

Abstract
Purpose
To develop a genomic signature that predicts benefit from trastuzumab in human epidermal growth factor receptor 2–positive breast cancer.
Patients and Methods
DASL technology was used to quantify mRNA in samples from 1,282 patients enrolled onto the Combination Chemotherapy With or Without Trastuzumab in Treating Women With Breast Cancer (North Central Cancer Treatment Group N9831 [NCCTG-N9831]) adjuvant trastuzumab trial. Cox proportional hazard ratios (HRs), adjusted for significant clinicopathologic risk factors, were used to determine the association of each gene with relapse-free survival (RFS) for 433 patients who received chemotherapy alone (arm A) and 849 patients who received chemotherapy plus trastuzumab (arms B and C). Network and pathway analyses were used to identify key biologic processes linked to RFS. The signature was built by using a voting scheme.
Results
Network and functional ontology analyses suggested that increased RFS was linked to a subset of immune function genes. A voting scheme model was used to define immune gene enrichment based on the expression of any nine or more of 14 immune function genes at or above the 0.40 quantile for the population. This model was used to identify immune gene–enriched tumors in arm A and arms B and C. Immune gene enrichment was linked to increased RFS in arms B and C (HR, 0.35; 95% CI, 0.22 to 0.55; P < .001), whereas arm B and C patients who did not exhibit immune gene enrichment did not benefit from trastuzumab (HR, 0.89; 95% CI, 0.62 to 1.28; P = .53). Enriched immune function gene expression as defined by our predictive signature was not associated with increased RFS in arm A (HR, 0.90; 95% CI, 0.60 to 1.37; P = .64).
Conclusion
Increased expression of a subset of immune function genes may provide a means of predicting benefit from adjuvant trastuzumab.
INTRODUCTION
The Combination Chemotherapy With or Without Trastuzumab in Treating Women With Breast Cancer (North Central Cancer Treatment Group N9831 [NCCTG-N9831]) phase III adjuvant trastuzumab trial was one of several large studies that helped define the standard of care for patients with early-stage human epidermal growth factor receptor 2 (HER2) –positive breast cancer.1,2 Although these trials have demonstrated the efficacy of trastuzumab in the adjuvant setting, 20% to 25% of all patients with HER2-positive breast tumors develop tumor relapse despite HER2-targeted therapy. Several potential mechanisms have been proposed to account for differential response to HER2-targeted therapy, including overexpression of epidermal growth factor receptor (EGFR), cMYC, or ERBB3, mutational activation of PI3K, and mutational loss of PTEN.3 We recently reported that EGFR4 and soluble HER25 are prognostic, albeit not predictive of benefit from trastuzumab in patients from the NCCTG-N9831 trial. Analysis of their samples failed to demonstrate a link between increased disease-free survival and cMYC,6 ERBB3, or PTEN.7,8 Preliminary analyses have been presented from several clinical trials that addressed the role of PI3K mutations in HER2-positive breast cancer in the adjuvant, metastatic, and neoadjuvant settings.9,10 Although the preliminary findings are not invariably consistent in these trials, the data suggest that such mutations may be prognostic but are not predictive of benefit from HER2-targeted therapy. Therefore, a need exists to identify genomic and/or biologic features that are linked to risk of relapse after adjuvant trastuzumab treatment.
We report here whole transcriptome (DASL [cDNA-mediated annealing, selection, extension, and ligation]) analysis of 1,282 samples from early-stage HER2-positive breast tumors from the NCCTG-N9831 adjuvant trastuzumab trial. Our goal was to use these data to identify the basic biologic principles that underlie clinical outcome (relapse-free survival [RFS]) and to develop a genomic signature that would be useful for prediction of benefit from trastuzumab and potentially informative of novel therapeutic strategies. We report here that durable response to chemotherapy plus trastuzumab, but not chemotherapy alone, in the NCCTG-N9831 trial is strongly linked to expression of a subset of genes that are involved in the regulation of immune function. Several studies have linked tumor-infiltrating lymphocytes to chemotherapeutic response in breast tumors.11–14 It has been reported that there is an association between tumor-infiltrating lymphocytes and patient outcome in the Fluorouracil, Epirubicin, and Cyclophosphamide With Either Docetaxel or Vinorelbine, With or Without Trastuzumab, As Adjuvant Treatments of Breast Cancer (FinHER) adjuvant trastuzumab trial10,15 and the Neoadjuvant Study of Pertuzumab and Herceptin in an Early Regimen Evaluation (NeoSphere) and Combination Chemotherapy With or Without Capecitabine and/or Trastuzumab Before Surgery in Treating Women With Stage I, Stage II, or Stage III Breast Cancer (GeparQuattro) neoadjuvant trials.10,15,16 Our observations represent, to the best of our knowledge, the first demonstration of a cohort of immune function genes that seem to predict benefit from adjuvant trastuzumab in patients with HER2-positive breast tumors.
PATIENTS AND METHODS
Patients
In all, 3,505 patients were enrolled onto NCCTG-N9831 and were randomly assigned to three arms. All patients received anthracycline plus cyclophosphamide. Arm A patients received paclitaxel alone, arm B patients received paclitaxel followed by trastuzumab, and arm C patients received paclitaxel and concurrent trastuzumab (Data Supplement) after completion of the anthracycline plus cyclophosphamide therapy. From these patients, 1,282 samples (arm A, 433; arm B, 477; arm C, 372) were evaluable for DASL gene expression profiling (Fig 1). There were some differences in the clinicopathologic characteristics between the 1,282 patients included in this analysis and the 2,223 patients who were excluded (Data Supplement); however, the differences in outcomes among the three arms for the 1,282 included patients (Data Supplement) were similar to those reported for the trial as a whole.1 Because the point of this analysis is to determine the biologic basis that underlies benefit from trastuzumab, we pooled the 849 patients who received trastuzumab (arms B and C, hereafter arms B/C). Note that patients who were enrolled onto arm A after April 25, 2004, were allowed to cross over to arm C on April 25, 2005, so information for patients enrolled after April 25, 2004, was censored on April 25, 2005, which resulted in a considerable number of patients being censored within the first year. The Mayo Clinic institutional review board and the Correlative Science Committee of the North American Breast Cancer Group approved this translational study. Signed informed consent was obtained from all patients.
Fig 1.

CONSORT diagram describing the process whereby 1,282 samples were selected for downstream analyses. The Combination Chemotherapy With or Without Trastuzumab in Treating Women With Breast Cancer (North Central Cancer Treatment Group N9831 [NCCTG-N9831]) trial registered 3,505 patients of whom 1,282 (arm A, 433; arm B, 477; arm C, 372) were evaluable for DASL (cDNA-mediated annealing, selection, extension, and ligation) gene expression profiling. The median follow-up time was 6 years and 11 months and included all follow-up available through March 22, 2012. All tumors included in this article were tested for human epidermal growth factor receptor 2 (HER2) protein overexpression by immunohistochemistry and/or gene amplification by fluorescent in situ hybridization at a central laboratory (Mayo Clinic, Rochester, MN), and some tumors were excluded after central review of HER2 status. The largest cause of exclusion was insufficient tissue. Quality control (QC) failure after DASL analysis eliminated a small number of samples.
