

Abstract
Background. The emergence of artemisinin-resistant Plasmodium falciparum in Southeast Asia threatens malaria treatment efficacy. Mutations in a kelch protein encoded on P. falciparum chromosome 13 (K13) have been associated with resistance in vitro and in field samples from Cambodia.
Methods. P. falciparum infections from artesunate efficacy trials in Bangladesh, Cambodia, Laos, Myanmar, and Vietnam were genotyped at 33 716 genome-wide single-nucleotide polymorphisms (SNPs). Linear mixed models were used to test associations between parasite genotypes and parasite clearance half-lives following artesunate treatment. K13 mutations were tested for association with artemisinin resistance, and extended haplotypes on chromosome 13 were examined to determine whether mutations arose focally and spread or whether they emerged independently.
Results. The presence of nonreference K13 alleles was associated with prolonged parasite clearance half-life (P = 1.97 × 10−12). Parasites with a mutation in any of the K13 kelch domains displayed longer parasite clearance half-lives than parasites with wild-type alleles. Haplotype analysis revealed both population-specific emergence of mutations and independent emergence of the same mutation in different geographic areas.
Conclusions. K13 appears to be a major determinant of artemisinin resistance throughout Southeast Asia. While we found some evidence of spreading resistance, there was no evidence of resistance moving westward from Cambodia into Myanmar.
Keywords: malaria, artemisinin resistance, Southeast Asia, Plasmodium falciparum, kelch
(See the editorial commentary by Sibley on pages 667–9,and the major article by Taylor et al on pages 680–8.)
Malaria treatment efficacy and elimination rely on the continued efficacy of artemisinin-based combination therapies (ACTs). Plasmodium falciparum resistance to artemisinin derivatives was first documented in western Cambodia [1–3] and is now present on both sides of the Thailand-Myanmar border [4, 5], as well as in northern Cambodia near the border with Thailand and in southern Vietnam bordering Cambodia [6]. Recently, mutations in a kelch protein (K13), located within a region on P. falciparum chromosome 13 identified in our previous genome-wide association study (GWAS) [7], were associated with increased resistance in an artemisinin-resistant parasite line selected in the laboratory and with delayed parasite clearance in clinical isolates from Cambodia [8].
While mutations in this gene appear to provide a promising marker for artemisinin resistance, replication and validation studies are needed to estimate the association of these putative markers with delayed parasite clearance in other parasite populations. We performed a replication GWAS to confirm the K13 gene as a major determinant of clinical artemisinin resistance and to identify secondary loci that may contribute to resistance. We also estimated the prevalence of mutations within the resistance domain of the K13 protein (ie, the propeller domain), their association with parasite clearance, and their origins, in samples from Bangladesh, Cambodia, Laos, Myanmar, and Vietnam.
METHODS
Study Samples
Our previous GWAS included samples from completed efficacy studies of artesunate and ACTs conducted in Cambodia, Thailand, and Bangladesh from 2008 to 2009 as described previously [2, 7, 9, 10] (Supplementary Methods). The replication data set included samples from efficacy studies conducted from 2010 to 2011 in Cambodia, Myanmar, Laos, and Vietnam, also described previously [4, 11, 12] (Supplementary Methods). Samples from both the previous and replication data sets were used to estimate the distribution of mutations within the K13 gene, their association with parasite clearance half-life, and their origins. All samples used in this study were collected with informed consent from patients or their parents or guardians and were approved for use by the University of Maryland School of Medicine Institutional Review Board, as well as by local ethics boards and committees described in source publications.
Parasite Genotypes
DNA extracted from leukocyte-depleted venous blood was sequenced using an Illumina Genome Analyzer II as part of the MalariaGen Community Project (Data release v2.0). Samples that were unable to be sequenced were genotyped using a P. falciparum–specific NimbleGen 4.2M probe custom DNA microarray (accession no. GSE56390) that types a total of 33 716 SNPs [13]. Samples that were included in our previous GWAS had undergone genotyping at 8079 SNPs using a P. falciparum–specific Affymetrix molecular inversion probe DNA microarray [7], and the informative subset of SNPs from this array were also typed on the NimbleGen array. All 303 successfully sequenced samples had the same 33 716 SNPs called, as well as 12 SNPs within the K13 gene, from among 86 158 quality-controlled SNPs identified in short-read sequence alignments against the P. falciparum 3D7 reference. SNPs not within the list of high-quality SNPs, including some K13 mutations, were called using processed read counts from the short-read sequence alignments against the P. falciparum 3D7 reference. The genotype data used in the analysis have been submitted to PlasmoDB (http://www.plasmodb.org) and the National Institutes of Health Gene Expression Omnibus for public access (http://www.ncbi.nlm.gov/geo/).
