

Abstract
Hepatic drug metabolizing enzymes and transporters play a crucial role in determining the fate of drugs, and alterations in liver function can place individuals at greater risk for adverse drug reactions (ADRs). We have shown that nonalcoholic steatohepatitis (NASH) leads to changes in the expression and localization of enzymes and transporters responsible for the disposition of numerous drugs. The purpose of this study was to determine the effect of NASH on methotrexate (MTX) disposition and the resulting toxicity profile. Sprague Dawley rats were fed either a control or methionine-choline-deficient diet for 8 weeks to induce NASH, then administered a single ip vehicle, 10, 40, or 100 mg/kg MTX injection followed by blood, urine, and feces collection over 96 h with terminal tissue collection. At the onset of dosing, Abcc1–4, Abcb1, and Abcg2 were elevated in NASH livers, whereas Abcc2 and Abcb1 were not properly localized to the membrane, similar to that previously observed in human NASH. NASH rodents receiving 40–100 mg/kg MTX exhibited hepatocellular damage followed by initiation of repair, whereas damage was absent in controls. NASH rodents receiving 100 mg/kg MTX exhibited slightly greater renal toxicity, indicating multiple organ toxicity, despite the majority of the dose being excreted by 6 h. Intestinal toxicity in NASH however, was strikingly less severe than controls, and coincided with reduced fecal MTX excretion. Because MTX-induced gastrointestinal toxicity limits the dose escalation necessary for cancer remission, these data suggest a greater risk for life-threatening MTX-induced hepatic and renal toxicity in NASH in the absence of overt gastrointestinal toxicity.
Keywords: ABC transporters, methotrexate, hepatotoxicity, adverse drug reactions, hepatobiliary disposition
Abbreviations
- ALL
acute lymphoblastic leukemia
- FFPE
formalin-fixed, paraffin-embedded
- H&E
hematoxylin and eosin
- MCD
methionine-choline-deficient
- MTX
methotrexate
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PCNA
proliferating cell nuclear antigen
- SCN
single cell necrosis
Adverse drug reactions (ADRs) are defined as an unintended, harmful reaction to a medication at the recommended dose (Wilke et al., 2007). In 2011, the USFDA recorded 573,111 serious ADR outcomes causing hospitalization or disability, of which 98,518 resulted in death (USFDA, 2012). The substantial burden of ADRs warrants investigation of mechanisms underlying these interindividual variations in response to a drug. Hepatic drug metabolizing enzymes and transporters are often a focus of ADR investigations. Perturbations in the expression or function of enzymes and transporters can significantly affect elimination of, and exposure to a drug. Although polymorphisms account for a proportion of ADRs in the population, the contribution of underlying metabolic diseases, such as nonalcoholic fatty liver disease (NAFLD), to the occurrence of ADRs is not fully understood.
NAFLD is a spectrum of liver pathologies that initially presents as simple steatosis, which is largely considered benign. However, patients are then more susceptible to further hepatocellular damage in the form of oxidative stress, inflammation, increased lipid accumulation, and fibrosis, all of which facilitate progression to the more severe stage of nonalcoholic steatohepatitis (NASH) (Marra et al., 2008). NAFLD and NASH have been estimated to affect 17–33% and 5.7–17% of the adult United States population, respectively (McCullough, 2011). These diseases also are more prevalent in the obese and type 2 diabetes subpopulations with 30–50% and 40–70% estimated to have NASH, respectively (Anstee et al., 2011; Cheung and Sanyal, 2009). More alarming is the increasing prevalence of NAFLD and NASH in the pediatric population. Approximately 5% of normal to overweight and 38% of obese children are believed to have some form of NAFLD (Mencin and Lavine, 2011). Concomitant with the increasing prevalence of obesity, the incidence of NAFLD is expected to rise in the coming years. Due to its already substantial prevalence in the general population, patients with NAFLD and particularly NASH may be at significant risk for ADRs.
Our laboratory has previously demonstrated the effects of NASH on several drug metabolizing enzymes and transporters in both rodent models and human tissues. Progression of human NAFLD to the severe stage of NASH leads to alterations in the expression and activity of specific cytochrome P450 enzymes, sulfotransferases, and glutathione S-transferases (Fisher et al., 2009b; Hardwick et al., 2010, 2013). Additionally, hepatic uptake transporter expression and function are decreased in both human and rodent NASH, while efflux transporters are upregulated (Fisher et al., 2009a; Hardwick et al., 2011, 2012; Lake et al., 2011). More specifically, NASH results in the altered localization and dysfunction of the apical/canalicular transporter ABCC2/Abcc2 and upregulation of the basolateral/sinusoidal transporter ABCC3/Abcc3, which we have shown leads to increased plasma retention of acetaminophen metabolites and ezetimibe-glucuronide (Hardwick et al., 2012; Lickteig et al., 2007). However, it remains unclear if increased systemic exposure to drugs in NASH correlates with increased susceptibility to multiple organ toxicities.
In the current study, we determined the effect of NASH on the disposition and multiple organ toxicity profile of methotrexate (MTX). MTX is an antiproliferative agent used at low doses to treat rheumatoid arthritis and psoriasis, and at high doses to treat various cancers such as acute lymphoblastic leukemia (ALL). MTX, which is a substrate for multiple efflux transporters, was chosen due to its well-documented dose-limiting toxicity (Barker et al., 2011; Schmiegelow, 2009; Warren and Griffiths, 2008; Widemann and Adamson, 2006). Furthermore, others have identified a greater risk for liver injury in NASH patients receiving chronic, low-dose MTX (Langman et al., 2001). We hypothesized that as a result of the altered localization and expression of hepatic efflux transporters, rodents with NASH would exhibit increased systemic exposure to MTX and multiple organ toxicity. We have determined the disease-dependent expression and localization of hepatic and renal transporters, in addition to MTX dose-dependent effects on hepatic, renal, and intestinal toxicity in NASH. We present novel evidence of altered hepatobiliary disposition of MTX and a disrupted toxic response in NASH leading to a preferential shift from gastrointestinal toxicity in control rodents to the more severe hepatic and renal toxicity in NASH.