Genomic Analyses
Detailed methodologies for DASL analysis of gene expression and subsequent network and pathway analyses are given in the Data Supplement.
Statistical Analysis
A decision was made not to split the samples into separate training and validation sets for the signature development because of the limited power in the overall data set (204 recurrence events, with 89 in arm A and 115 in arms B/C). A split-sample approach, in which the data are divided into two cohorts for training and validation, fails to use all the information in the sample for signature development, yielding a noisy signature.17 For a preliminary validation of the signature, we used cross validation using a voting scheme.
Our analyses focused on genes that had a plausible biologic function with respect to trastuzumab response, as identified by network and functional ontology analysis. A voting scheme was used to develop a signature from a cohort of genes with a hazard ratio (HR) less than 1.0, adjusted-model P < .01, and interaction P < .05. Because it is likely that the contribution of individual genes within the biologic process might vary from tumor to tumor, we used a voting scheme to develop a signature. A tumor was designated as enriched for a biologic function if m or more of the genes in the functional group had one or more probes expressed above a quantile q threshold. To determine the best pair of m and q values, we searched a response surface that consisted of all quantile values of q between 0.25 and 0.75 by increments of 0.01. For each q/m pair, tumors were classified as enriched if they had m or more genes with at least one probe having an expression value above the q quantile for that probe across all samples. The q/m pair that was selected as best had the smallest P value for a comparison of RFS between women with enriched tumors (as determined by the voting scheme based on q/m values) who were treated with trastuzumab compared with women with enriched tumors who were not treated with trastuzumab.
Cross Validation of the Signature Development
A cross-validation method was used to assess whether the observed predictive nature of the signature was generalizable. Because the feature selection was based on identified biologic processes that differed between arm A and arms B/C, it was not possible to perform a complete cross validation of the entire process starting from feature selection. However, we were able to cross validate the development of the signature on the basis of the selected probes. Not being able to perform a full cross validation means that the cross validation results will still be biased.
A five-fold cross validation was replicated 100 times for determining the performance of the voting scheme for classifying tumors as enriched or not enriched and whether the resulting signature appears predictive of RFS. During each cross validation replicate, all patients were randomly assigned into five different cohorts. Four of the cohorts were then used to define the best set of q/m pairs by searching the q/m grid (Data Supplement). The q/m pairs determined in this fashion were then used to define the immune enrichment scores of the one fifth of the tumors that were left out. This procedure was repeated five times, leaving out one of the cohorts each time. The combined immune enrichment status from each cohort was then used in RFS analysis to determine how well the best q/m pair worked for that cross validation replicate. Replicating this analysis 100 times determined each tumor as immune enriched or not enriched.
Final Voting Scheme Values and Analysis
By using the selected q/m values, patients were grouped into enriched and nonenriched groups. Kaplan-Meier curves were used to summarize the RFS for each group and compared with a log rank test. Multivariable Cox proportional hazards regression models adjusted for predetermined prognostic factors and with treatment group, enriched status (determined by the voting scheme), and the treatment group–enriched status interaction term were used to determine whether the genomic signature was potentially predictive.
RESULTS
Gene Expression and Outcome Association
There was a total of 204 recurrent disease events: 89 in arm A and 115 in arms B/C. Multivariable Cox regression was used to identify genes significantly associated with RFS in arm A and arms B/C. We identified 473 genes that were associated with RFS at adjusted-model P < .01 in arm A (Data Supplement). We identified 485 genes significantly associated with RFS at adjusted-model P < .01 in arms B/C (Data Supplement).
Functional Analyses
Cytoscape Functional Interactome tools were used to construct four interactome models by using genes significantly associated with outcome (Fig 2). Each interactome map contained 10 to 12 highly interconnected modules (color coded) that were connected to other modules within the networks. Pathway enrichment statistics were used to assess the biologic significance of these four network models. The top-scoring pathways for each network are provided in Table 1. The most significant pathways associated with decreased RFS (HR > 1.0) in arm A were integrin signaling, coregulation of androgen receptor activity, and vascular smooth muscle contraction (Table 1). Pathways associated with increased RFS (HR < 1.0) in arm A included formation and maturation of mRNA transcript, ribosome, neuroactive ligand-receptor interaction, homologous recombination, and innate immunity signaling (Table 1).
Fig 2.

Network models reveal functional connections between genes associated with outcome in the Combination Chemotherapy With or Without Trastuzumab in Treating Women With Breast Cancer (North Central Cancer Treatment Group N9831 [NCCTG-N9831]) trial. The Cytoscape Functional Interactome tool integrates functional relationships defined by multiple bioinformatics tools, including protein-protein and gene-gene interaction data sets. This tool was used to define networks associated with either decreased relapse-free survival (RFS; panels A and C) or increased RFS (panels B and D) in arm A (panels A and B) or arms B/C (panels C and D). Networks were constructed by using genes with significant hazard ratios (P < .01), identified in the Data Supplement. Insertion of a single linker gene was allowed in network construction.
Table 1.