Phenotype
Parasite clearance half-lives were estimated using the parasite clearance estimator developed by the WorldWide Antimalarial Resistance Network [14] (Figure 1). Owing to less frequent assessment of parasitemia in the Bangladesh and Myanmar studies (parasitemia was assessed every 12 hours instead of every 6 hours, as in other studies), some patients had only 2 parasite counts available prior to clearance. To estimate clearance for these patients, the 0 parasitemia at 24 hours was replaced by the level of detection for microscopy (40 parasites/μL), and a straight line was fitted to the loge-transformed parasitemia values. As a result, the absolute value of the slope likely overestimated the half-life for patients at these sites.
Figure 1.
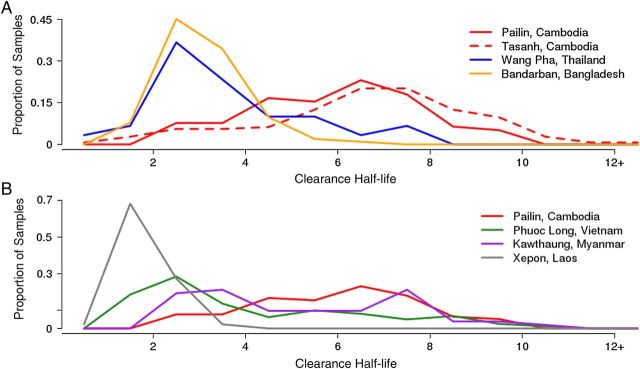
Distribution of parasite clearance half-life by geography in the previous genome-wide associated study (GWAS) and replication data sets. A, Previous GWAS data set. B, Replication data set. A large proportion of parasites from western Cambodia demonstrate delayed parasite clearance, as do a subset of parasites from Myanmar and Vietnam. Parasites from Laos and Bangladesh demonstrated mostly rapid parasite clearance.
SNP Imputation
A complete data set consisting of all 33 716 variable positions and all samples was imputed using BEAGLE V3.3.2 [15]. Only biallelic positions were included in the data set, to allow for use of genotype probabilities. Samples were grouped by geography, and imputation was performed separately within each group, to avoid population structure associated errors. A probability of 90% was used to call an imputed SNP.
Quality Control
A total of 18 470 invariant SNPs were removed at the imputation step, and an additional 9041 low-frequency SNPs (minor allele frequency [MAF], < 5%) were removed prior to analysis. 1336 SNPs with >20% undetermined calls were also excluded from the analysis. After imputation, all samples had <30% undetermined calls and were retained in the analysis. After quality control, the final meta-analysis included 4620 SNPs (including K13 SNPs) and 551 samples—308 from the previous GWAS and 243 from the replication data set. Of these samples, 303 had the whole genome sequence available, 110 from the previous GWAS and 193 from the replication data set.
Regression
Genome-wide mixed-model association (GEMMA) [16] was used to estimate the association between each SNP and parasite clearance half-life, with adjustment for age, log-transformed parasitemia at diagnosis, and the first 3 principal components [16] (Figure 2). Principal components were estimated from a matrix of pair-wise sample identity-by-state metrics [17]. An identity-by-state allele-sharing matrix was included as a random effect, to account for the correlation between genetically similar individuals that results from population structure. The GEMMA method was used to estimate coefficients and P values. A Bonferroni threshold (0.05/number of SNPs analyzed) was used to evaluate genome-wide significance. Quantile-quantile plots for P values were used to assess the robustness of modeling approaches in minimizing false-positive results due to population structure or other confounding.
Figure 2.
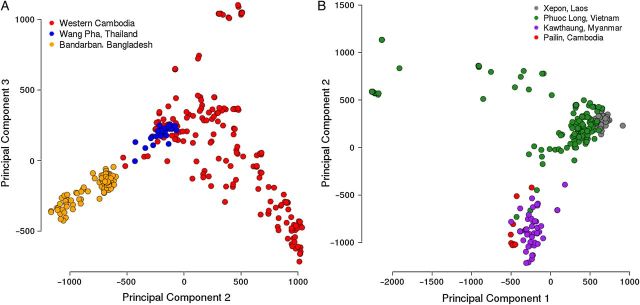
Population structure by geography in the previous genome-wide associated study (GWAS) and replication data sets. A, Previous GWAS data set. B, Replication data set. In the imputed data set representing the previous GWAS, most of the population structure was accounted for by the second principal component. As most of the samples from this data set were genotyped with the least dense single-nucleotide polymorphism array, the first principal component accounted for most of the missing data. In the replication data set, most of the population structure was accounted for by the first principal component.
