

Abstract
Background
A limitation of current antiplatelet therapies is their inability to separate thrombotic events from bleeding occurrences. Better understanding of the molecular mechanisms leading to platelet activation is of importance for the development of improved therapies. Recently, protein tyrosine phosphatases (PTPs) have emerged as critical regulators of platelet function.
Methods and Results
This is the first report implicating the dual-specificity phosphatase 3 (DUSP3) in platelet signaling and thrombosis. This phosphatase is highly expressed in human and mouse platelets. Platelets from DUSP3-deficient mice displayed a selective impairment of aggregation and granule secretion mediated through the collagen receptor glycoprotein VI (GPVI) and the C-type lectin-like receptor 2 (CLEC-2). DUSP3-deficient mice were more resistant to collagen- and epinephrine-induced thromboembolism, compared to wild-type mice, and showed severely impaired thrombus formation upon ferric chloride-induced carotid artery injury. Intriguingly, bleeding times were not altered in DUSP3-deficient mice. At the molecular level, DUSP3 deficiency impaired Syk tyrosine phosphorylation, subsequently reducing phosphorylation of PLCγ2 and calcium fluxes. To investigate DUSP3 function in human platelets, a novel small-molecule inhibitor of DUSP3 was developed. This compound specifically inhibited collagen and CLEC-2-induced human platelet aggregation, thereby phenocopying the effect of DUSP3 deficiency in murine cells.
Conclusions
DUSP3 plays a selective and essential role in collagen- and CLEC-2-mediated platelet activation and thrombus formation in vivo. Inhibition of DUSP3 may prove therapeutic for arterial thrombosis. This is the first time a PTP, implicated in platelet signaling, has been targeted with a small-molecule drug.
Keywords: platelets, signal transduction, thrombosis, collagen, inhibitors
Introduction
Antiplatelet therapy has been effective in reducing the mortality and morbidity of acute myocardial infarction, the most common cause of death in developed countries.1 However, FDA-approved antiplatelet agents have serious side effects, including gastrointestinal toxicity, neutropenia, thrombocytopenia, and the common bleeding.1 There also remains a considerable incidence of arterial thrombosis in patients receiving currently available antiplatelet therapy.1 A better understanding of the molecular mechanisms leading to platelet activation will be essential for the development of new therapeutics.
Platelet activation depends on rapid phosphorylation and dephosphorylation of key signaling proteins, in particular on tyrosine.2 While the repertoire of protein tyrosine kinases (PTKs) has been well described in platelet activation, the expression, regulation, specificity, and function of platelet-expressed protein tyrosine phosphatases (PTPs) are largely unknown. A recent proteomic analysis found that 14 out of 37 classical, phosphotyrosine (pY)-specific PTPs are expressed in human platelets.3 Expression and function of the dual-specificity phosphatases (DSPs),4, 5 the largest subgroup of the PTP superfamily, are unexplored.
DUSP3, also known as Vaccinia H1-related (VHR) phosphatase, is a DSP encoded by the DUSP3/Dusp3 gene. DUSP3 (185 amino acids; Mr 21 kDa), which only contains a catalytic (PTP) domain,6 has been reported to dephosphorylate the mitogen-activated protein kinases (MAPKs) ERK1/2 and JNK1/2.7 Additional reported substrates include EGFR and ErbB2.8 DUSP3 is implicated in cell cycle regulation, and its expression is altered in human cancer.9-11 However, since all of these studies were performed either in vitro, using recombinant proteins, or in cell lines, using transient overexpression or siRNA knockdown, the true physiological function of DUSP3 has remained elusive. We recently generated a full Dusp3-knockout (Dusp3-KO) mouse.12 Dusp3-KO mice were healthy, fertile, and showed no spontaneous phenotypic abnormality. However, DUSP3 deficiency prevented neo-angiogenesis and bFGF-induced microvessel outgrowth.12 In the present study, we identified DUSP3 as a key and non-redundant player in GPVI- and CLEC-2-mediated signaling pathways in mouse and human platelets. We show that DUSP3 deficiency limits platelet activation and arterial thrombosis. Moreover, we developed a specific small-molecule inhibitor of DUSP3, which was able to phenocopy DUSP3 deficiency in platelets.
Methods
Platelet RNA sampling and Microarray
Platelets from 256 healthy volunteers were isolated from citrate-anticoagulated blood. Donors were informed about the objectives of the study and signed an informed consent. The study was approved by the ethical committee review board of the Liège University Hospital. RNA extraction and microarray procedures are described in the Supplementary Material.
Mice
C57BL/6-Dusp3-KO were generated by homologous recombination.12 Heterozygous mice were mated to generate +/+ and −/− littermates used for experimentation (8-12 weeks old male mice). All experiments were approved by the local ethics committee.
Isolation of human and mouse platelets
Human platelets were prepared from peripheral blood freshly drawn from healthy donors as previously described.13 Mouse washed platelets (WPs) were prepared as previously described.14
Isolation of human and murine B and T cells
Human B and T cells were sorted from freshly collected blood using EasySep B and T cell–negative selection kits (Stemcell Technologies). Mouse B and T cells were sorted from spleens.
Platelet aggregation analyses
Light transmission was recorded during platelet aggregation induced by collagen, convulxin (CVX), collagen related peptide (CRP), rhodocytin, thrombin, U46619, or ADP in the presence of 2 mM CaCl2 on a Lumi-Aggregometer (Chrono-log).
Flow cytometry
WPs were stimulated for 15 min with different concentrations of collagen, CRP, thrombin, or ADP under non-stirring conditions. Saturating concentrations of FITC-conjugated P-selectin and PE-conjugated JON/A antibodies were added. Samples were analyzed on a FACSCantoII flow cytometer (BD Biosciences).
Electron microscopy
Platelet pellets were fixed for 60 min in 2.5% glutaraldehyde in Sörensen’s buffer (0.1 M, pH 7.4), post-fixed for 30 min with 1% osmium tetroxide, dehydrated in a series of ethanol concentrations, and embedded in Epon. Ultrathin sections were stained with uranyl acetate and lead citrate and examined on a Jeol-CX100II transmission electron microscope (60 kV).
Whole blood platelet aggregate formation under flow
Thrombus formation under flow conditions was assessed with anticoagulated mouse blood (4 U/mL heparin, 20 μM PPACK) as previously described.15 Area coverage from phase-contrast images was analyzed using ImagePro (Media Cybernetics).16 Area coverage by platelets stained with OG488-annexin A5 was determined with Quanticell (Visitech).
Ca2+ flux
Apyrase (0.5 U/mL)–treated murine WPs were loaded with 3.5 μM fura-2-acetoxymethyl ester in the presence of Pluronic F-127 for 15 min and fluorescence was recorded on an Aminco spectrofluorimeter (SLM Instruments) as described.17
Arterial thrombosis models
Pulmonary embolism was induced by injection of a mixture of collagen (170 μg/kg) and epinephrine (60 μg/kg) into the plexus retro-orbital veins of anesthetized mice (ketamine: 60 mg/kg; xylazine: 5 mg/kg). Time to death was monitored. Lungs were perfused with 4% formaldehyde solution and collected for histological studies.
Injury of carotid arteries of anesthetized mice was performed by applying a filter paper soaked in 10% ferric chloride (FeCl3) solution on the exposed artery for 5 min.18 Fluorescence of exogenously CFSE-labeled platelets was monitored using a BX61WI microscope (Olympus). Digital images were captured with a Hamamatsu 9100-13 EMCCD camera, using a Lambda DG-4 (Sutter instrument) light source and Slidebook software 5.5 (3i).
Mouse irradiation and bone marrow (BM) transplantation
Donor mice (7-8 weeks old) were euthanized by cervical dislocation. Tibia and fibula were collected and BMs were flushed with PBS. 10×106 single BM cells were transplanted to 4-5 weeks old lethally irradiated (866.3 cGy) recipient mice. Chimeric mice were used in the FeCl3 model 3-4 weeks after transplantation. Chimerism was evaluated by western blot of DUSP3 in lysates of peritoneal cavity cells.