Pathway Enrichment Statistics From Cytoscape Networks
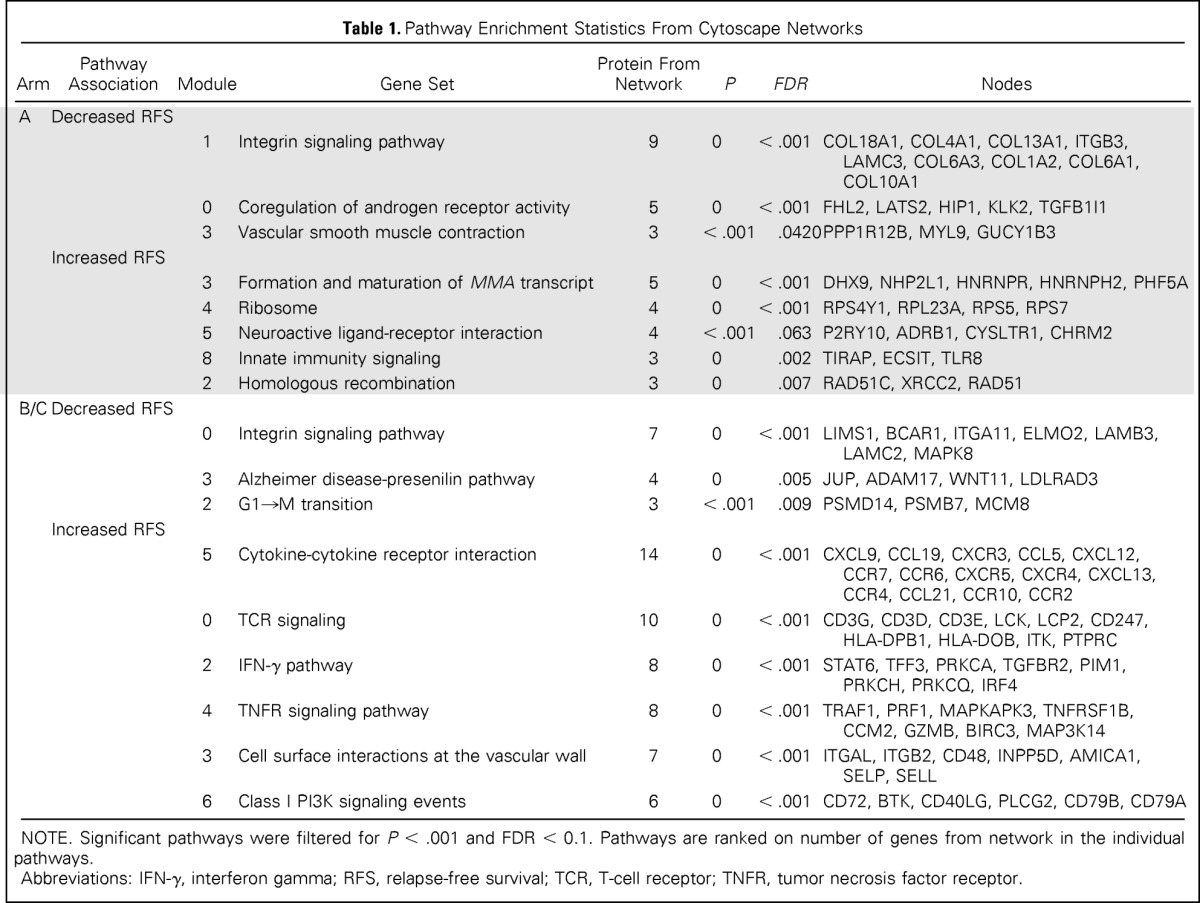
| Arm | Pathway Association | Module | Gene Set | Protein From Network | P | FDR | Nodes |
|---|---|---|---|---|---|---|---|
| A | Decreased RFS | ||||||
| 1 | Integrin signaling pathway | 9 | 0 | < .001 | COL18A1, COL4A1, COL13A1, ITGB3, LAMC3, COL6A3, COL1A2, COL6A1, COL10A1 | ||
| 0 | Coregulation of androgen receptor activity | 5 | 0 | < .001 | FHL2, LATS2, HIP1, KLK2, TGFB1I1 | ||
| 3 | Vascular smooth muscle contraction | 3 | < .001 | .0420 | PPP1R12B, MYL9, GUCY1B3 | ||
| Increased RFS | |||||||
| 3 | Formation and maturation of MMA transcript | 5 | 0 | < .001 | DHX9, NHP2L1, HNRNPR, HNRNPH2, PHF5A | ||
| 4 | Ribosome | 4 | 0 | < .001 | RPS4Y1, RPL23A, RPS5, RPS7 | ||
| 5 | Neuroactive ligand-receptor interaction | 4 | < .001 | .063 | P2RY10, ADRB1, CYSLTR1, CHRM2 | ||
| 8 | Innate immunity signaling | 3 | 0 | .002 | TIRAP, ECSIT, TLR8 | ||
| 2 | Homologous recombination | 3 | 0 | .007 | RAD51C, XRCC2, RAD51 | ||
| B/C | Decreased RFS | ||||||
| 0 | Integrin signaling pathway | 7 | 0 | < .001 | LIMS1, BCAR1, ITGA11, ELMO2, LAMB3, LAMC2, MAPK8 | ||
| 3 | Alzheimer disease-presenilin pathway | 4 | 0 | .005 | JUP, ADAM17, WNT11, LDLRAD3 | ||
| 2 | G1→M transition | 3 | < .001 | .009 | PSMD14, PSMB7, MCM8 | ||
| Increased RFS | |||||||
| 5 | Cytokine-cytokine receptor interaction | 14 | 0 | < .001 | CXCL9, CCL19, CXCR3, CCL5, CXCL12, CCR7, CCR6, CXCR5, CXCR4, CXCL13, CCR4, CCL21, CCR10, CCR2 | ||
| 0 | TCR signaling | 10 | 0 | < .001 | CD3G, CD3D, CD3E, LCK, LCP2, CD247, HLA-DPB1, HLA-DOB, ITK, PTPRC | ||
| 2 | IFN-γ pathway | 8 | 0 | < .001 | STAT6, TFF3, PRKCA, TGFBR2, PIM1, PRKCH, PRKCQ, IRF4 | ||
| 4 | TNFR signaling pathway | 8 | 0 | < .001 | TRAF1, PRF1, MAPKAPK3, TNFRSF1B, CCM2, GZMB, BIRC3, MAP3K14 | ||
| 3 | Cell surface interactions at the vascular wall | 7 | 0 | < .001 | ITGAL, ITGB2, CD48, INPP5D, AMICA1, SELP, SELL | ||
| 6 | Class I PI3K signaling events | 6 | 0 | < .001 | CD72, BTK, CD40LG, PLCG2, CD79B, CD79A |
NOTE. Significant pathways were filtered for P < .001 and FDR < 0.1. Pathways are ranked on number of genes from network in the individual pathways.
Abbreviations: IFN-γ, interferon gamma; RFS, relapse-free survival; TCR, T-cell receptor; TNFR, tumor necrosis factor receptor.
Among the trastuzumab-treated patients (arms B/C), integrin signaling, Alzheimer disease-presenilin pathway, and G→M cell cycle transition pathways were the most significant pathways linked to decreased RFS (HR > 1.0; Table 1). The most significant pathways associated with increased RFS (HR < 1.0) after adjuvant trastuzumab therapy (Table 1) included cytokine-cytokine receptor interaction, T-cell receptor signaling in CD8+ T cells, interferon gamma pathway, tumor necrosis factor receptor signaling pathway, cell surface interaction at the vascular endothelium, and class I PI3K signaling events. The observation that four of six significant pathways are linked to immunologic functions strongly suggests an association between immune response and increased RFS in trastuzumab-treated patients with HER2-positive breast tumors.
Gene Ontology (GO) Biologic Process terms were defined for each gene with a significant HR (adjusted-model P < .01), as described in the Data Supplement. Fisher's exact test was used to identify 13 GO Biologic Process descriptors that exhibited significantly different distribution in arm A and arms B/C at P < .01 (Table 2); the most significant was immune response (GO:0006955_BP). Ten of 13 biologic processes were linked to immune functions, including T-cell and B-cell responses, chemokine signaling and chemotaxis, and inflammation. These results suggest that a major immunologic component is predictive of RFS among trastuzumab-treated patients with early-stage HER2-positive breast cancer.
Table 2.