MATERIALS AND METHODS
Materials
MTX was purchased from Toronto Research Chemicals (Ontario, Canada). Sodium bicarbonate, acetic acid, ethylenediaminetetraacetic acid, and ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid were purchased from Sigma-Aldrich (St Louis, MO); Protocol 10% neutral-buffered formalin and sodium chloride from Fisher Scientific (Kalamazoo, MI); and Tris from Amresco (Solon, OH).
Animals
Male Sprague Dawley rats weighing 200–250 g (7–8 weeks old) were obtained from Harlan Laboratories (Indianapolis, IN), and acclimated to 12 h light and dark cycles in a University of Arizona AAALAC-certified animal facility for 1 week prior to experiments. Rats were fed either a control (Dyets, Bethlehem, PA) or methionine-choline-deficient (MCD) diet (Dyets, Bethlehem, PA) ad libitum for 8 weeks prior to MTX administration. Housing and experimental procedures were IACUC approved and in accordance with NIH guidelines for the care and use of experimental animals.
MTX disposition experiments
MTX was dissolved in 0.3 M sodium bicarbonate (pH 8.0). Control and MCD rodents were administered a single intraperitoneal dose of vehicle, 10, 40, or 100 mg/kg MTX. All experiments were performed in groups of eight rodents with mixed dosing (i.e., two vehicle, two 10 mg/kg, etc.). Rodents were placed in Nalgene metabolism cages with access to water and respective diets (powdered) ad libitum. Blood samples were collected at 6 h before dosing and 2, 4, 6, 12, 24, 48, and 96 h postdose via tail vein into preheparinized tubes. Samples were centrifuged at 10,000 × g for 2 min and plasma transferred to clean tubes. Urine and fecal samples were collected at −6 and 0 h before dosing, and at 6, 12, 18, 24, 36, 48, 60, 72, 84, and 96 h postdose into metabolism cage collection tubes. Cages were rinsed with 30 ml 100% ethanol and 30 ml deionized water after each collection. All samples were stored at −80°C until analysis. At 96 h postdose, all rodents were euthanized via CO2 asphyxiation. Histological liver and kidney slices for histolomorphologic examination were placed in 10% neutral-buffered formalin for 24 h, followed by 70% ethanol until paraffin embedding and hematoxylin and eosin (H&E) staining by the University of Arizona Histology Service Laboratory. The remaining liver and kidney tissue was snap frozen in liquid nitrogen and stored at −80°C. Small intestines of vehicle and 100 mg/kg MTX rodents were sectioned by forming a “Z” with the full length of the organ, differentiating the duodenum, jejunum, and ileum. Histological tissue samples were taken from the middle of each arm of the “Z,” flushed with sterile saline and fixed as described above onto nitrocellulose membranes for support. Colon samples were prepared by division into two approximately equal sections designating proximal and distal just below the cecum and above the rectum, respectively, and fixed as described.
Quantification of MTX
UHPLC-MS/MS analysis of MTX was adapted from the methods of Guo (Guo et al., 2007). Briefly, feces were homogenized in 2% formic acid (125 mg/ml buffer). Fecal supernatant and urine samples were diluted 3000-fold. Twenty microliters plasma was added to 40 μl methanol and centrifuged at 13,000 rpm for 10 min at 4°C. Twenty microliters plasma supernatant was added to 180 μl 5% methanol and filtered through 0.2 μm nylon. All samples were injected (20 μl) directly onto an Agilent 1260 UHPLC with an Agilent (Santa Clara, CA) Poroshell 120 EC C-18 (3 mm × 50 mm, 2.7 μm particle size) column (30°C). Chromatographic separation was achieved with a mobile phase of 0.1% formic acid (A) and 5mM ammonium formate in acetonitrile/water (v/v: 90/10, B) at a flow rate of 300 μl/min. Separation and timing was as follows: 10% B, 1 min; linear gradient to 45% B, 6 min; linear increase to 100% B, 9 min; decreased to 20% B, 11 min, hold 1 min. Mass identification was performed on an Agilent 6490 triple quadrupole mass spectrometer in multiple reaction monitoring mode with an electrospray ionization source in positive mode. MTX transitions monitored (4.3 min retention time): m/z 455.1 > 308.1, 455.1 > 175.1, and 455.1 > 134.1. Aminopterin was used as a surrogate standard and detected at m/z 441.1 > 294.1. Data were acquired and processed using Agilent MassHunter (v6.0) software. The method detection limit for MTX was 0.05 ng/ml based on S/N >3, whereas the calibration curve range was 0.5–750 ng/ml. Samples were quantified with a quadratic fit and 1/X weighting, R2 >0.99.