Tail bleeding
Mice were anesthetized with isoflurane. A 3 mm portion of the tail tip was excised and submerged in a 37°C water bath. Bleeding was monitored for 15 min.
Platelet activation, cell lysis, immunoprecipitation, and western blotting
Mouse WPs were activated with CRP or rhodocytin in Tyrode’s buffer for 30, 60, or 90 s under 400 rpm stirring conditions at 37°C. Western blotting and immunoprecipitations were performed according to standard procedures.19
Statistical analysis
Data are presented as mean ± SEM of at least three independent experiments. Data were analyzed using unpaired Student’s t-test or ANOVA and the Bonferroni multiple comparison test as indicated in each figure legend. Differences in survival were determined using Kaplan-Meier analysis (log-rank Mantel test). A p-value <0.05 was considered significant. Calculations were performed using GraphPad-Prism (GraphPad Software, Inc.).
Results
DUSP3 expression in platelets
Transcriptomic analysis of platelets from 256 healthy human individuals revealed that DUSP3-encoding mRNA is highly expressed in platelets (Figure 1A). Abundance of DUSP3 in human and mouse platelets was confirmed by western blot analysis (Figure 1B-1C). Expression levels of DUSP3 were substantially higher in platelets as compared to B and T lymphocytes (Figure 1B-1C), where DUSP3 function had been previously described.7 Thus, we set out to investigate the role of DUSP3 in platelets using both genetic deletion in mice and pharmacological inhibition of DUSP3 in isolated human platelets.
Figure 1.
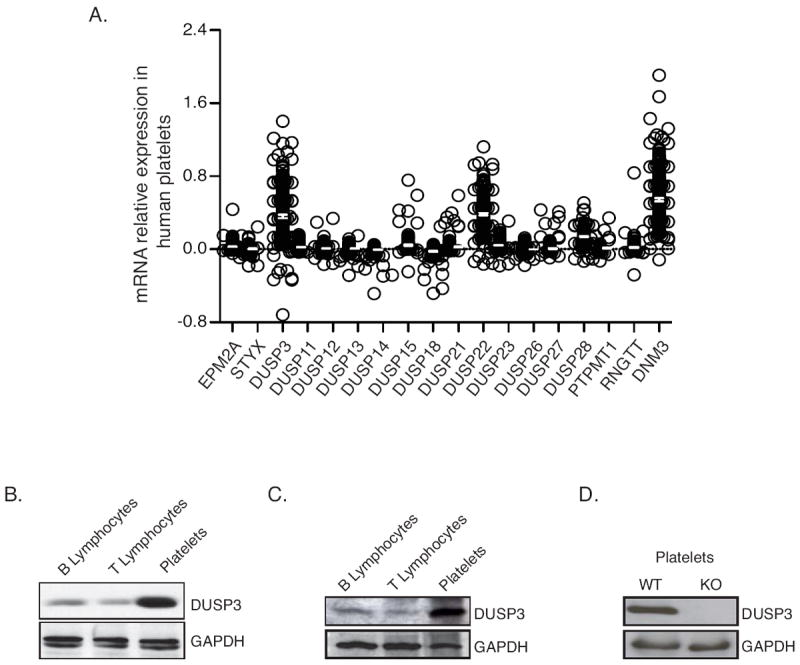
DUSP3 expression in human and mouse platelets. (A) Microarray data of mRNA expression of 17 atypical DSPs in human platelets isolated from 256 healthy volunteers. Each open circle represents one individual. DNM3 was used as positive control for platelet expressed mRNA. Data are presented as ratio of the fluorescence intensity for the DSP probe of interest and the mean fluorescence intensity for the housekeeping genes of each sample. A negative value corresponds to an expression bellow background level. Mean ± SEM are shown. (B-D) DUSP3 protein expression in human B and T lymphocytes and in platelets isolated from peripheral blood (B); in mouse splenic B and T cells and in washed platelets (C); and in WT and Dusp3-KO mouse washed platelets (D). Western blot analysis was performed using anti-human (B) and anti-mouse DUSP3 (C-D). GAPDH was used as loading control. Representative blots of three independent experiments are shown.
Activation and aggregation of DUSP3-deficient mouse platelets
Utilizing our previously generated Dusp3-KO mice,12 we confirmed that platelets isolated from these animals do not express DUSP3 (Figure 1D). Hematological parameters were normal, except for slight but significant differences in monocytes (p<0.05) and mean platelet volume (MPV) (p<0.0001) (Supplemental Table 1). Dusp3-KO mice did not show any spontaneous bleeding or thrombotic disorders. However, in platelet aggregation assays, DUSP3-deficient platelets failed to aggregate in response to low concentrations of collagen (0.5 μg/mL) and selective GPVI agonists, including CVX (5 ng/mL) and CRP (0.1 μg/mL) (Figure 2A-C). Additionally, Dusp3-KO platelets exhibited delayed aggregation induced by low concentrations of rhodocytin (2.5 and 5 nM), a selective CLEC-2 receptor agonist (Figure 2D). GPVI and CLEC-2 surface expression on Dusp3-KO platelets was similar to wild-type (WT) platelets (Figures S1A and S2A). In contrast, aggregation induced by ADP (5-50 μM), thromboxane A2 mimetic U46619 (0.75-2 μM), or thrombin (0.01-0.1 U/mL) occurred normally (Figure 2E and data not shown), indicating normal G-protein coupled receptor (GPCR)-mediated responses.
Figure 2.

DUSP3-deficient platelets exhibit impaired GPVI- and CLEC-2-mediated platelet aggregation. (A-E) Washed platelets prepared from WT or Dusp3-KO mice were stimulated with collagen (0.5 and 1 μg/mL) (A), CRP (0.1, 0.3, and 1 μg/mL) (B), CVX (5, 10, and 100 ng/mL) (C), rhodocytin (2.5, 5, and 10 nM) (D), or thromboxane A2 analog U46619 (1 μM), thrombin (0.05 U/mL), or ADP (20 μM) (E). Representative platelet aggregation curves of three individual experiments are shown.
To investigate the mechanism responsible for the impairment of collagen- and CRP-induced aggregation of DUSP3-deficient platelets, we analyzed their ability to release granule content by measuring P-selectin surface expression, and examined their capacity to activate integrin αIIbβ3 by using the JON/A antibody, which is specific for the high-affinity conformation of mouse αIIbβ3. In DUSP3-deficient compared to WT platelets, P-selectin expression was reduced after stimulation with low concentration of collagen (0.5 μg/mL) or various concentrations of CRP (0.1, 0.3, and 1 μg/mL) (Figure 3A). Integrin αIIbβ3 activation was reduced with low concentrations of collagen (0.5 μg/mL) and CRP (0.1 μg/mL) (Figure 3B). Electron microscopy analysis of resting DUSP3-deficient platelets revealed normal ultrastructure but a slightly increased number of α-granules (Figure 3C and 3D). When activated using CVX, degranulation remained incomplete among the few DUSP3-deficient platelet aggregates compared to WT (Figure 3C). These findings indicate that DUSP3 deficiency impairs GPVI- and CLEC-2-dependent mouse platelet activation and aggregation.
Figure 3.