Analysis of Biologic Process Defined by GO Terms Reveals Enrichment of Immune Function Terms in Arms B and C
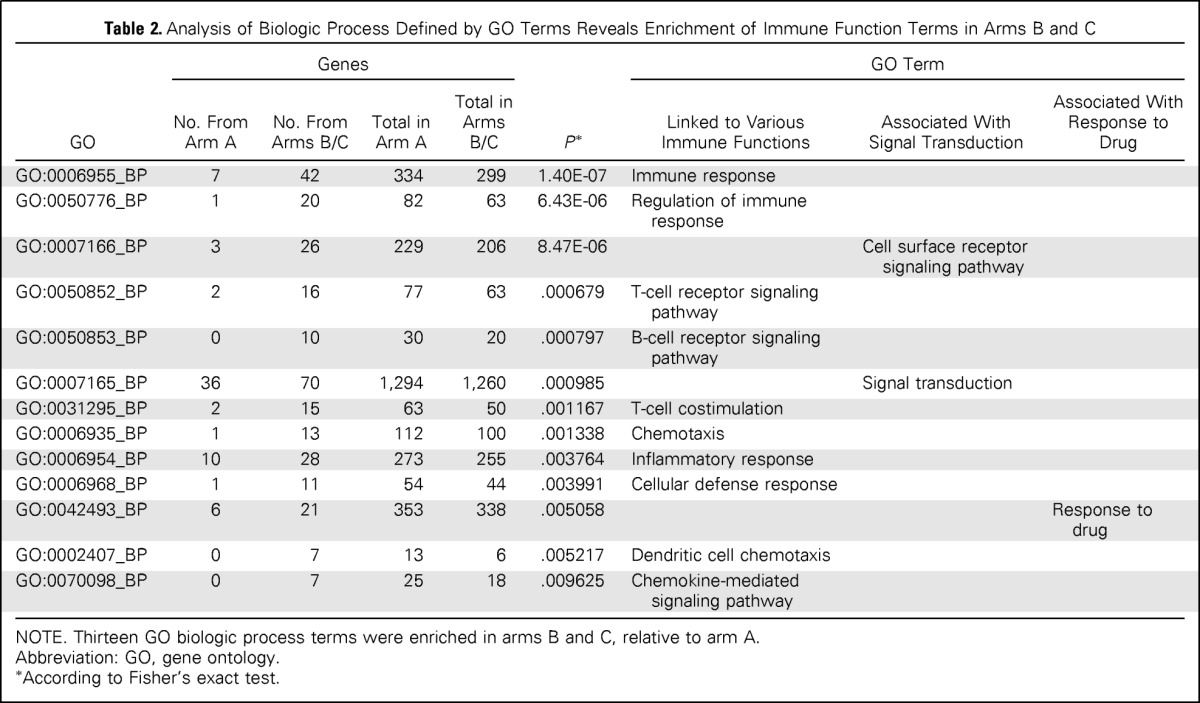
| GO | Genes |
P* | GO Term |
|||||
|---|---|---|---|---|---|---|---|---|
| No. From Arm A | No. From Arms B/C | Total in Arm A | Total in Arms B/C | Linked to Various Immune Functions | Associated With Signal Transduction | Associated With Response to Drug | ||
| GO:0006955_BP | 7 | 42 | 334 | 299 | 1.40E-07 | Immune response | ||
| GO:0050776_BP | 1 | 20 | 82 | 63 | 6.43E-06 | Regulation of immune response | ||
| GO:0007166_BP | 3 | 26 | 229 | 206 | 8.47E-06 | Cell surface receptor signaling pathway | ||
| GO:0050852_BP | 2 | 16 | 77 | 63 | .000679 | T-cell receptor signaling pathway | ||
| GO:0050853_BP | 0 | 10 | 30 | 20 | .000797 | B-cell receptor signaling pathway | ||
| GO:0007165_BP | 36 | 70 | 1,294 | 1,260 | .000985 | Signal transduction | ||
| GO:0031295_BP | 2 | 15 | 63 | 50 | .001167 | T-cell costimulation | ||
| GO:0006935_BP | 1 | 13 | 112 | 100 | .001338 | Chemotaxis | ||
| GO:0006954_BP | 10 | 28 | 273 | 255 | .003764 | Inflammatory response | ||
| GO:0006968_BP | 1 | 11 | 54 | 44 | .003991 | Cellular defense response | ||
| GO:0042493_BP | 6 | 21 | 353 | 338 | .005058 | Response to drug | ||
| GO:0002407_BP | 0 | 7 | 13 | 6 | .005217 | Dendritic cell chemotaxis | ||
| GO:0070098_BP | 0 | 7 | 25 | 18 | .009625 | Chemokine-mediated signaling pathway | ||
NOTE. Thirteen GO biologic process terms were enriched in arms B and C, relative to arm A.
Abbreviation: GO, gene ontology.
According to Fisher's exact test.
We identified 87 immune function genes, defined by the 10 immune function GO terms that were enriched in arms B/C (Table 2) and associated with increased RFS (HR < 1.0) at adjusted-model P < .01 (Data Supplement). To determine which of these probes was potentially predictive, we selected probes among the 87 immune function genes that had a significant (P < .05) interaction term (ie, probe by treatment group term). This resulted in a list of 14 genes (Data Supplement).
Voting Scheme Parameters
The response surface analysis resulted in two unique sets of q/m values. The first set (q = 40 and m = 9 [q40m9]) occurred 235 times (47%) and identified 226 enriched patients (52.2%) in arm A and 441 enriched patients (51.9%) in arms B/C. The second set (q = 58 and m = 8 [q58m8]) occurred 265 times (53%) and identified 139 enriched patients (32.1%) in arm A and 310 enriched patients (36.5%) in arms B/C. Because both sets of optimum q/m values occurred about the same number of time, q/m pair q40m9 was selected as the optimum.
Final Signature Analysis
On the basis of the optimum set of q/m values, a tumor was designated as immune enriched if any nine (m) or more of the 14 immune function genes were expressed at or above the 0.40 quantile (q) expression value for one or more probes. This signature was used to “bin” tumors in arm A and arms B/C into immune response–enriched (IRE) and nonimmune response–enriched (NIRE) groups. Baseline clinicopathologic characteristics (including hormone receptor status, nodal status, and tumor grade) were not significantly different in enriched and nonenriched tumor cohorts (Data Supplement), although the immune gene–enriched population tended to contain more tumors that were ≤ 2 cm in size. The difference in RFS between the IRE and NIRE tumors in arm A was not statistically significant (HR, 0.90; 95% CI, 0.60 to 1.37; P = .64; Fig 3A, blue and yellow curves). Patients with IRE tumors exhibited significantly increased RFS after adjuvant trastuzumab (black curve), compared with IRE patients who did not receive trastuzumab (blue curve; HR, 0.35; 95% CI, 0.22 to 0.55; P < .001). Furthermore, the RFS of trastuzumab-treated patients whose tumors were NIRE (red curve) was not significantly different from the RFS of IRE patients who received chemotherapy alone (HR, 0.89; 95% CI, 0.62 to 1.28; P = .53). A multivariable Cox model was evaluated that included the prognostic factors as adjusting variables, immune-enrichment status, treatment group, and an immune-enrichment status and treatment group interaction group term. In this model, the interaction term value was significant (P < .001). Figures 3B and 3C show the effect of the interaction on trastuzumab RFS. There is a difference in RFS for patients with IRE tumors treated with trastuzumab compared with those who received chemotherapy alone (HR, 0.36; 95% CI, 0.23 to 0.56; P < .001; Fig 3B). There is no difference in RFS for patients with NIRE tumors treated with trastuzumab and those who received chemotherapy alone (HR, 0.98; 95% CI, 0.68 to 1.41; P = .91; Fig 3C).
Fig 3.