All SNPs after imputation (minus invariant loci) were used to dichotomize every gene in the P. falciparum genome for the gene-based analysis. Using the 3D7 strain as a reference (v2.1.4), genes with any SNP with a nonreference call were coded as 1, and genes with all reference calls and no missing values were coded as 0. Genes with no nonreference calls and unknown SNP calls (missing data) were coded as missing. GEMMA was used with identical covariates and thresholds as described above.
Random Forests
Random forest analyses [18] were done using the randomForest package [19] of the R statistical computing environment [20]. Missing SNPs were imputed using the randomForest function na.roughfix, which replaces missing values with the most frequent nucleotide at that position (breaking ties at random). The number of variables tried at each split varied with each data set and ranged from 816 to 1797, and the number of trees in the forest was 100 000, except for the meta-analyses, in which the number of trees was 25 000. The importance of each SNP in predicting the phenotype was assessed on the basis of the percentage increase in mean-squared error.
Association With Parasite Clearance
t tests were used to estimate the difference in mean parasite clearance half-life between parasites with each K13 mutation and those with the wild-type allele at all polymorphic sites within the gene. Means and 95% confidence intervals were estimated using SAS v.9.2.
Linkage Disequilibrium
The program Haploview [21] was used to determine linkage disequilibrium (LD) in SNPs surrounding the K13 gene, as well as around SNPs associated in genome-wide analyses. LD windows were defined to include all SNPs upstream and downstream from the associated SNP or mutation of interest up to but not including the next SNP with a MAF of > 0.05 and an R2 of <0.3. For associated SNPs, parasite subpopulations were determined using STRUCTURE v.2.3.4, and LD windows were defined in parasites within each parasite subpopulation separately, to avoid false estimates of LD due to population structure. This approach yielded a range of LD windows for each SNP. A consensus LD window was determined by estimating the median window size upstream and downstream of the SNP after removing outliers.
For K13 mutations, LD windows were defined within groups of samples containing each mutation, and windows were not defined for mutations present in <6 samples. This approach produced a range of LD windows for each K13 mutation. A consensus window containing 80 SNPs with no missing data (MAL13:1659301 to MAL13:1799851) was created after removing large windows that resulted from the clonal nature of certain K13 groups.
Haplotype Network
For the haplotypes defined by LD described above, the program SplitsTree4 [22] was used to estimate the most parsimonious median joining network [23]. Only samples with complete haplotypes were included. To reduce the complexity of the network, we removed samples from populations (located in Bangladesh and Laos) without resistance-associated K13 mutations. The median joining network was created with 263 samples from Cambodia, Myanmar, and Vietnam. SplitsTree4 defaults (ε = 0; 2000 iterations) were used to create the network and was manually curated.
RESULTS
Genome-Wide Association
No SNPs achieved genome-wide significance in the imputed data set representing our previous GWAS (Supplementary Figure 1A), although the SNP from chromosome 10 (MAL10:688956) and 1 SNP from chromosome 13 (MAL13:1719976) that were significant in our previous study were among the top 5 hits in the current analysis. The random forest analysis of the imputed data set representing our previous GWAS also indicated that 2 of the significant SNPs from that study (MAL10:688956 and MAL13:1718319) were the best predictors of parasite clearance half-life, after the principal component accounting for most of the population structure (Supplementary Figure 1A).
In the analysis of the replication data set, 2 SNPs, 1 on chromosome 7 (MAL7:472852) and 1 on chromosome 9 (MAL9:130454), achieved genome-wide significance (Table 1), while the random forest analysis of the replication data set showed SNP MAL13:1886271 to be the best SNP predictor of clearance half-life (Supplementary Figure 1B).
Table 1.
Single-Nucleotide Polymorphisms (SNPs) Associated With Parasite Clearance Half-life
| Chromosome | SNP | Previous GWAS |
Replication GWAS |
Meta-analysis |
|||
|---|---|---|---|---|---|---|---|
| GEMMA P |
Random Forest Rank | GEMMA P |
Random Forest Rank | GEMMA P |
Random Forest Rank | ||
| 7 | MAL7:472852 | …a | …a | 5.17E-06 | 10 | …a | …a |
| 9 | MAL9:130454 | …a | …a | 6.91E-06 | >20 | …a | …a |
| 10 | MAL10:688956b | 3.41E-05 | 1 | 7.34E-02 | >20 | 7.27E-02 | >20 |
| 11 | MAL11:608589 | 2.76E-02 | >20 | 2.98E-04 | >20 | 6.39E-06 | >20 |
| 13 | MAL13:1718319b | 3.29E-03 | 2 | 8.68E-01 | >20 | 2.25E-01 | 4 |
| 13 | MAL13:1886271 | 1.35E-01 | >20 | 3.00E-03 | 1 | 5.47E-02 | 1 |
| 14 | MAL14:275834 | 7.50E-02 | >20 | …a | …a | 8.30E-06 | >20 |
| 14 | MAL14:3029267 | 1.13E-01 | >20 | 1.11E-03 | >20 | 8.10E-06 | >20 |
Significant associations are shown in bold.