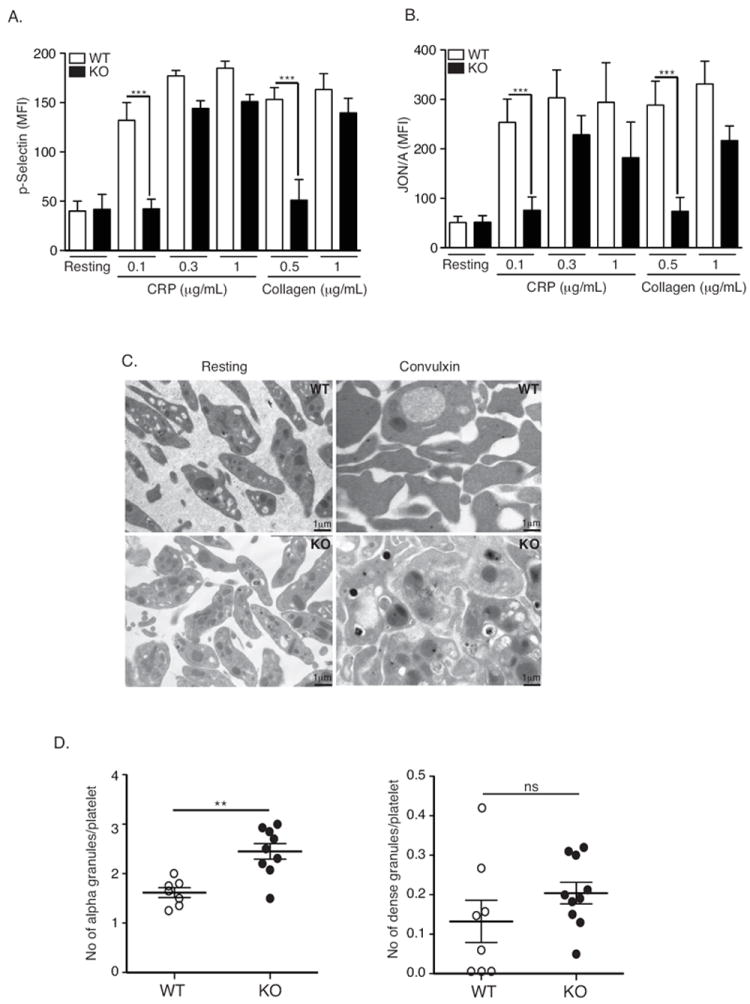
Impaired GPVI-mediated platelet activation in DUSP3-deficient platelets. (A-B) Washed platelets from WT or Dusp3-KO mice were stimulated with 0.1, 0.3, or 1 μg/mL CRP or 0.5 or 1 μg/mL collagen under non-stirring conditions, or left untreated. Surface expression of P-selectin and active integrin αIIbβ3 (JON/A) was quantified by flow cytometry. Mean fluorescence intensity histograms for P-selectin (A) and JON/A (B) are shown. Data were analyzed using ANOVA and the Bonferroni multiple comparison test and represent mean ± SEM of four independent experiments performed on platelet pools from three mice each; *p< 0.05, **p< 0.01. (C) Electron microscopy analysis of WT and Dusp3-KO washed platelets. Platelet ultrastructure was visualized in resting state or upon CVX stimulation (100 ng/mL). (D) Scatter plots of alpha and dense granules counted on five separated micrographs. Data were analyzed using unpaired Student t-test and represent mean ± SEM of three independent experiments performed on platelet pools from three mice; *p< 0.05, **p< 0.01.
GPVI and CLEC-2 signaling in DUSP3-deficient platelets
Earlier studies suggested that DUSP3 dephosphorylates ERK1/2 and JNK1/2 but not p38.7 Therefore, we evaluated activation of these MAPKs using phospho-specific antibodies at basal levels and after CRP stimulation. No differences in MAPK activation between DUSP3-deficient and WT platelets were found (Figure S3). We then analyzed global tyrosine phosphorylation and found decreased phosphorylation of a ~70-kDa band in DUSP3-deficient compared to WT platelets after CRP or rhodocytin stimulation (Figure 4A and 4C). Longer exposure of the pY blot revealed additional bands (at ~12, ~26, and ~40 kDa) with decreased phosphorylation in DUSP3-deficient compared to WT platelets after CRP stimulation (Figure 4B). We then tested whether the observed change in pY of the ~70-kDa band may correspond to pY levels in the tyrosine kinase Syk (Mr 72.1 kDa), a key signaling molecule in GPVI- and CLEC-2-mediated platelet activation. Indeed, pY of immunoprecipitated Syk was significantly reduced in DUSP3-deficient compared to WT platelets after GPVI and CLEC-2 stimulation (p<0.05) (Figures 4D-E and S4A-B). Probing total lysates (TLs) of DUSP3-deficient or WT platelets with phospho-Syk-specific antibodies revealed that, after activation with CRP, Syk phosphorylation was reduced on the activatory residues Tyr-525/526, while phosphorylation of the negative regulatory Tyr-323 was not affected (Figures 4F and S4F-G). In rhodocytin-stimulated platelets, Syk phosphorylation was reduced on both Tyr-525/526 and Tyr-323 in the absence of DUSP3 (Figures 4G and S4H-I).
Figure 4.
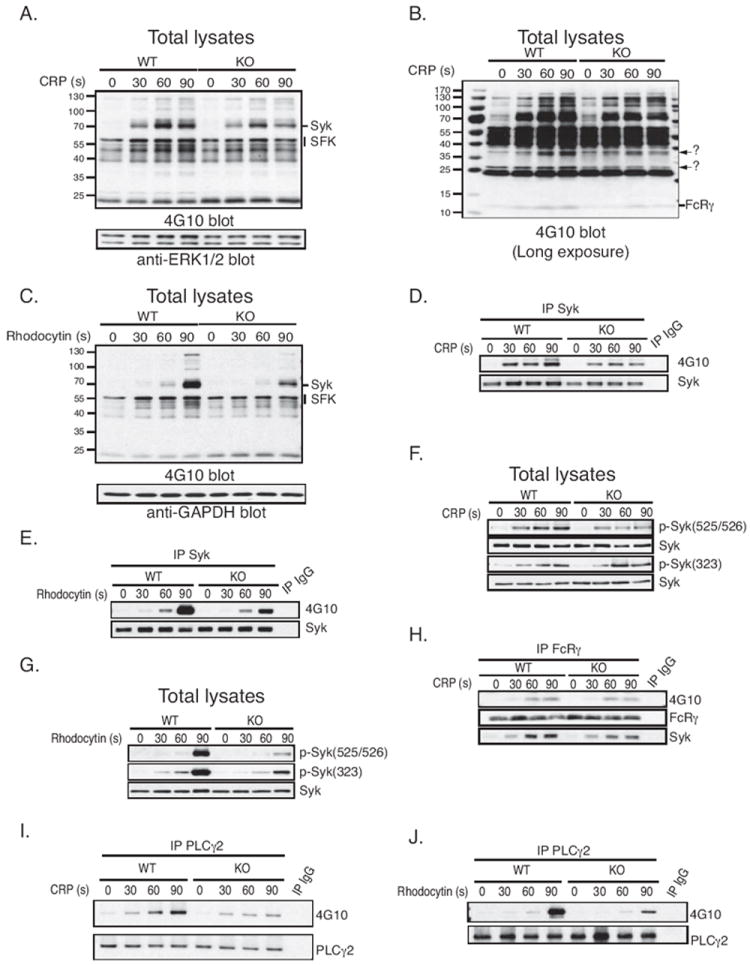
DUSP3-deficiency impairs Syk tyrosine phosphorylation. TLs were prepared from WT or Dusp3-KO mice platelets. Cells were non-activated, CRP- (0.3 μg/mL) or rhodocytin-activated (10 nM) for 30, 60, or 90 s. (A-C) Western blot analysis with anti-pY antibody (4G10) and with ERK1/2 (A) or GAPDH (C) as loading control. Arrows in (B) indicate unknown protein bands with attenuated pY levels in DUSP3-deficient platelets. (D-E) Representative pY blot of Syk immunoprecipitates from TLs of CRP- (D) or rhodocytin-activated (E) platelets. (F-G) Representative blot of Syk phosphorylation on Tyr-323 and Tyr-525/526 in CRP- (F) or rhodocytin-activated (G) platelets. Normalization was performed using total Syk. (H) pY and Syk western blots on FcRγ immunoprecipitates. (I-J) pY blot of PLCγ2 immunoprecipitates from TLs of CRP- (I) or rhodocytin-activated (J) platelets. Results shown are representative of 3 independent experiments performed on platelet pools from three mice each.