Immunoblot analysis
Whole cell lysates of rodent liver and kidney tissue were prepared as described previously (Hardwick et al., 2012). Immunoblots were performed as previously described (Hardwick et al., 2011, 2012) using the following monoclonal antibodies: Abcc1 (MRPm5), Abcc3 (M3II-9), Abcb1 (JSB-1), Abcc4 (Abcam, Inc., Cambridge, MA), Abcc2 (M2III-5), and Abcg2 (BXP-21) (Kamiya Biomedical Company, Seattle, WA). Quantification of relative protein expression was determined as reported (Hardwick et al., 2012) and normalized to total ERK (C-16 and C-14, Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Immunohistochemistry and Masson Trichrome staining
Immunohistochemistry (IHC) staining for Abcc2 and Abcb1 was performed on formalin-fixed, paraffin-embedded liver samples as described previously (Hardwick et al., 2012). IHC staining for proliferating cell nuclear antigen (PCNA) was performed via deparaffinization in xylene and rehydration in an ethanol gradient. Heat-mediated antigen retrieval was performed with Tris-EGTA buffer (pH 9.0), followed by endogenous peroxidase blocking as previously described (Hardwick et al., 2012), and overnight incubation with rabbit polyclonal antibody incubation (Abcam, Inc., Cambridge, MA) at 4°C. Antibody binding was detected via the MACH4 method and color development performed with Romulin AEC chromogen (Biocare Medical, Concord, CA). Masson Trichrome staining was performed using the Masson Trichrome Kit from Sigma-Aldrich (St Louis, MO) per manufacturer's instructions. All slides were imaged with a Leica DM4000B microscope, DFC450 camera, and Leica Application Suite software (Leica Microsystems, Wetzlar, Germany).
Statistical analysis
The data within this study did not conform to a normal distribution as determined by the Kurtosis and Shapiro-Wilk tests. Therefore, data were analyzed by Wilcoxon Rank Sum and presented as mean ± SEM. Asterisks (*) represent a disease effect due to diet administration and indicate a significant difference from control diet within each dosing group (i.e., control vehicle vs. MCD vehicle, etc.). Daggers (†) represent a drug effect due to MTX treatment and indicate a significant difference from vehicle within the same diet group (i.e., control vehicle vs. control 10 mg/kg MTX, etc.). All analyses were performed with Stata9 software, p ≤ 0.05 (Stata Corporation, College Station, TX).
RESULTS
For ease of explanation, rodent treatment groups will be referred to as follows: control diet with vehicle, 10, 40, or 100 mg/kg MTX—C0 (n = 5), C10 (n = 5), C40 (n = 5), and C100 (n = 5), respectively; MCD diet (NASH) with vehicle, 10, 40, or 100 mg/kg MTX—M0 (n = 5), M10 (n = 5), M40 (n = 5), and M100 (n = 6), respectively.
Hepatic Efflux Transporter Expression
Protein levels of Abcc1–4, Abcb1, and Abcg2 were assessed by immunoblot in control and MCD (NASH) liver lysates 96 h postdose. The results, expressed as relative to the control protein total ERK, are shown in Figure 1 with representative immunoblots depicting two samples from each group (A) and densitometric results of all rodents in the study (B). Each transporter has been implicated in the efflux of MTX, though Abcc2 is believed to be dominant in this function, whereas Abcc3 and Abcg2 may compensate in its absence (Vlaming et al., 2008, 2009b). Expression at baseline before rodents are exposed to drug is synonymous with vehicle treatment (C0 and M0), and represents basal disease effects. M0 rodents exhibited significantly higher levels of the basolateral transporters Abcc1, 3, and 4, as well as the canalicular transporters Abcc2, Abcb1, and Abcg2. MTX treatment resulted in a significant reduction in Abcc1–3 and Abcb1 protein levels in C100 rodents, whereas C10, C40, and C100 rodents had higher levels of Abcc4. Though MCD rodents exhibit higher levels of hepatic transporters at baseline, by 96 h after MTX administration a reduction in Abcc1–3, Abcb1, and Abcg2 protein was observed in all dose groups.
FIG. 1.

Hepatic transporter expression and localization. (A) Representative immunoblots of Abcc1–4, Abcb1, Abcg2, and total ERK in control and MCD rodents with two samples from each treatment group. (B) Relative protein levels of all samples normalized to total ERK; mean ± SEM. IHC staining of Abcc2 (C) and Abcb1 (D), 100× objective magnification. Asterisks (*) indicate p ≤ 0.05, control versus MCD; daggers (†) indicate p ≤ 0.05, MTX dose versus vehicle. Arrows indicate disrupted transporter localization.
Localization of Hepatic Efflux Transporters
IHC staining for the canalicular transporters Abcc2 and Abcb1 is shown in Figures 1C and 1D, respectively. Abcc2 staining diminished in control rodents with increasing dose of MTX, but was properly localized to the membrane. In MCD rodents, Abcc2 exhibited an irregular pattern suggestive of internalization of the protein (arrows, Fig. 1C). The altered staining pattern persisted throughout all MTX dose groups. A similar staining pattern was observed for Abcb1 in which positive staining was localized to the membrane in controls regardless of MTX dose received, and an aberrant staining pattern was evident in all MCD groups. Localization of Abcg2 was unaltered in M0 compared to C0 (Supplemental fig. 1), suggesting that the disrupted localization of Abcc2 and Abcb1 is a mechanism-based specific response rather than a generalized effect.
MTX-Induced Hepatocellular Damage
H&E liver sections were evaluated by a pathologist to assess three parameters: diet-induced, MTX-induced, and diet-dependent/MTX-induced changes. The tabulated results are shown in Table 1 with representative histology images in Figure 2A. As expected at baseline (vehicle), M0 rodents exhibited inflammation, lipid accumulation, and single cell necrosis (SCN) compared to C0. C40 rodents exhibited a slight increase in inflammation, whereas C40 and C100 presented with increased SCN. Overall, control rodents exhibited no overt hepatic damage as a result of MTX treatment. There was a slight attenuation in inflammation, lipid accumulation, and SCN in M10 and M40, but not M100, compared to M0. Additionally, it appeared as though the M40 and M100 groups incurred more damage; however, due to experimental design, there was sufficient time after the single injection of MTX for reparative mechanisms to begin. Nonetheless, M100 rodents incurred more severe damage compared to C100, as well as M10 and M40, in the form of fibrosis and biliary hyperplasia (arrows, Fig.2C and 2A, respectively).
