse metabolic functions. J Bacteriol. 1987;169:2424–31. doi: 10.1128/jb.169.6.2424-2431.1987. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howieson JG, Ewing MA, D'antuono MF. Selection for acid tolerance in Rhizobium meliloti. Plant Soil. 1988;105:179–88. doi: 10.1007/BF02376781. http://dx.doi.org/10.1007/BF02376781. [DOI] [Google Scholar]
- Jarvis BDW, Van Berkum P, Chen WX, Nour SM, Fernandez MP, Cleyet-Marel JC, Gillis M. Transfer of Rhizobium loti, Rhizobium huakuii, Rhizobium ciceri, Rhizobium mediterraneum, Rhizobium tianshanense to Mesorhizobium gen.nov. Int J Syst Evol Microbiol. 1997;47:895–8. [Google Scholar]
- Tighe SW, de Lajudie P, Dipietro K, Lindstrom K, Nick G, Jarvis BDW. Analysis of cellular fatty acids and phenotypic relationships of Agrobacterium, Bradyrhizobium, Mesorhizobium, Rhizobium and Sinorhizobium species using the Sherlock Microbial Identification System. Int J Syst Evol Microbiol. 2000;50:787–801. doi: 10.1099/00207713-50-2-787. PubMed http://dx.doi.org/10.1099/00207713-50-2-787. [DOI] [PubMed] [Google Scholar]
- Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen M, Angiuoli SV, Ashburner M, Axelrod N, Baldauf S, Ballard S, Boore JL, Cochrane G, Cole J, Dawyndt P, de Vos P, de Pamphilis C, Edwards R, Faruque N, Feldman R, Gilbert J, Gilna P, Glöckner FO, Goldstein P, Guralnick R, Haft D, Hancock D. et al. Towards a richer description of our complete collection of genomes and metagenomes “Minimum Information about a Genome Sequence” (MIGS) specification. Nat Biotechnol. 2008;26:541–7. doi: 10.1038/nbt1360. PubMed http://dx.doi.org/10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity GM, Bell JA, Lilburn T, Phylum XIV. In: Bergey's Manual of Systematic Bacteriology. Second Edition, Volume 2, Part B. Garrity GM, Brenner DJ, Krieg NR, Staley JT, editor. New York: Springer; 2005. Proteobacteria phyl. nov; p. 1. [Google Scholar]
- Garrity GM, Bell JA, Lilburn T. In: Bergey's Manual of Systematic Bacteriology. Second. Garrity GM, Brenner DJ, Kreig NR, Staley JT, editor. New York: Springer - Verlag; 2005. Class I. Alphaproteobacteria class. [Google Scholar]
- Kuykendall LD. In: Bergey's Manual of Systematic Bacteriology. Second. Garrity GM, Brenner DJ, Kreig NR, Staley JT, editor. New York: Springer - Verlag; 2005. Order VI. Rhizobiales ord. nov; p. 324. [Google Scholar]
- Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56:1–6. doi: 10.1099/ijs.0.64188-0. PubMed http://dx.doi.org/10.1099/ijs.0.64188-0. [DOI] [PubMed] [Google Scholar]
- Mergaert J, Swings J, Family IV. In: Bergy's Manual of Systematic Bacteriology. Second. Garrity GM, Brenner DJ, Kreig NR, Staley JT, editor. New York: Springer - Verlag; 2005. Phyllobacteriaceae; p. 393. [Google Scholar]
- Jarvis BDW, Van Berkum P, Chen XW, Nour SM, Fernandez MP, Cleyet-Marel JC, Gillis M. Transfer of Rhizobium loti, Rhizobium huakuii, Rhizobium ciceri, Rhizobium mediterraneum and Rhizobium tianshanense to Mesorhizobium gen. nov. Int J Syst Bacteriol. 1997;47:895–8. doi: 10.1099/00207713-47-3-895. http://dx.doi.org/10.1099/00207713-47-3-895. [DOI] [Google Scholar]
- Biological Agents. Technical rules for biological agents. TRBA. p. 466. http://www.baua.de.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. PubMed http://dx.doi.org/10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. PubMed http://dx.doi.org/10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. New York: Oxford University Press; 2000. [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91. doi: 10.2307/2408678. http://dx.doi.org/10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008;36:D475–9. doi: 10.1093/nar/gkn240. PubMed http://dx.doi.org/10.1093/nar/gkm884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringer JE. R factor transfer in Rhizobium leguminosarum. J Gen Microbiol. 1974;84:188–98. doi: 10.1099/00221287-84-1-188. PubMed http://dx.doi.org/10.1099/00221287-84-1-188. [DOI] [PubMed] [Google Scholar]
- DOE Joint Genome Institute user homepage. http://my.jgi.doe.gov/general/index.html.
- Bennett S, Solexa L. Pharmacogenomics. 2004. pp. 433–8. PubMed http://dx.doi.org/10.1517/14622416.5.4.433. [DOI] [PubMed]
- DOE Joint Genome Institute. http://www.jgi.doe.gov/
- Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics. 2010;Chapter 11:11–5. doi: 10.1002/0471250953.bi1105s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–94. PubMed http://dx.doi.org/10.1101/gr.8.3.175. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–85. doi: 10.1101/gr.8.3.175. PubMed http://dx.doi.org/10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. PubMed http://dx.doi.org/10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. PubMed http://dx.doi.org/10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 2010;7:455–7. doi: 10.1038/nmeth.1457. PubMed http://dx.doi.org/10.1038/nmeth.1457. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. PubMed http://dx.doi.org/10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8. doi: 10.1093/nar/gkm160. PubMed http://dx.doi.org/10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–41. doi: 10.1093/nar/gkg006. PubMed http://dx.doi.org/10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. PubMed http://dx.doi.org/10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–95. doi: 10.1016/j.jmb.2004.05.028. PubMed http://dx.doi.org/10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8. doi: 10.1093/bioinformatics/btp393. PubMed http://dx.doi.org/10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]