Syk is recruited to the GPVI/Fc receptor γ-chain (FcRγ) complex via phosphorylation of FcRγ-associated immunoreceptor tyrosine-based activation motifs (ITAMs) by Src-family kinases (SFKs), and is then activated via autophosphorylation.20, 21 We found that phosphorylation of FcRγ-associated ITAMs was reduced in DUSP3-deficient compared to WT platelets in response to CRP (Figures 4H and S4J). In agreement with this observation, recruitment of Syk to FcRγ was impaired in DUSP3-deficient platelets (Figures 4H and S4K). Additionally, inducible tyrosine phosphorylation in PLCγ2, a key signaling molecule downstream of Syk, was reduced in both CRP- and rhodocytin-stimulated DUSP3-deficient compared to WT platelets (Figures 4I-J and S4L-M). In contrast, activation of SFKs, including Lyn, Fyn, and Src, was not altered (Figure S5A and S5B), indicating that the reduced activation and recruitment of Syk in DUSP3-deficient platelets was not due to aberrant activation of SFKs.
Collagen-induced aggregation under flow, calcium fluxes, and phosphatidylserine exposure in DUSP3-deficient platelets
To further assess the role of DUSP3 in GPVI-dependent platelet responses, platelet aggregate formation and exposure of procoagulant phosphatidylserine (PS) on a collagen surface were analyzed in whole mouse blood under flow. The area covered by platelets was reduced by ~40% for blood from Dusp3-KO compared to WT mice (Figure 5A and 5B), which was in agreement with reduced GPVI activation in DUSP3-deficient platelets.22 Accordingly, overall PS exposure on adhered platelets was also diminished (Figure 5C and 5D). Because PS exposure requires Ca2+ influx,23 we investigated if Ca2+ flux was affected by DUSP3 deficiency. CVX-induced Ca2+ flux was greatly reduced (50%) in DUSP3-deficient compared to WT platelets (Figure 5E and 5F). Thapsigargin-induced Ca2+ increase occurred normally in DUSP3-deficient platelets (Figure S6A), suggesting intact intrinsic Orai 1-mediated store-operated Ca2+ entry. Additionally, there were no differences in Ca2+ rises induced by thrombin, ADP, or U46619 between WT and Dusp3-KO platelets (Figure S6B-D). These data further support a positive role of DUSP3 in GPVI-mediated platelet activation under physiological flow conditions.
Figure 5.
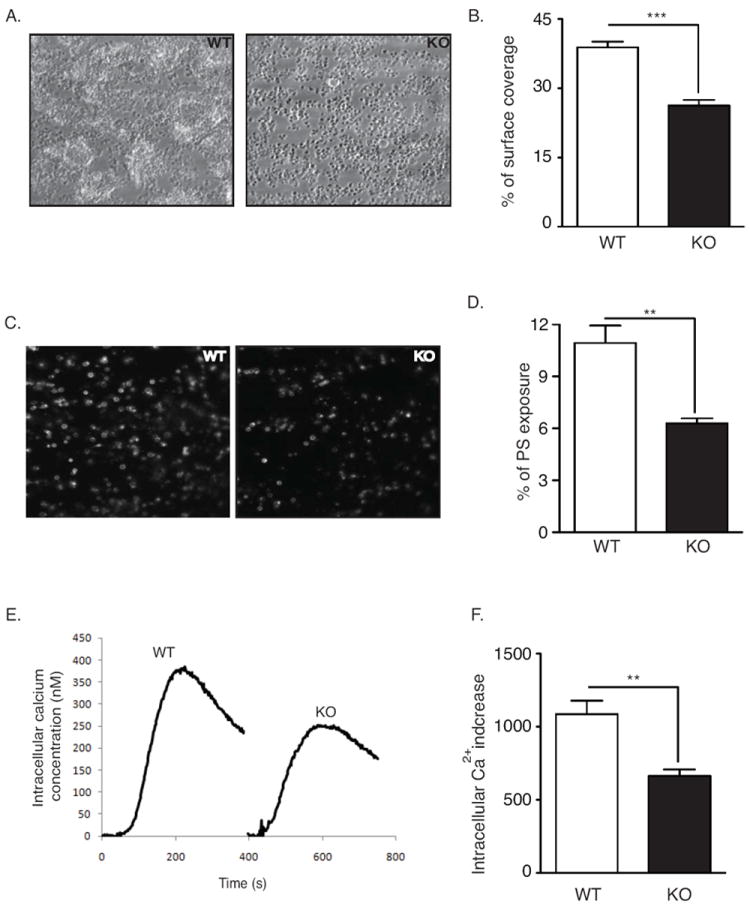
Impaired platelet aggregate formation on whole blood from DUSP3-deficient mice. (A-D) Anticoagulated blood from WT or Dusp3-KO mice was perfused over collagen-coated coverslip through a parallel-plate transparent flow chamber at a wall-shear rate of 1000 s-1 for 4 min. Representative phase-contrast images of fixed platelets (A) and percentages of surface coverage by platelets (B) are shown. Exposure of phosphatidylserine (PS) was detected by post-perfusion with heparin and OG488-labeled Annexin-V-containing rinsing buffer. Representative fluorescent images (C) and percentage of area coverage by labeled platelets (D) are shown. (E-F) Intracellular Ca2+ increase in WT and Dusp3-KO platelets upon CVX stimulation (50 ng/mL). Representative curves (E) and histogram depicting the area under the curve (F) are shown. WT values are arbitrarily set to 100%. Unpaired Student’s t-test was used for comparison. Results are representative of five independent experiments performed on platelet pools from three mice each. Data represent mean ± SEM of three independent experiments; **p<0.01, ***p<0.001.
DUSP3 deficiency and thrombus formation in vivo
To evaluate the importance of DUSP3 in platelet function in vivo, we used a model of pulmonary thromboembolism induced by intravenous injection of a mixture of collagen and epinephrine. About 80% of DUSP3-deficient compared to 45% of WT mice survived (Figure 6A). Analyses of lung sections revealed significantly decreased numbers of occluded microvessels in DUSP3-deficient compared to WT mice (p<0.001) (Figure 6B and 6C). We then examined thrombus formation in real-time by intravital microscopy in a model of FeCl3-induced injury of the carotid arteries. In this model, collagen is exposed to circulating blood, and thrombus formation highly depends on GPVI. In DUSP3-deficient mice, blood vessels were never occluded due to failure to form stable thrombi, while full occlusion occurred at 8-10 min after FeCl3 application in WT vessels (Figure 6D and 6G). To test if the defect in thrombus formation was specifically due to impaired platelet function, we generated chimeric mice by transferring Dusp3-KO bone marrow (BM) (KO>WT) or WT BM (WT>WT) to lethally irradiated WT mice. Successful transplantation was evaluated by quantification of DUSP3 expression in peritoneal cell lysates from KO>WT and WT>WT mice (Figure 6F). Similar to Dusp3-KO, we found that thrombus formation was severely impaired in blood vessels of KO>WT mice (Figure 6E and 6G), confirming that the thrombosis defect in DUSP3-deficient animals was due to platelet dysfunction. Importantly, tail bleeding time, a measure of primary hemostasis in vivo, was identical for WT and DUSP3-deficient mice (Figure 6H).
Figure 6.