Abbreviations: GEMMA, genome-wide mixed model association; GWAS, genome-wide association study.
a SNPs absent from analysis because of high levels of missing data or low minor-allele frequency
b SNPs identified in the previous GWAS.
In the meta-analysis of the previous GWAS and replication data sets, 3 different SNPs, 1 on chromosome 11 (MAL11:608589) and 2 on chromosome 14 (MAL14:275834 and MAL14:3029267) achieved genome-wide significance (Supplementary Figure 1C), and the random forest analysis showed SNP MAL13:1886271 to be the best SNP predictor of clearance half-life. Quantile-quantile plots indicated little residual confounding due to population structure or other potential confounding variables (eg, host immunity; Supplementary Figure 1).
As discussed below, in some instances different K13 mutations were found in different parasite populations, which may make them difficult to associate with the phenotype of interest in a GWAS while adjusting for population structure. To overcome this obstacle, we created a dichotomous variable for each gene within the parasite genome, including the K13 gene, representing the presence of any nonreference allele, using the P. falciparum 3D7 strain as the reference. In a gene-by-gene analysis, we estimated the association of nonreference alleles in each gene with parasite clearance half-life (Figure 3 and Supplementary Figure 2).
Figure 3.
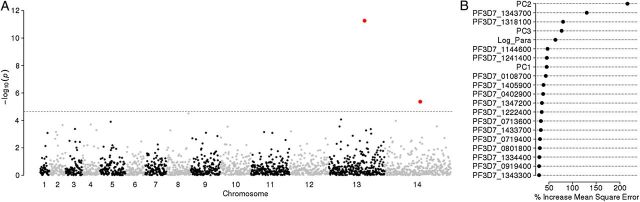
Manhattan and random forest plots for the gene-based genome-wide association study. Meta-analysis of the association between the presence of nonreference alleles in each gene in the genome and parasite clearance half-life. A, −Log10 P values for all genes, with both the K13 protein (PF3D7_1343700) and PF3D7_1442600 achieving genome-wide significance. B, The percentage increase in mean squared error for the top 20 single-nucleotide polymorphisms or covariates in the random forests analysis. The presence of nonreference alleles in the K13 gene was the best predictor of parasite clearance half-life after the second principal component, which accounted for most of the population structure.
In the gene-based analysis of the data set representing our previous GWAS, 1 gene on chromosome 13 (PF3D7_1342300) achieved genome-wide significance (Table 2). This gene encodes a putative tetratricopeptide repeat family protein that lies within a top-ranked signature of selection identified in our previous GWAS study.
Table 2.
Genes Associated With Parasite Clearance Half-life in Gene-Based Analysis
| Chromosome | Gene | Previous GWAS |
Replication GWAS |
Meta-analysis |
|||
|---|---|---|---|---|---|---|---|
| GEMMA P |
Random Forest Rank | GEMMA P |
Random Forest Rank | GEMMA P |
Random Forest Rank | ||
| 5 | PF3D7_0514600 | 9.77E-01 | >20 | 1.18E-05 | >20 | 1.27E-04 | >20 |
| 9 | PF3D7_0903100 | 7.61E-01 | >20 | 2.42E-05 | 16 | 2.05E-03 | >20 |
| 13 | PF3D7_1318100 | 3.22E-01 | 1 | 6.69E-01 | >20 | 5.40E-01 | 2 |
| 13 | PF3D7_1342300 | 1.39E-05 | >20 | 8.30E-01 | >20 | 3.05E-02 | >20 |
| 13 | PF3D7_1343700 | 2.47E-03 | 2 | 5.28E-10 | 1 | 5.46E-12 | 1 |
| 13 | PF3D7_1347200 | 2.45E-01 | >20 | 4.93E-03 | 2 | 4.10E-03 | 8 |
| 14 | PF3D7_1442600 | … | … | 3.55E-05 | 3 | 4.41E-06 | >20 |
Significant associations are shown in bold.
Abbreviations: GEMMA, genome-wide mixed model association; GWAS, genome-wide association study.
a Genes absent from analysis because of high levels of missing data.