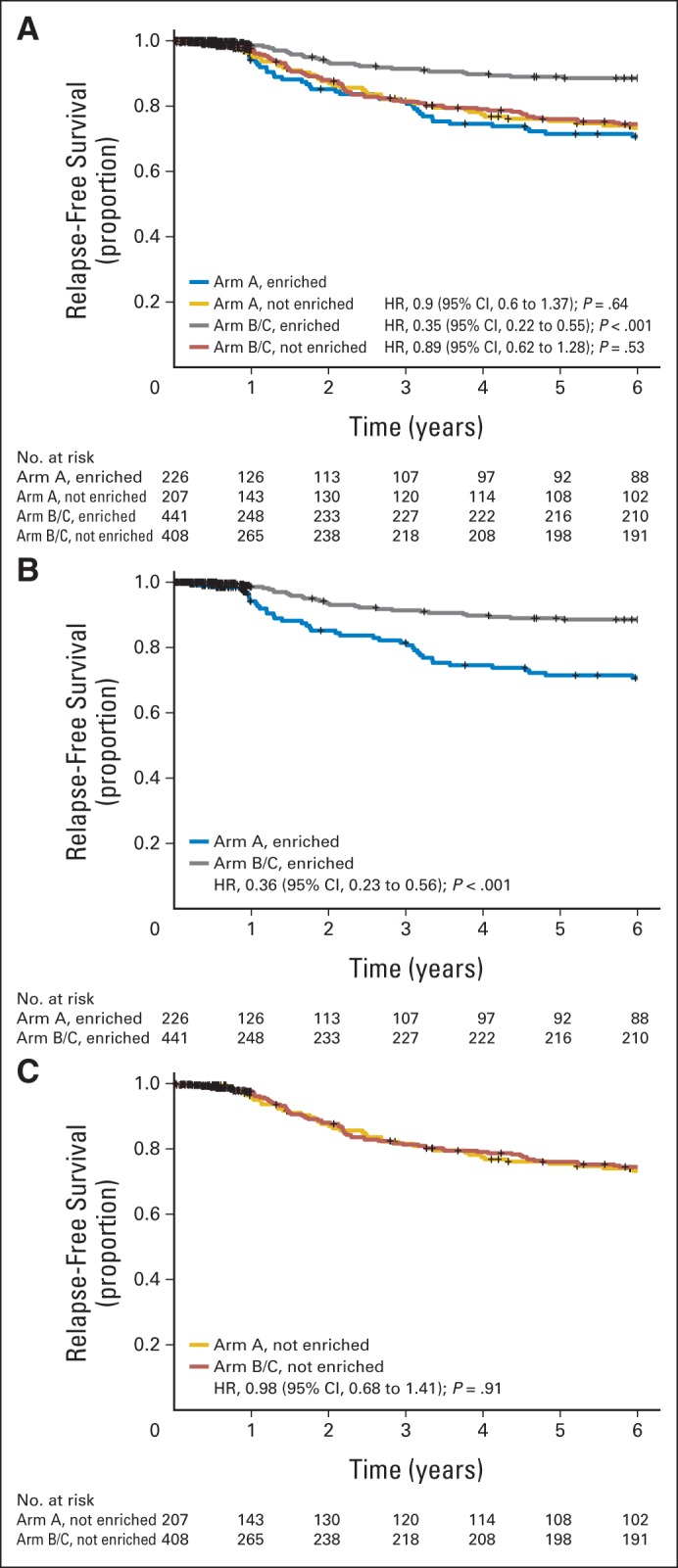
A cohort of immune function genes is strongly associated with outcome after trastuzumab treatment in the N9831 trial but has no effect on relapse-free survival (RFS) following chemotherapy alone. Tumors in arm A and arms B/C were “binned” into immune-enriched and not immune-enriched by using the voting model in which enrichment was defined by the m9q41 model. (A) RFS for enriched and not enriched subsets of tumors from both arms. (B) RFS for the enriched subset of tumors from both arms. (C) RFS for the not enriched subset of tumors from both arms. HR, hazard ratio.
Cross Validation Results
To validate the signature, we performed a five-fold cross validation, described in “Patients and Methods.” The immune enrichment status of the tumors in the “left out” groups for each iteration were combined, so that all samples within the study were assigned as enriched or nonenriched. The range of RFS HRs between the arm A and arms B/C IRE tumors in the 100 cross-validation instances was 0.24 to 0.40, with all 100 P values less than .001. The range of RFS HRs in the 100 cross-validation instances between arm A and arms B/C NIRE tumors was 0.84 to 1.05, with all 100 P values greater than .29. Appendix Figure A1 (online only) shows the RFS curves for each enrichment status and treatment group combination obtained from cross validation. The results shown in Appendix Figure A1 are similar to those in Figure 3A.
DISCUSSION
We report here findings from whole transcriptome analysis of the NCCTG-N9831 adjuvant trastuzumab trial of early-stage HER2-positive breast cancer. We used a system biology approach based on identification of processes and pathways associated with patient outcome, whereas Pogue-Geile et al18 built a predictive signature based on ESR1 and ERBB2 expression, amplification, and signaling data from the Doxorubicin and Cyclophosphamide Plus Paclitaxel With or Without Trastuzumab in Treating Women With Node-Positive Breast Cancer That Overexpresses HER2 (NSABP B-31) adjuvant trastuzumab trial. We had considered this approach but were discouraged by the observations that estrogen receptor (ER) status was prognostic but not predictive in the combined analysis of 10-year follow-up on patients in the NCCTG-N9831 and NSABP B-31 trials19 and ERBB2 mRNA, assayed by quantitative polymerase chain reaction in arms A and C of the NCCTG-N9831 trial, was not predictive of benefit from trastuzumab.20 Instead, we have used an empirical approach, informed by our network analysis, which suggested that immune function gene expression patterns are strongly linked to RFS in trastuzumab-treated patients.
Both network and functional ontology analysis revealed a strong association between the immune gene expression status of the tumor and RFS following adjuvant trastuzumab. Our data indicate that a subset of HER2-positive tumors likely manifests a high level of immunologic activity, as evidenced by expression of a diverse cohort of immune function genes. Although this immune-enriched subset of tumors is manifested in the samples in arm A, this feature appears to have no effect on RFS after chemotherapy alone. In contrast, tumors that were enriched for immune function genes exhibited increased RFS (relative to chemotherapy alone), whereas trastuzumab did not increase RFS in patients whose tumors did not exhibit increased immune function gene expression scores. These results suggest that benefit from adjuvant trastuzumab, beyond the benefit conveyed by chemotherapy alone, may reside largely within this cohort of immune-enriched tumors. However, because we were not able to fully cross validate the development process of the signature, our results are likely to be somewhat biased.
Both immune gene signatures and lymphocyte infiltration have been associated with clinical outcome and pathologic complete response in ER-positive and ER-negative breast tumors.21–25 Several studies of immune functions in small numbers of HER2-positive tumor samples have been published. The earlier studies did not involve trastuzumab treatment but generally reported a link between immune functions and favorable outcome in both adjuvant and neoadjuvant settings.26,27 An interesting report implicated an immune response signature in stromal cells isolated from HER2-positive tumors.22 Conversely, a recent report did not confirm an association between immune function genes and outcome in HER2-positive patients treated with chemotherapy alone,26 consistent with our conclusions.