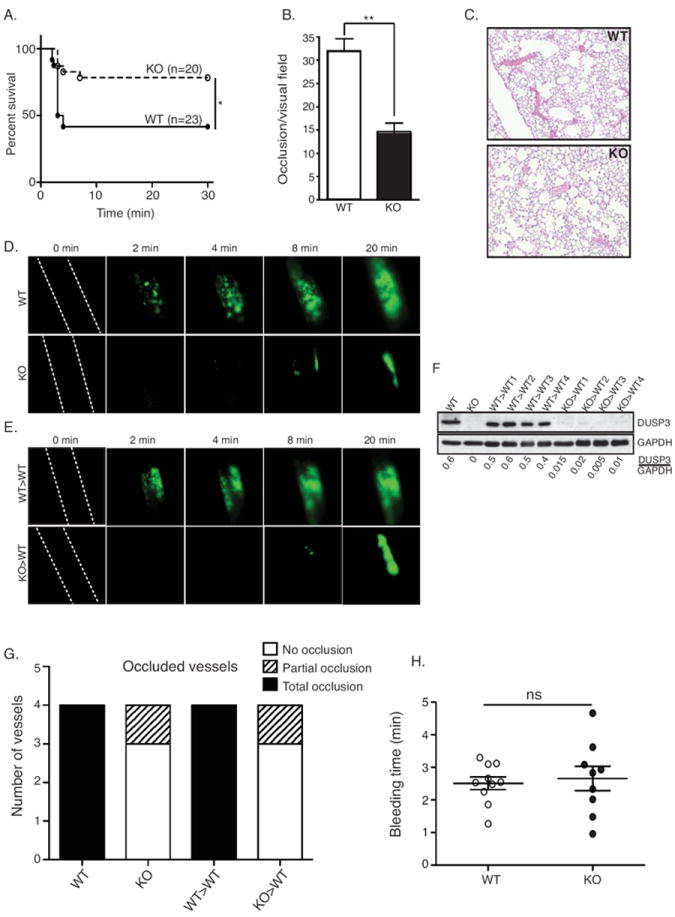
Impaired arterial thrombosis and preserved hemostasis in DUSP3-deficient mice. (A-C) Pulmonary thromboembolism induced by injection of a mixture of collagen and epinephrine. Mortality incidence rates were compared using Kaplan-Meir with log-rank test (n=20 for KO and n=23 for WT mice) (A), quantification of the number of occluded vessels per visual field on lung sections from WT and Dusp3-KO mice (B), and representative field on lung sections from WT and Dusp3-KO mice (C) are shown. Data (from three to six lung sections from three mice of each group) were analyzed using unpaired Student’s t-test; **p<0.01. (D-E) FeCl3 injury of carotid arteries. Representative snapshot images in WT and Dusp3-KO mice (D), and in WT mice transplanted with WT (WT>WT) or with Dusp3-KO (KO>WT) BM cells (E) are shown. (F) Western blot analysis of DUSP3 expression in peritoneal TLs from WT>WT and KO>WT BM-transplanted mice used in the FeCL3 assay. Normalization was performed using GAPDH. (G) Numbers of intact and partially occluded vessels are shown for four mice of each group. (H) Tail bleeding time of WT (dark circle) and Dusp3-KO (open circle) mice. Each dot/circle represent one mouse. Results are presented as mean ± SEM.
Pharmacological inhibition of DUSP3
In order to corroborate DUSP3 function in human platelets, we investigated the possibility of specifically inhibiting DUSP3 activity with small molecules. To identify DUSP3 inhibitors, we employed high-throughput screening (HTS), using a colorimetric phosphatase assay with p-nitrophenolphosphate (pNPP) as substrate, and screened 291,018 drug-like molecules.24 Of the 1,524 primary HTS hits (≥50% inhibition), 1,048 compounds were available from BioFocus DPI and ordered for confirmatory assays. The hits were tested in two reconfirmation single-dose screens in triplicate, using both the primary colorimetric assay and an orthogonal fluorescent assay with 3-O-methylfluorescein phosphate (OMFP) as substrate. Compounds with an average of ≥50% inhibition of DUSP3 activity were further tested in a 10-point dose-response assay in both colorimetric and fluorescent formats. IC50 values were determined, and 67 ‘cross-active’ compounds were identified with IC50 values <20 μM in both assays. Upon visual inspection of each molecule, 32 compounds were discarded from further consideration because of their known promiscuous PTP inhibitory activity. The remaining 35 compounds were taken into selectivity profiling studies for further prioritization. Compound selectivity for inhibiting DUSP3 over the related DUSP6 and three additional PTPs, HePTP, LYP, and STEP, was evaluated (Supplemental Table S2).
Based on the selectivity and potency of compounds, two scaffolds were selected for structure-activity relationship (SAR) studies: MLS-0103602 and MLS-0049585 (Supplemental Table S2). MLS-0103602 (IC50 = 0.37 μM) was the most potent inhibitor with some degree of selectivity for DUSP3; MLS-0049585 (IC50 = 2.68 μM) exhibited the best selectivity for DUSP3. Based on the benzothioamide structure of MLS-0103602, 37 analogs were tested and counterscreened. All analogs were at least an order of magnitude less potent than the original hit, with no improvement of selectivity, leading to the termination of this series (data not shown). In contrast, several analogs containing the N-(benzo[d]thiazol-2-yl)-5-phenyl-1,3,4-oxadiazol-2-amine structure of MLS-0049585 with similar or even better potency could be identified (Supplemental Table S3). The four most potent compounds were selected for testing in human platelets. Inhibition of platelet aggregation was assessed using platelets collected from three healthy donors. In these experiments, MLS-0437605 (Figure 7A) efficiently inhibited platelet aggregation in response to CRP and rhodocytin, but not after stimulation with thromboxane (Figure 7B and 7C). Tests on platelets from WT mice yielded similar results (Figure 7D). In contrast, MLS-0437605 only minimally affected aggregation of DUSP3-deficient platelets (Figure 7D). Selectivity was further evaluated against 10 additional PTPs (Table 1). In these assays, MLS-0437605 showed excellent selectivity for DUSP3 over the vast majority of PTPs tested. Importantly, there was good selectivity of MLS-0437605 for DUSP3 over DUSP22 (7-fold), another DSP that is highly expressed in platelets (Figure 1A). We next examined the effect of MLS-0437605 on GPVI- and CLEC-2-induced signaling in human platelets. Global tyrosine phosphorylation was analyzed on TLs from resting or activated platelets. MLS-0437605 caused a decrease in pY of a ~70-kDa band after stimulation with CRP or rhodocytin (Figure 7E and 7F). Tyrosine phosphorylation of immunoprecipitated Syk and PLCγ2 was also reduced by MLS-0437605 (Figure 7G and 7H). These data demonstrate that pharmacological inhibition of DUSP3 activity in human platelets similarly affects platelet signaling as DUSP3 deficiency in Dusp3-KO platelets.
Figure 7.