TABLE 1. Hepatic Pathology.
| Group | Inflammation | Lipid accumulation | Single cell necrosis | Fibrosis | Biliary hyperplasia |
|---|---|---|---|---|---|
| C0 | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) |
| C10 | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) |
| C40 | 0.20 (0.20) | 0.00 (0.00) | 0.60 (0.24) | 0.00 (0.00) | 0.00 (0.00) |
| C100 | 0.00 (0.00) | 0.00 (0.00) | 0.50 (0.16) | 0.00 (0.00) | 0.20 (0.20) |
| M0 | 2.50 (0.32) | 4.50 (0.16) | 1.40 (0.40) | 0.00 (0.00) | 0.00 (0.00) |
| M10 | 2.10 (0.24) | 3.40 (0.29) | 1.40 (0.40) | 0.00 (0.00) | 0.00 (0.00) |
| M40 | 1.20 (0.34) | 2.75 (0.14) | 1.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) |
| M100 | 1.67 (0.36) | 4.50 (0.13) | 1.25 (0.21) | 0.75 (0.21) | 1.33 (0.36) |
Note. Hepatic pathology grading (0–5, 5 being most severe) of control and MCD (NASH) rodents 96 h following a single ip injection of vehicle, 10, 40, or 100 mg/kg MTX. Data are shown as mean ± (SEM).
FIG. 2.
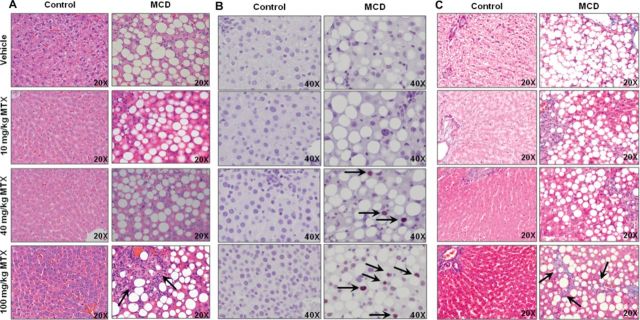
MTX-induced hepatocellular damage. Representative images of (A) H&E-stained liver, (B) IHC staining of PCNA, and (C) Masson Trichrome staining of fibrosis; objective magnification is as indicated. Arrows indicate biliary hyperplasia (A), positive PCNA staining (B), and fibrosis (C).
Hepatic Regeneration Following MTX-Induced Damage in NASH
Tissue collection was conducted 96 h after administration of a single MTX dose, thus allowing time for initiation of reparative mechanisms following any histological damage. Histopathologic examination suggested hepatocellular damage in M40 and M100 groups that was substantial enough for the initiation of a regenerative response. This was assessed via staining for PCNA. Representative images are shown in Figure 2B with arrows denoting positive nuclei. Coinciding with the histopathological analysis, nuclei positive for PCNA were observed only in the M40 and M100 groups near the periportal region.
MTX-Induced Hepatic Fibrosis in NASH
To further characterize the hepatic fibrosis revealed in the histomorphologic analysis of M100 rodents, Masson Trichrome staining was performed (Fig. 2C). No aberrant staining was observed in control groups at any dose, indicating no evidence of overt fibrosis due to MTX. However, positive fibrotic staining increased in MCD rodents with MTX dose. Strikingly, M100 livers had pronounced branching fibrosis (arrows, Fig. 2C), indicating significant disease-dependent, MTX-induced damage. No branching fibrosis was observed in other treatment groups.
Renal Efflux Transporter Expression
Protein levels of Abcc2, Abcc4, and Abcg2 were assessed by immunoblot in control and MCD kidney lysates 96 h postdose. The results, expressed as relative to the control protein total ERK, are shown in Figure 3 with representative immunoblots depicting two samples from each group (A) and densitometric results of all rodents in the study (B). Comparing M0 to C0, there were no changes in protein levels of any transporter, indicating no disease effects on renal transporters. Abcc2 protein was reduced in C10, C40, and C100 compared to C0, whereas Abcc2 was reduced in M10 and M100 compared to M0; Abcc2 also was reduced in M10 compared to C10. No significant alterations in Abcc4 protein levels were observed. Abcg2 protein levels were reduced in C100 compared to C0; however, no other alterations were noted.
FIG. 3.

Renal transporter expression and MTX-induced kidney damage. (A) Representative immunoblots of Abcc2, Abcc3, Abcg2, and total ERK in control and MCD rodents with two samples from each treatment group. (B) Relative protein levels of all samples normalized to total ERK; mean ± SEM. Representative H&E-stained kidney (C), 20× objective magnification. Asterisks (*) indicate p ≤ 0.05, control versus MCD; daggers (†) indicate p ≤ 0.05, MTX dose versus vehicle. Arrows indicate protein cast formation.