In the gene-based analysis of both the replication data set and the meta-analysis, the K13 gene was the top hit by several orders of magnitude (Table 2), suggesting a strong association between mutations in this gene and parasite clearance half-life. Three other genes reached significance in the gene-based analysis of the replication and meta-data sets: PF3D7_0514600, a ribose 5-phosphate epimerase; PF3D7_0903100, a retrieval receptor for endoplasmic reticulum membrane proteins; and PF3D7_1442600, a sporozoite-specific transmembrane protein (Supplementary Figure 2). The gene-based random forest analyses yielded similar results, with nonreference alleles in the K13 gene (PF3D7_1343700) and in ferredoxin (PF3D7_1318100) being the 2 best predictors of parasite clearance half-life, after population structure (Table 2 and Supplementary Figure 2).
Genes in Regions Associated Parasite Clearance Half-life
The LD windows containing SNPs associated with clearance half-life are shown in Supplementary Table 1. The LD window surrounding SNP MAL7:472852 contains the gene encoding the P. falciparum chloroquine resistance transporter, which has a strong signature of selection in the genome [7, 24]. The window on chromosome 9 also contains one of the genes (PF3D7_0903100) that was associated with clearance half-life in the gene-based analysis. The SNP on chromosome 11 is approximately 15 kb away from a gene (PF3D7_1115700) identified by Ariey et al in laboratory isolates made resistant to artemisinins [8], as well as in another study of in vitro artemisinin resistance [25]. The LD window associated with SNP MAL13:1718319 contains the K13 gene, as observed in our previous GWAS. In addition, 3 regions identified by Cheeseman et al [24] as being under recent selection in Cambodia overlap with LD windows containing SNPs associated with delayed clearance in our study (Supplementary Table 1).
Prevalence of K13 Mutations and Association With Parasite Clearance Half-life
Twelve mutations in the K13 gene were observed in 123 samples among the 303 samples with whole-genome sequence data, with 10 of the 12 occurring within the resistance domain of the protein (ie, the propeller domain) [8] (Figure 4A). Parasites contained only 1, if any, K13 mutation. The most prevalent mutation was C580Y, found in western Cambodia, Myanmar, and Vietnam. Two mutations outside of the resistance domains were observed in samples from Bangladesh. No mutations were observed in this gene in patient samples from Laos, where delayed parasite clearance has not been observed until recently [6, 12] (Figure 4B).
Figure 4.
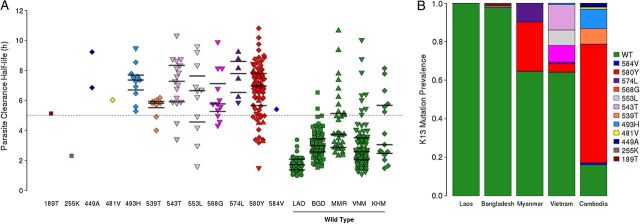
Prevalence of K13 mutations and association with parasite clearance half-life. A, Parasite clearance half-life for parasites with K13 mutations (colors) or wild-type alleles (green). Parasites with wild-type alleles are grouped by country (LAO = Laos, BGD = Bangladesh, MMR = Myanmar, VNM = Vietnam, KHM = Cambodia), and parasites with K13 mutations are designated by symbols (Laos = circles, Bangladesh = squares, Myanmar = triangle, Vietnam = inverted triangle, and Cambodia = diamond). Horizontal lines represent median half-lives and 95% confidence intervals for wild-type and K13 mutants represented more than twice in the data set. B, Prevalence of parasites with K13 mutations, by country.
Mean parasite clearance half-life was significantly longer in parasites with a mutation in the resistance domain, compared with parasites with reference-type alleles (Figure 4). While mean half-life varied by mutation, with the exception of R539T and Y493H most of the confidence intervals overlapped, raising the possibility that mean half-life may be similar between these different mutations. Several SNPs were observed in ≤2 infections and therefore could not be evaluated for association with delayed parasite clearance.
Origin of K13 Mutations
To determine the origins of K13 mutations from different geographic regions, we created a haplotype network tree based on 80 SNPs within LD flanking either side of the K13 gene (Figure 5). This haplotype analysis revealed both population-specific emergence of mutations, as well as independent emergence of the same mutation on different genetic backgrounds in different geographic sites. Three mutations in the K13 gene were found in multiple populations: 493H was present in 10 samples from Cambodia and 1 in Vietnam, 574L was found in 5 samples from Myanmar and 1 from Vietnam, and 580Y was found in 61 samples from Cambodia, 13 samples from Myanmar, and 6 samples from Vietnam. Three 493H haplotypes were found in our data set, with the single parasite from Vietnam sharing a common haplotype with the majority of samples from Cambodia, suggesting a common emergence and spread of this mutation. 574L appears to have originated twice, once in Vietnam and once in Myanmar. The widespread 580Y mutation seems to have emerged twice. One emergence appears to have a common Cambodia/Vietnam ancestor (as with 493H), and the second is a unique Myanmar event. All other mutations appear to have arisen independently.
Figure 5.