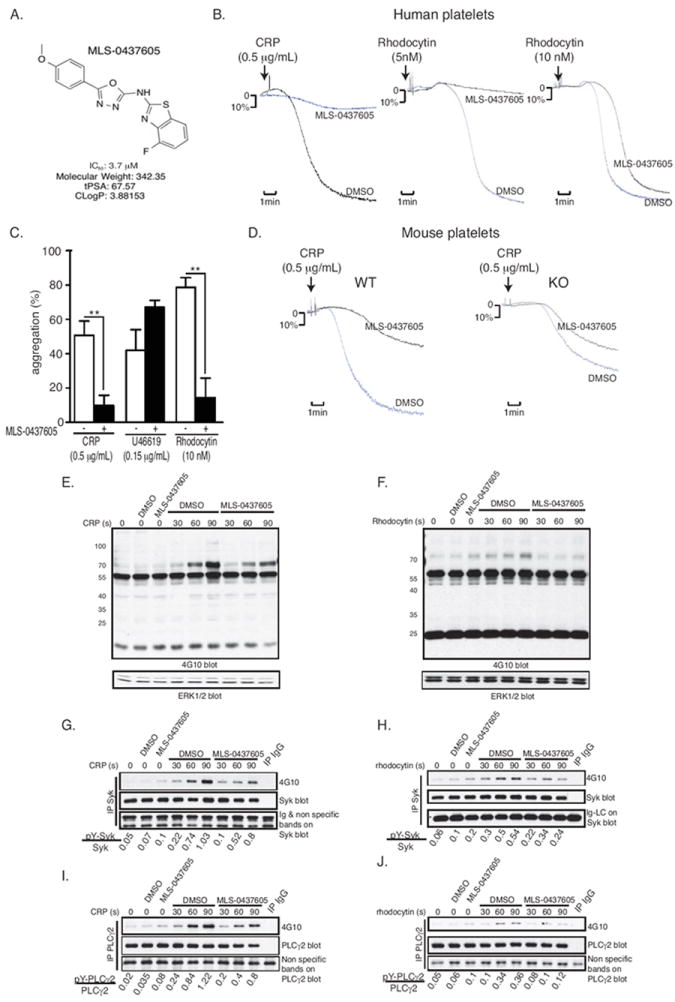
Specific DUSP3 inhibitor MLS-0437605 inhibits platelet activation in response to CRP and rhodocytin. (A) Chemical structure and key properties of MLS-0437605. (B-D) Washed human platelets (B/C) or WT and Dusp3-KO mice washed platelets (D) were pre-incubated for 30 min with DMSO (vehicle) or with MLS-0437605 (3.7 μM), before stimulated with CRP (0.5 μg/mL), rhodocytin (5 or 10 nM), or U46619 (0.15 μg/mL). Representative platelet aggregation curves (B/D) and quantification of platelet aggregation from three healthy human donors (C) are shown. Results were analyzed using one-way ANOVA Bonferroni multiple comparison test and are presented as mean ± SEM; **p<0.01. (E-J) TLs were prepared from vehicle- or MLS-0437605-pretreated human platelets. Cells were non-activated or activated with CRP or rhodocytin for the indicated times. Western blot analysis was performed with 4G10 antibody for global pY of CRP (E) and rhodocytin (F) activated platelets. ERK1/2 was used as a loading control. (G-H) Representative pY blot of Syk immunoprecipitates from TLs of CRP- (G) or rhodocytin-activated (H) platelets. (I-J) pY blot of PLCγ2 immunoprecipitates from TLs of CRP- (I) or rhodocytin-activated (J) platelets. Data are representative of two individual healthy donors.
Table 1.
Selectivity of DUSP3 inhibitor MLS-0437605
| IC50, μM | |
|---|---|
| DUSP3 | 3.7 |
| PTP-SL | 13 |
| DUSP22 | 26 |
| HePTP | 38 |
| LYP | 49 |
| TCPTP | 55 |
| CD45 | >100 |
| LAR | >100 |
| STEP | >100 |
| PTP1B | >100 |
| DUSP6 | >100 |
Discussion
This is the first study implicating a member of the PTP subfamily of dual-specificity phosphatases in GPVI- and CLEC-2-induced signaling. Motivated by our finding that DUSP3 is highly expressed in human and mouse platelets, we utilized Dusp3-KO mice to study the role of this phosphatase in hemostasis and thrombosis. DUSP3-deficient mice were more resistant to pulmonary thromboembolism than their WT littermates. Thrombus formation was strongly impaired in the model of FeCl3-induced injury of carotid artery in a platelet-specific manner. Intriguingly, DUSP3-deficient mice did not bleed spontaneously and showed normal tail bleeding times. These findings suggest that DUSP3 plays a key role in arterial thrombosis, but is dispensable for primary hemostasis.
Ex vivo, upon platelet stimulation with low concentrations of collagen, CVX, CRP, or rhodocytin, DUSP3 deficiency resulted in defective platelet aggregation, granule secretion, and integrin αIIbβ3 inside-out activation. In contrast, platelet activation mediated by GPCR agonists was not affected. DUSP3 deficiency led to a reduction of thrombus formation on collagen-coated surface under arterial shear, as well as lower PS exposure at the surface of adhered platelets. These data indicate that both GPVI- and CLEC-2-mediated platelet activation are impaired in DUSP3-deficient platelets.25-27 DUSP3 was dispensable for integrin αIIbβ3 outside-in signaling, as indicated by unaltered fibrin clot retraction (data not shown). Dusp3-KO mice exhibited levels of thrombus formation comparable to the previously reported GPVI-KO/FcRγ-KO28-30 CLEC-2-KO,31 CLEC-2-depleted,26 GPVI-depleted,32 and CLEC-2/GPVI-depleted mice.27 Similar to our findings in DUSP3-deficient mice, GPVI-KO and CLEC-2-KO mice do not exhibit prolonged bleeding time.28, 31, 33 DUSP3-deficient mice were also protected against pulmonary thromboembolism induced by a mixture of collagen and epinephrine, similar to GPVI-KO mice.33
At the molecular level, phosphorylation of the previously reported DUSP3 substrates ERK1/2 and JNK1/27 was not affected by DUSP3 deficiency, suggesting that signaling defects in DUSP3-deficient platelets are independent of the ERK1/2 and JNK1/2 pathways. However, we cannot exclude the possibility of functional or compensatory redundancies between DUSP3 and other phosphatases.
GPVI and CLEC-2 signaling pathways share many similarities, including the activation of Syk, PLCγ2, and adapter proteins such as LAT and SLP-76.34 However, there is also a significant difference: in GPVI-stimulated platelets, SFKs initiate signaling through phosphorylation of the FcRγ-associated ITAMs, leading to binding and activation of Syk;20, 21 in contrast, signaling through CLEC-2 depends on phosphorylation of CLEC-2 by Syk in an SFK-independent manner.35 Because DUSP3 deficiency limits platelet activation in response to both GPVI and CLEC-2 stimulation, SFK function is likely not controlled by DUSP3, which is also supported by our data showing that pY in SFKs is not altered in Dusp3-KO platelets. On the contrary, Syk may be directly or indirectly targeted by DUSP3. Intriguingly, however, DUSP3 deficiency decreased pY levels in Syk. Further, no hyperphosphorylated protein could be identified in pY blots of TLs from DUSP3-deficient platelets. This raises the question whether phosphoserine (pS) or phosphothreonine (pT) in Syk or other protein(s) may be targeted by DUSP3, a dual-specificity phosphatase able to dephosphorylate both pY and pS/pT. Given the limited recognition sites of available pS/pT antibodies, future studies using quantitative phospho-proteomics analysis will be necessary to address this question.
Platelet binding to von Willebrand factor (vWF) via GPIbα allows engagement of the collagen receptors GPVI and α2β1, leading to platelet arrest and subsequent platelet thrombus formation. The vWF-GPIb axis also induces GPVI dimerization, resulting in direct enhancement of GPVI interaction with collagen.36 However, platelets from Dusp3-KO mice exhibit normal binding to vWF-coated surface under flow (Supplementary Figure S7), suggesting intact GPIb signaling in these animals. Interestingly, a recent study by Nieswandt’s group showed that combined depletion of GPVI and CLEC-2 was sufficient to abrogate arterial thrombosis in mice.27 Thus, the defects observed in DUSP3-deficient platelets on CLEC-2- and GPVI-induced signaling are sufficient to explain the impaired thrombus formation in Dusp3-KO mice.