MTX-Induced Effects on Renal Histology
H&E kidney sections were evaluated as described for liver. The tabulated results are shown in Table 2 with representative histology images in Figure 3C. Both control and MCD rodents exhibited dose-dependent increases in nephropathy, tubular degeneration, necrosis, and regeneration; however, C100 and M100 groups tended to exhibit less severe cellular damage at 96 h following MTX administration as a result of an observed increase in regeneration. Evaluation of the kidney tissue at 96 h postdose appeared to be after an acute necrotic event that was followed by transition to a subacute period in which regeneration and dilated tubules are more common. Protein cast formation was observed only in C100 and M100 (arrows, Fig. 3C). In the 10 and 40 mg/kg MTX treatment groups, controls tended to exhibit slightly more damage, while M100 rodents exhibited greater tubular degeneration and necrosis. Additionally, M100 rodents exhibited a reduction in regeneration compared to C100. These data suggest greater overall damage in M100 compared to C100, accompanied by a reduced capacity for repair.
TABLE 2. Renal Pathology.
| Group | Nephropathy | Tubular degeneration | Necrosis | Regeneration | Protein casts | Tubule dilation |
|---|---|---|---|---|---|---|
| C0 | 1.00 (0.00) | 0.20 (0.20) | 0.20 (0.20) | 0.60 (0.24) | 0.00 (0.00) | 0.00 (0.00) |
| C10 | 1.00 (0.32) | 0.50 (0.22) | 0.40 (0.24) | 1.00 (0.32) | 0.00 (0.00) | 0.00 (0.00) |
| C40 | 2.20 (0.34) | 1.50 (0.52) | 0.70 (0.20) | 1.90 (0.24) | 0.00 (0.00) | 0.00 (0.00) |
| C100 | 0.80 (0.20) | 0.00 (0.00) | 0.80 (0.12) | 3.30 (0.60) | 0.50 (0.27) | 2.00 (0.20) |
| M0 | 0.00 (0.00) | 0.20 (0.20) | 0.00 (0.00) | 0.20 (0.20) | 0.00 (0.00) | 0.00 (0.00) |
| M10 | 0.90 (0.19) | 0.20 (0.20) | 0.10 (0.10) | 0.40 (0.24) | 0.00 (0.00) | 0.00 (0.00) |
| M40 | 1.40 (0.29) | 0.70 (0.30) | 0.50 (0.22) | 1.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) |
| M100 | 0.67 (0.21) | 1.17 (0.33) | 1.00 (0.26) | 1.42 (0.27) | 0.42 (0.20) | 1.00 (0.37) |
Note. Renal pathology grading (0–5, 5 being most severe) of control and MCD (NASH) rodents 96 h following a single ip injection of vehicle, 10, 40, or 100 mg/kg MTX. Data are shown as mean ± (SEM).
High-Dose MTX-Induced Intestinal Damage in Control Rodents
During sample collection, the first group of C100 rodents exhibited diarrhea, the classic manifestation of MTX-induced gastrointestinal toxicity, sooner and more severely than M100 rodents. On average, C100 rodents presented with diarrhea between 60 and 72 h postdose, whereas M100 rodents presented later, or not at all. Following this initial observation, intestinal samples were collected from the remaining C100, M100, and vehicle rodents and prepared for qualitative pathological analysis (n = 2). In Figure 4, representative images are shown for duodenum, jejunum, and ileum (A) and colon (B). C100 rodents tended to exhibit increased villous atrophy and fusion, inflammation, and mucosal collapse in the duodenum versus M100. Although damage to the jejunum tended to be somewhat comparable between C100 and M100, C100 rodents presented with more villous fusion and erosion. In the ileum, greater villous atrophy and fusion with dilated crypts was observed in C100. MTX-induced damage in control rodents was more pronounced in the colon. C100 rodents appeared to incur greater epithelial loss, inflammation, vacuolization, and erosion in both the proximal and distal colon. This qualitative analysis suggests greater gastrointestinal damage in control rodents as a result of high-dose MTX treatment.
FIG. 4.

High-dose MTX-induced intestinal damage. Representative images of H&E-stained (A) duodenum, jejunum, ileum, and (B) proximal and distal colon in control and MCD rodents, 20× objective magnification.
MTX Disposition
MTX levels in plasma, feces, and urine are shown in Figures 5A, 5B, and 5C, respectively, with cumulative urine in Figure 5D. Due to the onset of MTX-induced diarrhea, urine and feces samples could reliably be assessed only up to 24 h following MTX administration. Time intervals were chosen for analysis based on a minimum of n = 3 available samples at each time point. M40 rodents exhibited increased plasma MTX at 2, 4, and 6 h, whereas plasma MTX was increased in M100 at 6 h only. Fecal MTX excretion was significantly reduced in M40 at 6 h and in M10 and M100 at 24 h. Though cumulative urinary excretion of MTX was not different between controls and MCD up to 24 h, the majority of MTX that was excreted appeared to occur in the initial 6 h in M100 rodents, and was reduced in M100 compared to C100 at 18 h.
FIG. 5.

MTX disposition. MTX levels in (A) plasma, (B) feces, and (C) urine at indicated time points; mean ± SEM. (D) Cumulative total urinary MTX. Asterisks (*) indicate p ≤ 0.05, control versus MCD.
DISCUSSION
At low doses, MTX is used to treat inflammatory conditions, whereas high-dose MTX is reserved for the treatment of several types of cancers (Cole and Kamen, 2000; Widemann and Adamson, 2006). MTX is considered a cornerstone of many multidrug chemotherapeutic treatment regimens, and often requires gradual dose escalation to achieve disease remission (Schmiegelow, 2009). However, MTX incurs severe dose-limiting toxicity (hematopoietic, hepatic, gastrointestinal, and renal) that restricts dose escalation in the clinic, and is believed to be the result of delayed clearance and increased exposure (Schmiegelow, 2009; Shibayama et al., 2006; Widemann and Adamson, 2006). The current studies were designed to reflect the low doses of MTX such as that used to treat inflammatory disease, and high doses such as in the treatment of ALL. However, high-dose MTX used in the treatment of ALL is typically dosed based on body surface area. When considering the average body surface area of pediatric patients aged 1–10 years and conversion to mg/kg, the highest MTX dose used in the current study is under the range expected. MTX is primarily eliminated unchanged via urinary excretion; however, severe gastrointestinal toxicity is believed to be due to prolonged enterohepatic recycling (Kato et al., 2009; Kitamura et al., 2008; Schmiegelow, 2009). The known, dose-limiting toxicity profile of MTX makes it a useful probe to study the toxic effects of disrupted hepatobiliary disposition in NASH.