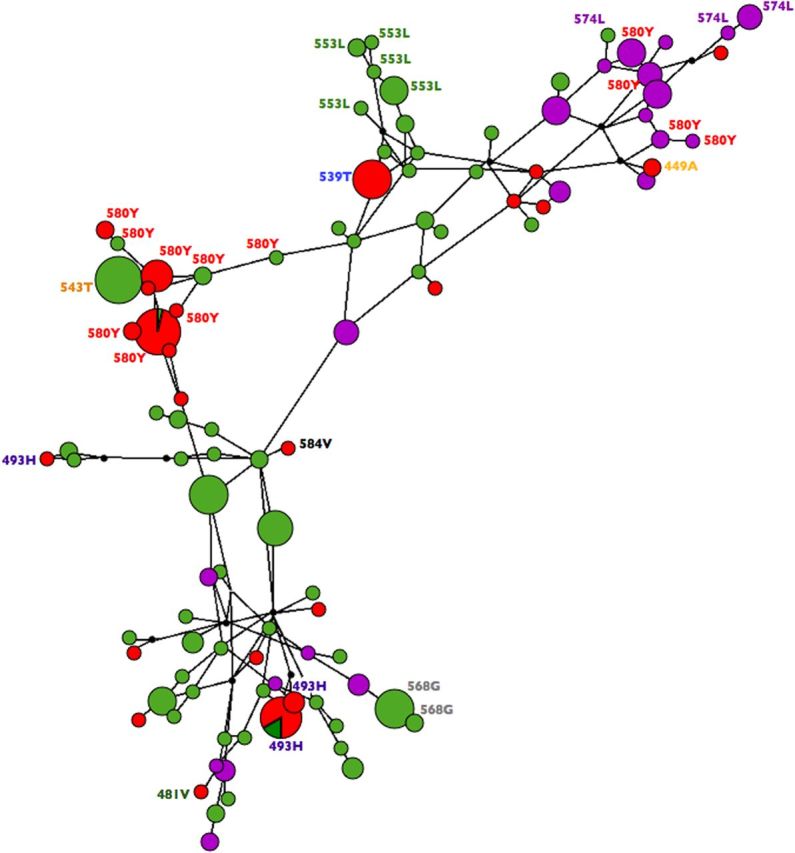
Median joining haplotype network of K13 mutations and single-nucleotide polymorphisms within linkage disequilibrium of the K13 gene. Haplotype prevalence is proportional to size of the circle, with circle color corresponding to population of origin: Cambodia (red), Myanmar (purple), or Vietnam (green). Individual mutations are labeled by amino acid position, and wild-type haplotypes are not labeled.
DISCUSSION
We previously conducted a GWAS [7] that identified a region on P. falciparum chromosome 13 associated with artemisinin resistance. Others have identified a gene within this genomic region, encoding a kelch protein (K13), that contains mutations that have been shown to confer resistance in an artemisinin-resistant parasite line selected in the laboratory and that are associated with delayed parasite clearance in clinical isolates from Cambodia [8]. Here we have shown that, in addition to western Cambodia, K13 mutations are present in southern Myanmar, southern Vietnam, and Bangladesh and that infections with parasites containing mutations within the resistance domain of the K13 protein show delayed parasite clearance, compared with infections with wild-type parasites (Figure 4A). In our gene-based GWAS, the presence of nonreference alleles within the K13 gene was by far the most strongly associated with prolonged parasite clearance half-life. An analysis of haplotypes surrounding the K13 gene suggests independent emergences of most K13 mutations. Exceptions are the Cambodia/Vietnam 580Y and Cambodia/Vietnam 493H mutations; in both cases, their frequency distribution suggests a Cambodian origin, where the mutations are most prevalent, with subsequent spread to Vietnam. Contrary to the widely assumed scenario, we found no evidence of westward spread of artemisinin resistance from Cambodia to Myanmar. The discovery and validation of K13 mutations outside of Cambodia that are associated with artemisinin resistance has critical implications for the treatment and elimination of P. falciparum malaria in Southeast Asia.
Our previous GWAS, which included primarily samples from Cambodia, identified a region of chromosome 13 that contains the gene encoding the K13 protein. Reanalysis of individual SNPs within this same data set after imputation showed that 3 of the 4 SNPs identified in our previous study were among the top SNPs associated with parasite clearance half-life. While being among the top 5 hits, no SNP from the previous GWAS achieved genome-wide significance in the current study, likely owing to a more stringent significance threshold resulting from evaluation of a larger number of SNPs. The signal on chromosome 13 from this data set is likely driven by the high frequency of the K13 580Y mutation in the samples from Cambodia, although 580Y itself was not detected, likely owing to the small number of samples within our previous data set (approximately one third) that had the whole genome sequence available from which to call K13 SNPs. In contrast, <5% of the samples from our replication data set were from Cambodia, with the rest being from Vietnam, Myanmar, and Laos. Analysis of individual SNPs within the replication and meta-data sets did not show any significant associations between SNPs in LD with the K13 gene and delayed parasite clearance. This result is likely attributable to the presence of mostly population-specific K13 mutations within these geographic regions, which are difficult to detect using SNP-based GWAS because they are not independent of the population structure. These results highlight some of the limitations of GWAS and the usefulness of gene-based and/or region-based association methods in these situations, as was recently done in a human GWAS of malaria susceptibility [26].