Preclinical data strongly support both an innate and acquired immune component of anti-HER2–targeted therapy.28,29 Preliminary reports of relevant translational data from three early-stage breast cancer trastuzumab trials have been presented in abstract form. The Neoadjuvant Herceptin (NOAH) trial30 reported that a plasma cell gene set was predictive of complete pathologic response in 114 patients randomly assigned to receive chemotherapy or chemotherapy plus trastuzumab. The FinHER trial reported that lymphocyte infiltration was associated with increased disease-free survival in 209 patients randomly assigned to receive adjuvant trastuzumab or no trastuzumab for 9 weeks after chemotherapy.10 Likewise, a preliminary report from the NeoSphere trial16 reported that metagene signatures involving immune function genes were predictive of complete pathologic response to neoadjuvant anti-HER2 therapy. Thus, it appears that the data from these preliminary analyses are entirely consistent with and support our conclusion that there is an association between immune function and benefit from trastuzumab in the adjuvant setting and suggest that a similar association may prevail in the neoadjuvant setting.
The potential clinical significance of our results, within the context of identification of patients who are likely to benefit (increased RFS) from adjuvant trastuzumab, is considerable. Identification of patients who are unlikely to benefit from trastuzumab on the basis of evaluation of the immune status of the tumor before initiation of therapy may have even greater significance. Patients with low immune function gene expression scores might be enrolled onto trials to test the efficacy of HER2-targeted regimens that combine trastuzumab with some other therapeutic agent. Alternatively, these patients might be the focus of future clinical trials designed to evaluate therapeutic approaches that might enhance the immune activity within HER2-positive tumors and thereby sensitize the tumors to biologic therapies. An obvious strategy involves attenuation of immune-suppressive signaling pathways (eg, by targeting entities such as the PD1/CD279 receptor and/or its ligand PDL1/CD274).16 An alternative strategy involves enrolling patients with nonenriched tumors onto clinical trials to test the effects of modification of the immunoglobulin backbone of anti-HER2 antibodies to enhance immunoreactivity.31 These alternative approaches to harness the immune system gene expression profiles to predict and potentially enhance benefit from adjuvant trastuzumab could have considerable significance for those patients with HER2-positive breast cancer who are likely to develop tumor relapse after adjuvant anti-HER2 therapy.
Supplementary Material
Acknowledgment
The authors thank Christopher Kolbert and the Mayo Clinic Genome Facility for outstanding technical support and Daniel J. Sargent, MD, (Mayo Clinic) for critical reading of the manuscript and discussion of the results.
Glossary Terms
- Cox proportional hazards regression model:
a statistical model for regression analysis of censored survival data, examining the relationship of censored survival distribution to one or more covariates. This model produces a baseline survival curve, covariate coefficient estimates with their standard errors, risk ratios, 95% CIs, and significance levels.
- epidermal growth factor receptor (EGFR):
a member of a family of receptors (HER2, HER3, HER4 are other members of the family) that binds to the EGF, TGF-α, and other related proteins, leading to the generation of proliferative and survival signals within the cell. EGFR (also known as HER1) also belongs to the larger family of tyrosine kinase receptors and is generally overexpressed in several solid tumors of epithelial origin.
- genomic signatures:
the expression of a set of genes in a biologic sample (eg, blood, tissue) using microarray technology.
- metagene:
a single aggregate measure of the expression of a group of genes that usually show coordinated expression in a set of samples. The metagene value may be defined as the arithmetic mean or the weighted average or some other mathematical combination of the genes of interest.
- trastuzumab:
a humanized anti-ErbB2 monoclonal antibody approved for treating patients whose breast cancers overexpress the ErbB2 protein or demonstrate ErbB2 gene amplification. It is currently being tested in combination with other therapies.
Appendix
DASL Analysis of mRNA Abundance
Individual tumor blocks, procured between 2000 and 2005, were examined microscopically, and tissue punches were obtained from demarcated areas of invasive tumor by using a 1-mm biopsy punch with plunger (Fisher Scientific, Pittsburgh, PA). Total RNA was extracted from at least one 1-mm tissue punch. Punches were deparaffinized in Citrisolv (Fisher Scientific) at room temperature for 30 minutes. The Citrisolv was aspirated, and the tissue was washed with 100% ethanol, vortexed, and centrifuged twice. Ethanol was removed, and the tissue was dried at 37°C for 10 minutes. The samples were then incubated in Proteinase K Digestion buffer and proteinase K (1 μg/μL) overnight (for at least 8 hours) at 56°C. The digested tissue was incubated for 15 minutes at 80°C and centrifuged (14,000 rpm) for 2 minutes at room temperature. The supernatant was collected, and the RNA extraction, including DNase I treatment, was completed by using the RNeasy formalin-fixed paraffin-embedded (FFPE) kit on an automated QIAcube platform according to the manufacturer's instructions (QIAGEN, Valencia, CA). The concentration of the purified RNA was determined by using a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE). Purified total RNA was stored at −80°C. A similar RNA extraction procedure using QIAGEN′s RNeasy FFPE kit has been shown to yield RNA suitable for gene expression profiling of FFPE breast tumors (Ton CC, et al: Breast Cancer Res Treat 125:879-883, 2011). Labeling and hybridizations to BeadChips (HumanRef v4 Beadchip; Illumina, San Diego, CA) were performed as previously described (Ton CC, et al: Breast Cancer Res Treat 125:879-883, 2011; Bibikova M, et al: Am J Pathol 165:1799-1807, 2004; Li HR, et al: Cancer Res 66:4079-4088, 2006; Reinholz MM, et al: BMC Med Genomics 3:60, 2010) with slight modifications. Samples (200 ng RNA) were randomized across 17 plates and subsequently to 136 chips according to date and order of RNA extraction, clinicopathologic characteristics, year on study, and treatment arm. The nonbackground corrected expression values from BeadStudio underwent a quality-control evaluation by using the metrics of proportion of probes detected at P < .05, interquartile range, and skewness (Mahoney DW, et al: BMC Res Notes 6:33, 2013). In addition, a stress metric that quantified the amount of transformation required for an array to be normalized was applied. The replicated patient sample with the lowest stress value was used for analysis. Samples with a stress value more than log2(1.5) were deemed to be poor quality and were removed. The remaining data were normalized by using quantile normalization. A detailed description of the quality assessment protocols applied to these samples has been published elsewhere (Mahoney DW, et al: BMC Res Notes 6:33, 2013).
Intra- and interplate technical replicates were performed by using randomly selected patient samples from the Combination Chemotherapy With or Without Trastuzumab in Treating Women With Breast Cancer (North Central Cancer Treatment Group N9831 [NCCTG-N9831]) trial and Universal Human Reference RNA (UHRR) control samples (Ambion Life Technologies, Grand Island, NY). UHRR samples were analyzed in duplicate on every plate, with correlation coefficients of more than 0.9 for both UHRR and patient samples (Data Supplement) well within US Food and Drug Administration and National Cancer Institute guidelines recommending that coefficient of variation values be less than 15% to be considered a precise assay (US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine: Guidance for Industry: Bioanalytical Method Validation, 2001).