Currently, we evaluated the dose-dependent, toxicity profile of MTX in NASH. Our previous studies suggest disrupted hepatobiliary drug disposition in NASH via altered localization of Abcc2 (limits biliary excretion and thus intestinal exposure) and increased Abcc3 expression, which facilitates excretion into systemic blood (Hardwick et al., 2011, 2012; Lickteig et al., 2007). We hypothesized that NASH rodents would develop increased MTX-induced hepatic and renal toxicity and plasma retention due to internalization and upregulation of canalicular and basolateral efflux transporters, respectively. Consequently, we have identified increased hepatic and renal toxicity in NASH, as well as reduced gastrointestinal toxicity, which coincides with reduced fecal elimination of MTX.
At low doses, the anti-inflammatory properties of MTX were evident through the observed reduction in hepatic inflammation, lipid accumulation, and SCN in NASH rodents. High-dose MTX (100 mg/kg) resulted in significant damage compared to vehicle. MTX-induced hepatic branching fibrosis in M100 rodents. This effect is typically observed in patients receiving chronic low-dose MTX for the treatment of inflammatory diseases (Barker et al., 2011); however, we have observed such damage in NASH rodents after only a single MTX dose. Additional evidence demonstrating increased hepatocellular damage exclusive to NASH livers was observed via PCNA staining. M40 and M100 rodents exhibited positive nuclear staining, whereas none was observed in control rodents at any MTX dose. These data suggest NASH rodents incurred more hepatocellular damage, and had begun repair by 96 h postdose.
MTX efflux transport has been shown to be dominated by Abcc2, Abcc3, and Abcg2; although Abcc1, Abcc4, and Abcb1 may serve as compensatory routes of excretion (Chen et al., 2002; Vlaming et al., 2008, 2009b; Wang et al., 2011). Abcc2 has been shown to compensate in the absence of Abcg2; however, Abcg2 can only partly mediate biliary elimination of MTX when Abcc2 is absent, suggesting dominance of Abcc2 in MTX biliary excretion (Vlaming et al., 2009a). Furthermore, Abcc3 facilitates the increase in plasma MTX levels when Abcc2 is absent (Vlaming et al., 2009b, 2011). Both the dominant and compensatory transporters responsible for MTX efflux were upregulated in M0 rodents. This is similar to the expression profile present when animals were first exposed to drug. However, two canalicular transporters (Abcc2 and Abcb1) exhibited disrupted localization in NASH, which coincided with diminished biliary excretion of MTX.
Disrupted localization of two canalicular transporters and concomitant upregulation of several sinusoidal transporters suggests that NASH rodents may have greater systemic exposure to MTX. However, we observed a significant elevation in plasma MTX in M40 rodents only, although M10 rodents exhibited a trend for increased plasma levels. It is likely that collection of plasma samples sooner following dosing would have revealed more details of toxicokinetic differences between control and NASH rodents. Using currently available analytical methods, we were unable to reliably extract tissue samples for analysis of MTX concentrations. It is also possible that increased tissue retention of MTX, particularly in the liver, may underlie the lack of a significant change in plasma MTX concentrations. Furthermore, shunting of MTX to metabolic clearance via aldehyde oxidase metabolism to a 7-hydroxy metabolite is known to lead to renal and hepatic toxicity (Smeland et al., 1994; Vlaming et al., 2011; Widemann and Adamson, 2006). Though we were unable to provide a reliable measure of 7-hydroxymethotrexate using our current analytical methods, it is quite possible that formation of the toxic metabolite and/or its retention in tissue could explain the observed increases in hepatic as well as renal toxicity in NASH.
We noted a stark difference in MTX target organ toxicity between C100 and M100 rodents. In addition to greater hepatotoxicity, M100 rodents tended towards increased renal toxicity in the form of tubular degeneration and necrosis. M100 rodents also appeared to have a reduced capacity for regeneration following renal injury. In contrast, the majority of MTX urinary excretion occurred by 6 h in NASH rodents, suggesting renal retention to be an unlikely cause of increased renal toxicity in NASH. Unlike hepatic transporters, we did not identify any NASH-induced effects on renal transporter expression. However, MTX induced numerous dose-dependent effects on hepatic transporter expression in NASH, while MTX effects on renal transporter expression in NASH were minimal. Together, these data further suggest increased liver and kidney exposure to MTX in NASH as compared to control rodents.
In contrast to greater hepatic and renal toxicity in NASH, C100 rodents exhibited increased intestinal damage compared to M100. These data coincide with reduced fecal elimination of MTX in NASH, and may likely be a result of altered localization of biliary transporters. Although administration of leucovorin may prevent severe hepatic and renal toxicity in high-dose MTX treatment, a portion of patients are resistant to leucovorin rescue and still experience fatal toxicity (Schmiegelow, 2009; Widemann and Adamson, 2006). Though gastrointestinal toxicity in the form of diarrhea may be an indicator of a patient susceptible to severe MTX toxicity (Schmiegelow, 2009), our data suggest this indicator may be unreliable in NASH. Specifically, in the absence of noticeable diarrhea, NASH rodents exhibited severe hepatotoxicity and increased renal toxicity compared to controls. Although gastrointestinal and nephrotoxicity may be reversed with early intervention, severe hepatotoxicity due to prolonged exposure poses a significant threat (Schmiegelow, 2009; Widemann and Adamson, 2006). The current data indicate the initiation of reparative mechanisms in damaged NASH livers; however, these efforts appear to result in the generation of fibrosis and may predispose to cirrhosis.