In our gene-based GWAS of the replication and meta-data sets, the K13 gene was most significantly associated with the phenotype, by several orders of magnitude, suggesting that the protein encoded by this gene is indeed the major determinant of artemisinin resistance. K13 did not achieve genome-wide significance (P = 2.4E-03) in the gene-based analysis of our previous GWAS data set, likely owing to reduced power stemming from the small number of samples that had sequence reads available from which to call K13 SNPs. Instead, a gene in LD with K13 achieved significance in this data set. While K13 was most strongly associated with the phenotype in gene-based analyses, 4 other genes were also shown to be associated with delayed parasite clearance in the GEMMA or random forest analyses. It is possible that these genes, or other genes identified within the LD windows surrounding significant SNPs from the SNP-based analysis, play a secondary or compensatory role in artemisinin resistance and should be investigated further. Of note is the PF3D7_1115700 gene encoding a cysteine proteinase falcipain 2a, which lies approximately 15 kb upstream from a significant SNP on chromosome 11. This gene was identified in the same study that reported the discovery of K13 [8] and, when knocked out in another in vitro study, was shown to decrease artemisinin activity by preventing hemoglobin digestion [25].
While mean and median half-life varied by mutation, most of the confidence intervals overlapped, suggesting that different K13 mutations may not confer different levels of resistance. However, genetic transformation experiments will be necessary to evaluate different levels of resistance and/or the fitness cost of different mutations. Transfection may also be a useful approach to explore the role of possible secondary or compensatory mutations.
The presence of multiple, population-specific mutations responsible for artemisinin resistance has implications for both surveillance and containment of resistant parasites. Other drug resistance loci, such as the genes encoding the P. falciparum chloroquine resistance transporter, dihydrofolate reductase, and dihydropteroate synthase, which have just 1 or a small number of mutations responsible for resistance, are amenable to quick and simple tests that can be done with minimal laboratory infrastructure [27–29]. The presence of a large number of unique mutations in the K13 propeller protein, each of which can confer resistance, will initially require gene sequencing or other sophisticated techniques that can genotype multiple SNPs at a time, making it more difficult to base surveillance in regional laboratories. It is also likely that additional K13 mutations with a phenotypic effect are yet to be identified.
The independent emergence of K13 mutations in multiple geographic locations suggests that efforts to eliminate artemisinin-resistant malarial parasites in one region may have a limited impact on the emergence of resistance in neighboring regions, particularly if selection pressure for resistance is sustained by continued availability of artemisinin monotherapies and counterfeit or substandard medicines. The finding of multiple origins of artemisinin resistance complicates containment strategies, which have mostly focused on preventing westward spread of resistance from its initial focus in western Cambodia, and highlights the need to map K13 mutations throughout the malaria-endemic world.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the laboratory staff at the HHMI/CVD Malaria Group at University of Maryland School of Medicine, for technical support; Ms Nicole Eddington Johnson and Ms Amanda Icenroad, for administrative support; the Malaria Programme laboratory and bioinformatics staff at the Wellcome Trust Sanger Institute, including Daniel Mead, Eleanor Drury, Susana Campino, Magnus Manske, and James Stalker, who helped generate and compile the whole-genome sequencing data; laboratory staff at the University of Notre Dame, including Becky Miller and Asako Tan, who helped design the NimbleGen SNP array; and all participants in the drug efficacy trials and their families, for their contribution to the work.
Y. S. died in December 2012. She was in integral part of the AFRIMS Cambodia team who conducted the artesunate efficacy study in Tasanh, Cambodia.