Finally, platelets are anucleate cells that are not amenable to RNA interference or recombinant DNA technologies. Thus, in order to corroborate our findings in human cells, we utilized a chemical genomics approach. Specifically, a small-molecule inhibitor of DUSP3 was identified via HTS of a large chemical library and subsequent SAR studies. Previously reported DUSP3 inhibitors suffer from either poor selectivity, lack of efficacy, or both,37-41 or cause immediate spontaneous aggregation of platelets (data not shown).42 Thus, we developed a novel, specific, and efficacious inhibitor, which we used to inhibit DUSP3 function in human washed platelets. Similar to DUSP3 deficiency in murine cells, inhibition of DUSP3 activity in human platelets led to suppression of platelet aggregation, specifically in response to CRP and rhodocytin, but not in response to the GPCR agonist thromboxane. MLS-0437605 is a drug-like compound43 and may serve as the basis for the development of potential therapeutics targeting DUSP3 for the treatment of arterial thrombosis.
Conclusion
We demonstrated that DUSP3 is a key signaling molecule for GPVI- and CLEC-2-induced platelet activation. We developed a specific small-molecule inhibitor of DUSP3, which efficiently inhibited human platelet activation in vitro. Given that Dusp3-KO mice remain healthy, do not exhibit any spontaneous phenotype, and do not suffer from increased bleeding events, our findings may lead to a novel antiplatelet therapy.
Supplementary Material
Acknowledgments
We thank the GIGA-animal, GIGA-genotranscriptomics and GIGA-imaging core facilities for their assistance and technical help.
Funding Sources: This work was supported by the Belgian National Fund for Scientific Research (F.R.S.-FNRS: PDR N° T.0105.13), the University of Liège (Fonds Spéciaux pour la Recherche to CO and SR), the Deutsche Forschungsgemeinschaft (Grant SFB1009 A09 to JAE), the Cardiovascular Centre of the Maastricht University Medical Centre (to JWMH), the American Heart Association (Innovative Research Grant 14IRG18980075 to LT), and Grants by the National Institutes of Health (5R01AI035603 to TM, U54 HG005033 to JC Reed/CPCCG, and 1R21CA132121 and 1R03MH084230 to LT).
Footnotes
Disclosures: None.
References
- 1.Michelson AD. Antiplatelet therapies for the treatment of cardiovascular disease. Nat Rev Drug Discov. 2010;9:154–169. doi: 10.1038/nrd2957. [DOI] [PubMed] [Google Scholar]
- 2.Watson SP, Auger JM, McCarty OJ, Pearce AC. Gpvi and integrin alphaiib beta3 signaling in platelets. J Thromb Haemost. 2005;3:1752–1762. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 3.Karisch R, Fernandez M, Taylor P, Virtanen C, St-Germain JR, Jin LL, Harris IS, Mori J, Mak TW, Senis YA, Ostman A, Moran MF, Neel BG. Global proteomic assessment of the classical protein-tyrosine phosphatome and “redoxome”. Cell. 2011;146:826–840. doi: 10.1016/j.cell.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Tautz L, Critton DA, Grotegut S. Protein tyrosine phosphatases: Structure, function, and implication in human disease. Methods Mol Biol. 2013;1053:179–221. doi: 10.1007/978-1-62703-562-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishibashi T, Bottaro DP, Chan A, Miki T, Aaronson SA. Expression cloning of a human dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1992;89:12170–12174. doi: 10.1073/pnas.89.24.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerignoli F, Rahmouni S, Ronai Z, Mustelin T. Regulation of map kinases by the vhr dual-specific phosphatase: Implications for cell growth and differentiation. Cell Cycle. 2006;5:2210–2215. doi: 10.4161/cc.5.19.3267. [DOI] [PubMed] [Google Scholar]
- 8.Wang JY, Yeh CL, Chou HC, Yang CH, Fu YN, Chen YT, Cheng HW, Huang CY, Liu HP, Huang SF, Chen YR. Vaccinia h1-related phosphatase is a phosphatase of erbb receptors and is down-regulated in non-small cell lung cancer. J Biol Chem. 2011;286:10177–10184. doi: 10.1074/jbc.M110.163295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahmouni S, Cerignoli F, Alonso A, Tsutji T, Henkens R, Zhu C, Louis-dit-Sully C, Moutschen M, Jiang W, Mustelin T. Loss of the vhr dual-specific phosphatase causes cell-cycle arrest and senescence. Nat Cell Biol. 2006;8:524–531. doi: 10.1038/ncb1398. [DOI] [PubMed] [Google Scholar]
- 10.Henkens R, Delvenne P, Arafa M, Moutschen M, Zeddou M, Tautz L, Boniver J, Mustelin T, Rahmouni S. Cervix carcinoma is associated with an up-regulation and nuclear localization of the dual-specificity protein phosphatase vhr. BMC Cancer. 2008;8:147. doi: 10.1186/1471-2407-8-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnoldussen YJ, Lorenzo PI, Pretorius ME, Waehre H, Risberg B, Maelandsmo GM, Danielsen HE, Saatcioglu F. The mitogen-activated protein kinase phosphatase vaccinia h1-related protein inhibits apoptosis in prostate cancer cells and is overexpressed in prostate cancer. Cancer Res. 2008;68:9255–9264. doi: 10.1158/0008-5472.CAN-08-1224. [DOI] [PubMed] [Google Scholar]
- 12.Amand M, Erpicum C, Bajou K, Cerignoli F, Blacher S, Martin M, Dequiedt F, Drion P, Singh P, Zurashvili T, Vandereyken M, Musumeci L, Mustelin T, Moutschen M, Gilles C, Noel A, Rahmouni S. Dusp3/vhr is a pro-angiogenic atypical dual-specificity phosphatase. Mol Cancer. 2014;13:108. doi: 10.1186/1476-4598-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oury C, Toth-Zsamboki E, Vermylen J, Hoylaerts MF. P2x(1)-mediated activation of extracellular signal-regulated kinase 2 contributes to platelet secretion and aggregation induced by collagen. Blood. 2002;100:2499–2505. doi: 10.1182/blood-2002-03-0812. [DOI] [PubMed] [Google Scholar]
- 14.Oury C, Kuijpers MJ, Toth-Zsamboki E, Bonnefoy A, Danloy S, Vreys I, Feijge MA, De Vos R, Vermylen J, Heemskerk JW, Hoylaerts MF. Overexpression of the platelet p2x1 ion channel in transgenic mice generates a novel prothrombotic phenotype. Blood. 2003;101:3969–3976. doi: 10.1182/blood-2002-10-3215. [DOI] [PubMed] [Google Scholar]
- 15.Gilio K, van Kruchten R, Braun A, Berna-Erro A, Feijge MA, Stegner D, van der Meijden PE, Kuijpers MJ, Varga-Szabo D, Heemskerk JW, Nieswandt B. Roles of platelet stim1 and orai1 in glycoprotein vi- and thrombin-dependent procoagulant activity and thrombus formation. J Biol Chem. 2010;285:23629–23638. doi: 10.1074/jbc.M110.108696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munnix IC, Kuijpers MJ, Auger J, Thomassen CM, Panizzi P, van Zandvoort MA, Rosing J, Bock PE, Watson SP, Heemskerk JW. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: Regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. 2007;27:2484–2490. doi: 10.1161/ATVBAHA.107.151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heemskerk JW, Sage SO. Calcium signalling in platelets and other cells. Platelets. 1994;5:295–316. doi: 10.3109/09537109409006439. [DOI] [PubMed] [Google Scholar]
- 18.Kuijpers MJ, Munnix IC, Cosemans JM, Vlijmen BV, Reutelingsperger CP, Egbrink MO, Heemskerk JW. Key role of platelet procoagulant activity in tissue factor-and collagen-dependent thrombus formation in arterioles and venules in vivo differential sensitivity to thrombin inhibition. Microcirculation. 2008;15:269–282. doi: 10.1080/10739680701653517. [DOI] [PubMed] [Google Scholar]