Considering the current prevalence of obesity and NAFLD together, determining the effects of NASH on drug disposition and susceptibility to multiple organ toxicities is of significant value. Furthermore, studies have shown that an increasing proportion of oncology patients, including pediatric patients, are presenting with obesity at the time of cancer diagnosis, thus expanding the susceptible patient pool (Butturini et al., 2007). The current data suggest that it is therefore likely that NASH patients receiving MTX may indeed be at greater risk for the life-threatening MTX-induced hepatic and renal toxicity in the absence of overt gastrointestinal toxicity.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health [AI083927, HD062489, ES007091, ES006694].
Supplementary Material
REFERENCES
- Anstee Q. M., Daly A. K., Day C. P. Genetic modifiers of non-alcoholic fatty liver disease progression. Biochim. Biophys. Acta. 2011;1812:1557–1566. doi: 10.1016/j.bbadis.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Barker J., Horn E. J., Lebwohl M., Warren R. B., Nast A., Rosenberg W., Smith C., International Psoriasis, C. Assessment and management of methotrexate hepatotoxicity in psoriasis patients: Report from a consensus conference to evaluate current practice and identify key questions toward optimizing methotrexate use in the clinic. J. Eur. Acad. Dermatol. Venereol. 2011;25:758–764. doi: 10.1111/j.1468-3083.2010.03932.x. [DOI] [PubMed] [Google Scholar]
- Butturini A. M., Dorey F. J., Lange B. J., Henry D. W., Gaynon P. S., Fu C., Franklin J., Siegel S. E., Seibel N. L., Rogers P. C., et al. Obesity and outcome in pediatric acute lymphoblastic leukemia. J. Clin. Oncol. 2007;25:2063–2069. doi: 10.1200/JCO.2006.07.7792. [DOI] [PubMed] [Google Scholar]
- Chen Z. S., Lee K., Walther S., Raftogianis R. B., Kuwano M., Zeng H., Kruh G. D. Analysis of methotrexate and folate transport by multidrug resistance protein 4 (ABCC4): MRP4 is a component of the methotrexate efflux system. Cancer Res. 2002;62:3144–3150. [PubMed] [Google Scholar]
- Cheung O., Sanyal A. J. Recent advances in nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 2009;25:230–237. doi: 10.1097/mog.0b013e3283294a18. [DOI] [PubMed] [Google Scholar]
- Cole P. D., Kamen B. A. High-dose methotrexate is lethal to rats. Why give it to children. Pediatr. Hematol. Oncol. 2000;17:609–613. doi: 10.1080/08880010050211312. [DOI] [PubMed] [Google Scholar]
- Fisher C. D., Lickteig A. J., Augustine L. M., Oude Elferink R. P., Besselsen D. G., Erickson R. P., Cherrington N. J. Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur. J. Pharmacol. 2009a;613:119–127. doi: 10.1016/j.ejphar.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. D., Lickteig A. J., Augustine L. M., Ranger-Moore J., Jackson J. P., Ferguson S. S., Cherrington N. J. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2009b;37:2087–2094. doi: 10.1124/dmd.109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P., Wang X., Liu L., Belinsky M. G., Kruh G. D., Gallo J. M. Determination of methotrexate and its major metabolite 7-hydroxymethotrexate in mouse plasma and brain tissue by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2007;43:1789–1795. doi: 10.1016/j.jpba.2006.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Ferreira D. W., More V. R., Lake A. D., Lu Z., Manautou J. E., Slitt A. L., Cherrington N. J. Altered UDP-glucuronosyltransferase and sulfotransferase expression and function during progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2013;41:554–561. doi: 10.1124/dmd.112.048439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Fisher C. D., Canet M. J., Lake A. D., Cherrington N. J. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2010;38:2293–2301. doi: 10.1124/dmd.110.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Fisher C. D., Canet M. J., Scheffer G. L., Cherrington N. J. Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2011;39:2395–2402. doi: 10.1124/dmd.111.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick R. N., Fisher C. D., Street S. M., Canet M. J., Cherrington N. J. Molecular mechanism of altered ezetimibe disposition in nonalcoholic steatohepatitis. Drug Metab. Dispos. 2012;40:450–460. doi: 10.1124/dmd.111.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S., Ito K., Kato Y., Wakayama T., Kubo Y., Iseki S., Tsuji A. Involvement of multidrug resistance-associated protein 1 in intestinal toxicity of methotrexate. Pharm. Res. 2009;26:1467–1476. doi: 10.1007/s11095-009-9858-6. [DOI] [PubMed] [Google Scholar]
- Kitamura Y., Hirouchi M., Kusuhara H., Schuetz J. D., Sugiyama Y. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3) J. Pharmacol. Exp. Ther. 2008;327:465–473. doi: 10.1124/jpet.108.140475. [DOI] [PubMed] [Google Scholar]