Disclaimer. P. R. is a staff member of the World Health Organization (WHO) and alone is responsible for the views expressed in this publication; they do not necessarily represent the decisions, policy, or views of the WHO. M. M. F., D. B., Y. S., C. L., S. D. T., and D. L. S. report being employees of the US government, and the views expressed in this article are theirs alone, and do not necessarily reflect the official policy or positions of the US Department of the Army, the Department of Defense, the US or Cambodian governments.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (grant R01AI101713 to S. T.-H. and grant U19AI10820.), the World Health Organization Global Malaria Programme, the Doris Duke Charitable Foundation, and the Howard Hughes Medical Institute, for genomic analyses; the Wellcome Trust, through core funding of the Wellcome Trust Sanger Institute (grant 098051), for sequencing analyses; the Wellcome Trust Centre for Human Genetics (grant 090532/Z/09/Z), the Resource Centre for Genomic Epidemiology of Malaria (grant 090770/Z/09/Z), and the Wellcome Trust Mahidol University Oxford Tropical Medicine Research Programme, through core funding from the Wellcome Trust; and the Centre for Genomics and Global Health, through the Medical Research Council (grant G0600718).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Amaratunga C, Sreng S, Suon S, et al. Artemisinin-resistant Plasmodium falciparum in Pursat province, western Cambodia: a parasite clearance rate study. Lancet Infect Dis. 2012;12:851–8. doi: 10.1016/S1473-3099(12)70181-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359:2619–20. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 4.Kyaw MP, Nyunt MH, Chit K, et al. Reduced susceptibility of Plasmodium falciparum to artesunate in southern Myanmar. PLoS One. 2013;8:e57689. doi: 10.1371/journal.pone.0057689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phyo AP, Nkhoma S, Stepniewska K, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–6. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashley EA, Dhorda M, Fairhurst RM, et al. Tracking Resistance to Artemisinin Collaboration (TRAC). Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411–23. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takala-Harrison S, Clark TG, Jacob CG, et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci U S A. 2013;110:240–5. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ariey F, Witkowski B, Amaratunga C, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bethell D, Se Y, Lon C, et al. Artesunate dose escalation for the treatment of uncomplicated malaria in a region of reported artemisinin resistance: a randomized clinical trial. PLoS One. 2011;6:e19283. doi: 10.1371/journal.pone.0019283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Starzengruber P, Swoboda P, Fuehrer HP, et al. Current status of artemisinin-resistant falciparum malaria in South Asia: a randomized controlled artesunate monotherapy trial in Bangladesh. PLoS One. 2012;7:e52236. doi: 10.1371/journal.pone.0052236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hien TT, Thuy-Nhien NT, Phu NH, et al. In vivo susceptibility of Plasmodium falciparum to artesunate in Binh Phuoc Province, Vietnam. Malar J. 2012;11:355. doi: 10.1186/1475-2875-11-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayxay M, Khanthavong M, Chanthongthip O, et al. No evidence for spread of Plasmodium falciparum artemisinin resistance to Savannakhet Province, Southern Laos. Am J Trop Med Hyg. 2012;86:403–8. doi: 10.4269/ajtmh.2012.11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacob CG, Tan JC, Miller BA. A microarray platform and novel SNP calling algorithm to evaluate Plasmodium falciparum field samples of low DNA quantity. BMC Genomics. 2014;15:719. doi: 10.1186/1471-2164-15-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flegg JA, Guerin PJ, White NJ, Stepniewska K. Standardizing the measurement of parasite clearance in falciparum malaria: the parasite clearance estimator. Malar J. 2011;10:339. doi: 10.1186/1475-2875-10-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 18.Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- 19.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 20.Vienna, Austria: R Foundation for Statistical Computing; 2008. A language and environment for statistical computing [computer program] [Google Scholar]
- 21.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 22.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–67. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 23.Bandelt HJ, Forster P, Sykes BC, Richards MB. Mitochondrial portraits of human populations using median networks. Genetics. 1995;141:743–53. doi: 10.1093/genetics/141.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheeseman IH, Miller BA, Nair S, et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012;336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klonis N, Crespo-Ortiz MP, Bottova I, et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A. 2011;108:11405–10. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Band G, Le QS, Jostins L, et al. Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet. 2013;9:e1003509. doi: 10.1371/journal.pgen.1003509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Djimde A, Doumbo OK, Cortese JF, et al. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:257–63. doi: 10.1056/NEJM200101253440403. [DOI] [PubMed] [Google Scholar]
- 28.Kublin JG, Dzinjalamala FK, Kamwendo DD, et al. Molecular markers for failure of sulfadoxine-pyrimethamine and chlorproguanil-dapsone treatment of Plasmodium falciparum malaria 5. J Infect Dis. 2002;185:380–8. doi: 10.1086/338566. [DOI] [PubMed] [Google Scholar]
- 29.Plowe CV, Djimde A, Bouare M, Doumbo O, Wellems TE. Pyrimethamine and proguanil resistance-conferring mutations in Plasmodium falciparum dihydrofolate reductase: polymerase chain reaction methods for surveillance in Africa. Am J Trop Med Hyg. 1995;52:565–8. doi: 10.4269/ajtmh.1995.52.565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.