Statistical Analysis of Cox Hazard Ratios
The primary end point was relapse-free survival (RFS), which was defined as the time from random assignment to first local, regional, or distant recurrence, or the development of a new contralateral primary breast cancer. Multivariable Cox models (adjusting for nodal status, tumor size, hormone receptor status, age, and tumor grade) were used to evaluate the association between RFS and probe expression for all genes. We assessed the association separately within each patient group to understand biologic processes that might be involved with response to trastuzumab. Probes meeting the filtering criteria and having an adjusted-model P < .01 were considered to be significantly associated with RFS for the purpose of the functional analysis. Cox proportional models, which included the prognostic factors as adjusting variables, were evaluated on the set of all patients and included probe, treatment group, and probe-treatment group interaction terms to identify probes that were potentially predictive of trastuzumab response.
Functional Analysis
Cox hazard ratios were determined for all genes from the DASL analysis by using time to event (RFS) as a continuous variable. The Cytoscape Functional Interactome tool (Matthews L, et al: Nucleic Acids Res 37:D619-D622, 2009) was used to define network associations among genes with Cox hazard ratios with adjusted-model P < .01. Functional processes associated with network components were deduced from the pathway enrichment statistics function within the Cytoscape Functional Interactome tool.
Enrichment of Gene Ontology Biologic Process Terms
Functional ontology enrichment was determined by analysis of Gene Ontology Biologic Process terms using Fisher's exact test. Individual gene ontology (GO) terms apply to many genes, and individual genes may have many associated GO terms. This one-to-many relationship between genes and GO terms was downloaded from the BioMart portal at Ensembl (http://useast.ensembl.org/biomart/martview/). The Ensembl human gene annotation version 70 (v70) was used to identify genes. A script developed in-house was used to assign each gene into all possible GO terms to which it belongs. This was done on both the genes with significant hazard ratios (HRs) and all genes in the v70 annotation. For each of the GO terms, a Fisher's exact test was performed on a 2 × 2 contingency table with (1) the number of genes with significant HRs belonging to the GO term from arm A; (2) the number of genes with significant HRs belonging to the GO term from arms B/C; (3) the numbers of genes excluding those in (1) from all v70 genes that were assigned to the GO term; and (4) the numbers of genes excluding those in (2) from all v70 genes that were assigned to the GO term.
Fig A1.
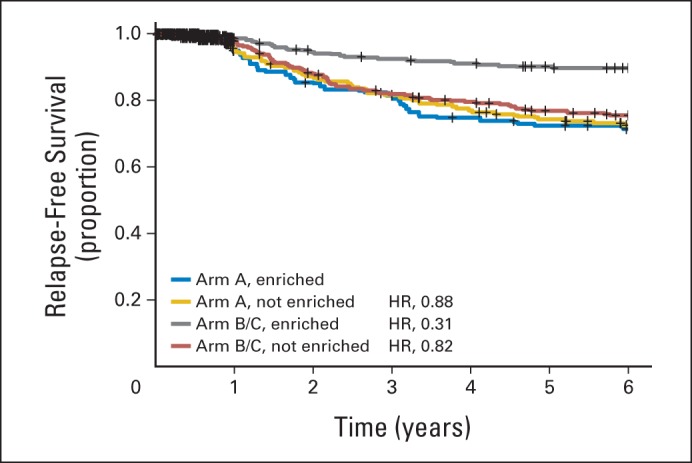
Cross validation of the immune function score model. The data were randomly split into five cohorts, and the optimal q/m combination was selected based on four cohorts. This q/m relationship was then used to determine whether a tumor was immune enriched (IRE) or not immune enriched (NIRE) in the remaining cohort. Each tumor was classified 100 times (once for each cross validation). The curves show the results of the observed relapse-free survival based on these 100 different cross-validation sets; hence, there is a total of 128,200 observations (arm A: IRE, 18,117 and NIRE, 25,183; arms B/C: IRE, 36,877 and NIRE, 48,023). HR, hazard ratio.
Footnotes
See accompanying editorial on page 673
Supported in part by Grant No. CA129949 from the National Cancer Institute, the Breast Cancer Research Foundation, and the 26.2 with Donna Foundation. Additional support for infrastructure was provided by Grants No. CA25225 and CA114740 from the National Institutes of Health, and Grant No. CA15083 from the Mayo Clinic Comprehensive Cancer Center and the Mayo Foundation. Tissues were processed by Wilma Lingle, Michael Zschunke, and Greg Reinholz of the Mayo Clinic Biospecimens Acquisition and Processing facility.
Clinical trial information: NCT00005970.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Edith A. Perez, E. Aubrey Thompson, Jeanette E. Eckel-Passow, Amylou C. Dueck, Jin Jen, Jian-Bing Fan, Monica M. Reinholz
Collection and assembly of data: Edith A. Perez, S. Keith Anderson, Amylou C. Dueck, Jin Jen, George W. Sledge, Eric P. Winer, Julie R. Gralow
Data analysis and interpretation: Edith A. Perez, E. Aubrey Thompson, Karla V. Ballman, S. Keith Anderson, Yan W. Asmann, Krishna R. Kalari, Amylou C. Dueck, Kathleen S. Tenner, Xochiquetzal J. Geiger, Ann E. McCullough, Beiyun Chen, Robert B. Jenkins, Monica M. Reinholz
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Genomic Analysis Reveals That Immune Function Genes Are Strongly Linked to Clinical Outcome in the North Central Cancer Treatment Group N9831 Adjuvant Trastuzumab Trial
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Edith A. Perez
No relationship to disclose
E. Aubrey Thompson
No relationship to disclose
Karla V. Ballman
No relationship to disclose
S. Keith Anderson
Patents, Royalties, Other Intellectual Property: Royalties from US Patent Number 20090298082, December 3, 2009, Biomarker Panels for Predicting Prostate Cancer Outcomes
Yan W. Asmann
No relationship to disclose
Krishna R. Kalari
No relationship to disclose
Jeanette E. Eckel-Passow
No relationship to disclose
Amylou C. Dueck
No relationship to disclose
Kathleen S. Tenner
Stock or Other Ownership: Merck (I)
Jin Jen
No relationship to disclose
Jian-Bing Fan
Employment: Illumina
Stock or Other Ownership: Illumina
Xochiquetzal J. Geiger
No relationship to disclose
Ann E. McCullough
No relationship to disclose
Beiyun Chen
No relationship to disclose
Robert B. Jenkins
No relationship to disclose
George W. Sledge
Leadership: Syndax
Stock or Other Ownership: Syndax
Honoraria: Genentech, Symphogen
Consulting or Advisory Role: Symphogen
Eric P. Winer
Consulting or Advisory Role: Verastem
Research Funding: Genentech/Roche (Inst), Novartis (Inst)
Travel, Accommodations, Expenses: Genentech/Roche
Julie R. Gralow
Research Funding: Roche/Genetech (Inst), Novartis (Inst), Amgen (Inst)
Monica M. Reinholz
Employment: Ventana Medical Systems
REFERENCES