- Lake A. D., Novak P., Fisher C. D., Jackson J. P., Hardwick R. N., Billheimer D. D., Klimecki W. T., Cherrington N. J. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2011;39:1954–1960. doi: 10.1124/dmd.111.040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langman G., Hall P. M., Todd G. Role of non-alcoholic steatohepatitis in methotrexate-induced liver injury. J. Gastroenterol. Hepatol. 2001;16:1395–1401. doi: 10.1046/j.1440-1746.2001.02644.x. [DOI] [PubMed] [Google Scholar]
- Lickteig A. J., Fisher C. D., Augustine L. M., Aleksunes L. M., Besselsen D. G., Slitt A. L., Manautou J. E., Cherrington N. J. Efflux transporter expression and acetaminophen metabolite excretion are altered in rodent models of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2007;35:1970–1978. doi: 10.1124/dmd.107.015107. [DOI] [PubMed] [Google Scholar]
- Marra F., Gastaldelli A., Svegliati Baroni G., Tell G., Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- McCullough A. J. Epidemiology of the metabolic syndrome in the USA. J. Dig. Dis. 2011;12:333–340. doi: 10.1111/j.1751-2980.2010.00469.x. [DOI] [PubMed] [Google Scholar]
- Mencin A. A., Lavine J. E. Advances in pediatric nonalcoholic fatty liver disease. Pediatr. Clin. North Am. 2011;58:1375–1392. doi: 10.1016/j.pcl.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Schmiegelow K. Advances in individual prediction of methotrexate toxicity: A review. Br. J. Haematol. 2009;146:489–503. doi: 10.1111/j.1365-2141.2009.07765.x. [DOI] [PubMed] [Google Scholar]
- Shibayama Y., Ushinohama K., Ikeda R., Yoshikawa Y., Motoya T., Takeda Y., Yamada K. Effect of methotrexate treatment on expression levels of multidrug resistance protein 2, breast cancer resistance protein and organic anion transporters Oat1, Oat2 and Oat3 in rats. Cancer Sci. 2006;97:1260–1266. doi: 10.1111/j.1349-7006.2006.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeland E., Bremnes R. M., Andersen A., Jaeger R., Eide T. J., Huseby N. E., Aarbakke J. Renal and hepatic toxicity after high-dose 7-hydroxymethotrexate in the rat. Cancer Chemother. Pharmacol. 1994;34:119–124. doi: 10.1007/BF00685928. [DOI] [PubMed] [Google Scholar]
- USFDA. Adverse Events Reporting System Statistics. Patient Outcomes by Year. 2012. Available at: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm070461.htm10.1093/toxsci/kfu156.html. Accessed January 13, 2014.
- Vlaming M. L., Pala Z., van Esch A., Wagenaar E., de Waart D. R., van de Wetering K., van der Kruijssen C. M., Oude Elferink R. P., van Tellingen O., Schinkel A. H. Functionally overlapping roles of Abcg2 (Bcrp1) and Abcc2 (Mrp2) in the elimination of methotrexate and its main toxic metabolite 7-hydroxymethotrexate in vivo. Clin. Cancer Res. 2009a;15:3084–3093. doi: 10.1158/1078-0432.CCR-08-2940. [DOI] [PubMed] [Google Scholar]
- Vlaming M. L., Pala Z., van Esch A., Wagenaar E., van Tellingen O., de Waart D. R., Oude Elferink R. P., van de Wetering K., Schinkel A. H. Impact of Abcc2 (Mrp2) and Abcc3 (Mrp3) on the in vivo elimination of methotrexate and its main toxic metabolite 7-hydroxymethotrexate. Clin. Cancer Res. 2008;14:8152–8160. doi: 10.1158/1078-0432.CCR-08-1609. [DOI] [PubMed] [Google Scholar]
- Vlaming M. L., van Esch A., Pala Z., Wagenaar E., van de Wetering K., van Tellingen O., Schinkel A. H. Abcc2 (Mrp2), Abcc3 (Mrp3), and Abcg2 (Bcrp1) are the main determinants for rapid elimination of methotrexate and its toxic metabolite 7-hydroxymethotrexate in vivo. Mol. Cancer Ther. 2009b;8:3350–3359. doi: 10.1158/1535-7163.MCT-09-0668. [DOI] [PubMed] [Google Scholar]
- Vlaming M. L., van Esch A., van de Steeg E., Pala Z., Wagenaar E., van Tellingen O., Schinkel A. H. Impact of abcc2 [multidrug resistance-associated protein (MRP) 2], abcc3 (MRP3), and abcg2 (breast cancer resistance protein) on the oral pharmacokinetics of methotrexate and its main metabolite 7-hydroxymethotrexate. Drug Metab. Dispos. 2011;39:1338–1344. doi: 10.1124/dmd.111.038794. [DOI] [PubMed] [Google Scholar]
- Wang Z., Zhou Q., Kruh G. D., Gallo J. M. Dose-dependent disposition of methotrexate in Abcc2 and Abcc3 gene knockout murine models. Drug Metab. Dispos. 2011;39:2155–2161. doi: 10.1124/dmd.111.041228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren R. B., Griffiths C. E. Systemic therapies for psoriasis: Methotrexate, retinoids, and cyclosporine. Clin. Dermatol. 2008;26:438–447. doi: 10.1016/j.clindermatol.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Widemann B. C., Adamson P. C. Understanding and managing methotrexate nephrotoxicity. Oncologist. 2006;11:694–703. doi: 10.1634/theoncologist.11-6-694. [DOI] [PubMed] [Google Scholar]
- Wilke R. A., Lin D. W., Roden D. M., Watkins P. B., Flockhart D., Zineh I., Giacomini K. M., Krauss R. M. Identifying genetic risk factors for serious adverse drug reactions: Current progress and challenges. Nat. Rev. Drug Discov. 2007;6:904–916. doi: 10.1038/nrd2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.