- 19.Senis YA, Tomlinson MG, Ellison S, Mazharian A, Lim J, Zhao Y, Kornerup KN, Auger JM, Thomas SG, Dhanjal T, Kalia N, Zhu JW, Weiss A, Watson SP. The tyrosine phosphatase cd148 is an essential positive regulator of platelet activation and thrombosis. Blood. 2009;113:4942–4954. doi: 10.1182/blood-2008-08-174318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watson SP, Asazuma N, Atkinson B, Berlanga O, Best D, Bobe R, Jarvis G, Marshall S, Snell D, Stafford M, Tulasne D, Wilde J, Wonerow P, Frampton J. The role of itam- and itim-coupled receptors in platelet activation by collagen. Thromb Haemost. 2001;86:276–288. [PubMed] [Google Scholar]
- 21.Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the src family kinases fyn and lyn with the collagen receptor glycoprotein vi-fc receptor gamma chain complex on human platelets. J Exp Med. 1998;188:267–276. doi: 10.1084/jem.188.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieswandt B, Brakebusch C, Bergmeier W, Schulte V, Bouvard D, Mokhtari-Nejad R, Lindhout T, Heemskerk JW, Zirngibl H, Fassler R. Glycoprotein vi but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001;20:2120–2130. doi: 10.1093/emboj/20.9.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Yao Y, Mason KD, O’Reilly LA, Henley KJ, Ono A, Hsiao S, Willcox A, Roberts AW, Huang DC, Salem HH, Kile BT, Jackson SP. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114:663–666. doi: 10.1182/blood-2009-01-200345. [DOI] [PubMed] [Google Scholar]
- 24.Bobkova EV, Liu WH, Colayco S, Rascon J, Vasile S, Gasior C, Critton DA, Chan X, Dahl R, Su Y, Sergienko E, Chung TD, Mustelin T, Page R, Tautz L. Inhibition of the hematopoietic protein tyrosine phosphatase by phenoxyacetic acids. ACS Med Chem Lett. 2011;2:113–118. doi: 10.1021/ml100103p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siljander P, Farndale RW, Feijge MA, Comfurius P, Kos S, Bevers EM, Heemskerk JW. Platelet adhesion enhances the glycoprotein vi-dependent procoagulant response: Involvement of p38 map kinase and calpain. Arterioscler Thromb Vasc Biol. 2001;21:618–627. doi: 10.1161/01.atv.21.4.618. [DOI] [PubMed] [Google Scholar]
- 26.May F, Hagedorn I, Pleines I, Bender M, Vogtle T, Eble J, Elvers M, Nieswandt B. Clec-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood. 2009;114:3464–3472. doi: 10.1182/blood-2009-05-222273. [DOI] [PubMed] [Google Scholar]
- 27.Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, Vogtle T, Remer K, Braun A, Bosl M, Watson SP, Nieswandt B. Combined in vivo depletion of glycoprotein vi and c-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol. 2013;33:926–934. doi: 10.1161/ATVBAHA.112.300672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato K, Kanaji T, Russell S, Kunicki TJ, Furihata K, Kanaji S, Marchese P, Reininger A, Ruggeri ZM, Ware J. The contribution of glycoprotein vi to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102:1701–1707. doi: 10.1182/blood-2003-03-0717. [DOI] [PubMed] [Google Scholar]
- 29.Dubois C, Panicot-Dubois L, Merrill-Skoloff G, Furie B, Furie BC. Glycoprotein vi-dependent and -independent pathways of thrombus formation in vivo. Blood. 2006;107:3902–3906. doi: 10.1182/blood-2005-09-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konishi H, Katoh Y, Takaya N, Kashiwakura Y, Itoh S, Ra C, Daida H. Platelets activated by collagen through immunoreceptor tyrosine-based activation motif play pivotal role in initiation and generation of neointimal hyperplasia after vascular injury. Circulation. 2002;105:912–916. doi: 10.1161/hc0802.105256. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, Kashiwagi H, Tomiyama Y, Yatomi Y, Umemura K, Shin Y, Hirashima M, Ozaki Y. Essential in vivo roles of the c-type lectin receptor clec-2: Embryonic/neonatal lethality of clec-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of clec-2-deficient platelets. J Biol Chem. 2010;285:24494–24507. doi: 10.1074/jbc.M110.130575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massberg S, Gawaz M, Gruner S, Schulte V, Konrad I, Zohlnhofer D, Heinzmann U, Nieswandt B. A crucial role of glycoprotein vi for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197:41–49. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lockyer S, Okuyama K, Begum S, Le S, Sun B, Watanabe T, Matsumoto Y, Yoshitake M, Kambayashi J, Tandon NN. Gpvi-deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb Res. 2006;118:371–380. doi: 10.1016/j.thromres.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Navarro-Nunez L, Langan SA, Nash GB, Watson SP. The physiological and pathophysiological roles of platelet clec-2. Thromb Haemost. 2013;109:991–998. doi: 10.1160/TH13-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Severin S, Pollitt AY, Navarro-Nunez L, Nash CA, Mourao-Sa D, Eble JA, Senis YA, Watson SP. Syk-dependent phosphorylation of clec-2: A novel mechanism of hem-immunoreceptor tyrosine-based activation motif signaling. J Biol Chem. 2011;286:4107–4116. doi: 10.1074/jbc.M110.167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loyau S, Dumont B, Ollivier V, Boulaftali Y, Feldman L, Ajzenberg N, Jandrot-Perrus M. Platelet glycoprotein vi dimerization, an active process inducing receptor competence, is an indicator of platelet reactivity. Arterioscler Thromb Vasc Biol. 2012;32:778–785. doi: 10.1161/ATVBAHA.111.241067. [DOI] [PubMed] [Google Scholar]
- 37.Usui T, Kojima S, Kidokoro S, Ueda K, Osada H, Sodeoka M. Design and synthesis of a dimeric derivative of rk-682 with increased inhibitory activity against vhr, a dual-specificity erk phosphatase: Implications for the molecular mechanism of the inhibition. Chem Biol. 2001;8:1209–1220. doi: 10.1016/s1074-5521(01)00089-8. [DOI] [PubMed] [Google Scholar]
- 38.Ueda K, Usui T, Nakayama H, Ueki M, Takio K, Ubukata M, Osada H. 4-isoavenaciolide covalently binds and inhibits vhr, a dual-specificity phosphatase. FEBS Lett. 2002;525:48–52. doi: 10.1016/s0014-5793(02)03065-x. [DOI] [PubMed] [Google Scholar]
- 39.Hirai G, Tsuchiya A, Koyama Y, Otani Y, Oonuma K, Dodo K, Simizu S, Osada H, Sodeoka M. Development of a vaccinia h1-related (vhr) phosphatase inhibitor with a nonacidic phosphate-mimicking core structure. Chem Med Chem. 2011;6:617–622. doi: 10.1002/cmdc.201100107. [DOI] [PubMed] [Google Scholar]
- 40.Park H, Jung SK, Jeong DG, Ryu SE, Kim SJ. Discovery of vhr phosphatase inhibitors with micromolar activity based on structure-based virtual screening. Chem Med Chem. 2008;3:877–880. doi: 10.1002/cmdc.200700348. [DOI] [PubMed] [Google Scholar]
- 41.Shi Z, Tabassum S, Jiang W, Zhang J, Mathur S, Wu J, Shi Y. Identification of a potent inhibitor of human dual-specific phosphatase, vhr, from computer-aided and nmr-based screening to cellular effects. Chembiochem. 2007;8:2092–2099. doi: 10.1002/cbic.200700397. [DOI] [PubMed] [Google Scholar]
- 42.Wu S, Vossius S, Rahmouni S, Miletic AV, Vang T, Vazquez-Rodriguez J, Cerignoli F, Arimura Y, Williams S, Hayes T, Moutschen M, Vasile S, Pellecchia M, Mustelin T, Tautz L. Multidentate small-molecule inhibitors of vaccinia h1-related (vhr) phosphatase decrease proliferation of cervix cancer cells. J Med Chem. 2009;52:6716–6723. doi: 10.1021/jm901016k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipinski C, Lombardo F, Dominy B, Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.