- 1.Perez EA, Romond EH, Suman VJ, et al. Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-postive breast cancer: Joint analysis of data from NCCTG N9831 and NSABP B-31. J Clin Oncol. 2011;29:3366–3373. doi: 10.1200/JCO.2011.35.0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez EA, Suman VJ, Davidson NE, et al. Sequential versus concurrent trastuzumab in adjuvant chemotherapy for breast cancer. J Clin Oncol. 2011;29:4491–4497. doi: 10.1200/JCO.2011.36.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arteaga CL, Sliwkowski MX, Osborne CK, et al. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 4.Cheng H, Ballman K, Vassilakopoulou M, et al. EGFR expression is associated with decreased benefit from trastuzumab in the NCCTG N9831 (Alliance) trial. Br J Cancer. 2014;111:1065–1071. doi: 10.1038/bjc.2014.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreno-Aspitia A, Hillman DW, Dyar SH, et al. Soluble human epidermal growth factor receptor 2 (HER2) levels in patients with HER2-positive breast cancer receiving chemotherapy with or without trastuzumab: Results from North Central Cancer Treatment Group adjuvant trial N9831. Cancer. 2013;119:2675–2682. doi: 10.1002/cncr.28130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez EA, Jenkins RB, Dueck AC, et al. C-MYC alterations and association with patient outcome in early-stage HER2-positive breast cancer from the North Central Cancer Treatment Group N9831 adjuvant trastuzumab trial. J Clin Oncol. 2011;29:651–659. doi: 10.1200/JCO.2010.30.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez EA, Dueck AC, McCullough AE, et al. Impact of PTEN protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2-positive breast cancer in the North Central Cancer Treatment Group N9831 Trial. J Clin Oncol. 2013;31:2115–2122. doi: 10.1200/JCO.2012.42.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez EA, Ballman KV, Reinholz MM, et al. Impact of quantitative measurement of HER2, HER3, HER4, EGFR, ER and PTEN protein expression on benefit to adjuvant trastuzumab in early-stage HER2+ breast cancer patients in NCCTG N9831. Cancer Res. 2011:71. (abstr PD05-03) [Google Scholar]
- 9.Gianni L, Bianchini G, Kiermaier A, et al. Neoadjuvant pertuzumab (P) and trastuzumab (H): Biomarker analysis of a 4-arm randomized phase II study (NeoSphere) in patients (pts) with HER2-positive breast cancer (BC) Cancer Res. 2011:71. (abstr S5-1) [Google Scholar]
- 10.Loi S, Michiels S, Salgado R, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann Oncol. 2014;25:1544–1550. doi: 10.1093/annonc/mdu112. [DOI] [PubMed] [Google Scholar]
- 11.Denkert C, Loibl S, Noske A, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28:105–113. doi: 10.1200/JCO.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- 12.Guo X, Fan Y, Lang R, et al. Tumor infiltrating lymphocytes differ in invasive micropapillary carcinoma and medullary carcinoma of breast. Mod Pathol. 2008;21:1101–1107. doi: 10.1038/modpathol.2008.72. [DOI] [PubMed] [Google Scholar]
- 13.West NR, Milne K, Truong PT, et al. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011;13:R126. doi: 10.1186/bcr3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaguchi R, Tanaka M, Yano A, et al. Tumor-infiltrating lymphocytes are important pathologic predictors for neoadjuvant chemotherapy in patients with breast cancer. Hum Pathol. 2012;43:1688–1694. doi: 10.1016/j.humpath.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Loi S. Tumor-infiltrating lymphocytes, breast cancer subtypes and therapeutic efficacy. Oncoimmunology. 2013;2:e24720. doi: 10.4161/onci.24720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gianni L, Bianchini G, Valagussa P, et al. Adaptive immune system and immune checkpoints are associated with response to pertuzumab (P) and trastuzumab (H) in the NeoSphere study. Cancer Res. 2012:72. (abstr S6-7) [Google Scholar]
- 17.Subramanian J, Simon R. An evaluation of resampling methods for assessment of survival risk prediction in high-dimensional settings. Stat Med. 2011;30:642–653. doi: 10.1002/sim.4106. [DOI] [PubMed] [Google Scholar]
- 18.Pogue-Geile KL, Kim C, Jeong JH, et al. Predicting degree of benefit from adjuvant trastuzumab in NSABP trial B-31. J Natl Cancer Inst. 2013;105:1782–1788. doi: 10.1093/jnci/djt321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: Planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32:3744–3752. doi: 10.1200/JCO.2014.55.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez EA, Butler SM, Dueck AC, et al. The relationship between quantitative HER2 gene expression by the 21 gene RT-PCR assay and adjuvant trastuzumab (H) benefit in NCCTG (Alliance) N9831. J Clin Oncol. 2013;31(suppl 15s):11s. abstr 520) [Google Scholar]
- 21.Desmedt C, Haibe-Kains B, Wirapati P, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14:5158–5165. doi: 10.1158/1078-0432.CCR-07-4756. [DOI] [PubMed] [Google Scholar]
- 22.Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 23.Staaf J, Ringnér M, Vallon-Christersson J, et al. Identification of subtypes in human epidermal growth factor receptor 2-positive breast cancer reveals a gene signature prognostic of outcome. J Clin Oncol. 2010;28:1813–1820. doi: 10.1200/JCO.2009.22.8775. [DOI] [PubMed] [Google Scholar]
- 24.Bianchini G, Qi Y, Alvarez RH, et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol. 2010;28:4316–4323. doi: 10.1200/JCO.2009.27.2419. [DOI] [PubMed] [Google Scholar]
- 25.Iwamoto T, Pusztai L, Matsuoka J, et al. ER + /HER2+ and ER−/HER2+ breast cancers are molecularly distinct but immune gene signatures are prognostic and predictive in both groups. Ann Oncol. 2012;23:ix74. (abstr 1720) [Google Scholar]
- 26.Alexe G, Dalgin GS, Scanfeld D, et al. High expression of lymphocyte-associated genes in node-negative HER2+ breast cancers correlates with lower recurrence rates. Cancer Res. 2007;67:10669–10676. doi: 10.1158/0008-5472.CAN-07-0539. [DOI] [PubMed] [Google Scholar]
- 27.Ignatiadis M, Singhal SK, Desmedt C, et al. Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: A pooled analysis. J Clin Oncol. 2012;30:1996–2004. doi: 10.1200/JCO.2011.39.5624. [DOI] [PubMed] [Google Scholar]
- 28.Park S, Jiang Z, Mortenson ED, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell. 2010;18:160–170. doi: 10.1016/j.ccr.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stagg J, Loi S, Divisekera U, et al. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A. 2011;108:7142–7147. doi: 10.1073/pnas.1016569108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bianchini G, Prat A, Pickl M, et al. Response to neoadjuvant trastuzumab and chemotherapy in ER+ and ER- HER2-positive breast cancers: Gene expression analysis. J Clin Oncol. 2011;29(suppl):52s. abstr 529. [Google Scholar]
- 31.Junttila TT, Parsons K, Olsson C, et al. Superior in vivo efficacy of a fucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010;70:4481–4489. doi: 10.1158/0008-5472.CAN-09-3704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.