

Abstract
Aim: Paraquat, a quaternary nitrogen herbicide, is a highly toxic prooxidant resulting in multi-organ failure including the heart although the underlying mechanism still remains elusive. This study was designed to examine the role of the cellular fuel sensor AMP-activated protein kinase (AMPK) in paraquat-induced cardiac contractile and mitochondrial injury. Results: Wild-type and transgenic mice with overexpression of a mutant AMPK α2 subunit (kinase dead, KD), with reduced activity in both α1 and α2 subunits, were administered with paraquat (45 mg/kg) for 48 h. Paraquat elicited cardiac mechanical anomalies including compromised echocardiographic parameters (elevated left ventricular end-systolic diameter and reduced factional shortening), suppressed cardiomyocyte contractile function, intracellular Ca2+ handling, reduced cell survival, and overt mitochondrial damage (loss in mitochondrial membrane potential). In addition, paraquat treatment promoted phosphorylation of AMPK and autophagy. Interestingly, deficiency in AMPK attenuated paraquat-induced cardiac contractile and intracellular Ca2+ derangement. The beneficial effect of AMPK inhibition was associated with inhibition of the AMPK-TSC-mTOR-ULK1 signaling cascade. In vitro study revealed that inhibitors for AMPK and autophagy attenuated paraquat-induced cardiomyocyte contractile dysfunction. Conclusion: Taken together, our findings revealed that AMPK may mediate paraquat-induced myocardial anomalies possibly by regulating the AMPK/mTOR-dependent autophagy.
Keywords: paraquat, AMPK deficiency, autophagy, myocardium, contractile function
Autophagy is a highly coordinated intracellular lysosomal-mediated catabolic process that degrades damaged or dysfunctional proteins, cytoplasmic components, and intracellular organelles (Klionsky and Emr, 2000; Levine and Klionsky, 2004). It is essential to maintain cellular homeostasis in which removal of dysfunctional components and replacement with newly synthesized ones occurs dynamically. Autophagy can be turned on in response to starvation, protein aggregation, oxidative stress, and the damage that affects distinct cytoplasmic organelles (Levine and Klionsky, 2004; Niso-Santano et al., 2011). It has been suggested that basal level of autophagy can be cardioprotective and play a pivotal role in the maintenance of cardiac geometry and function (Kuma et al., 2004; Nakai et al., 2007; Nemchenko et al., 2011). Not surprisingly, autophagy can serve as a double-edged sword with both beneficial and detrimental effects in heart. Whereas certain studies have shown that autophagy may be protective in a number of heart diseases including ischemia-reperfusion injury and pressure overload-induced cardiac hypertrophy (Kassiotis et al., 2009; Nakai et al., 2007; Nemchenko et al., 2011; Xu et al., 2013a), others suggest that excessive or uncontrolled autophagy may lead to loss of functional protein, depletion of essential molecules, oxidative stress, collapse of cellular catabolic machinery, and ultimately autophagic cell death in the heart (Nemchenko et al., 2011; Xie et al., 2011).
Paraquat (1,1′-dimethyl-4,4′-bipyridinium dichloride), a widely used non-selective herbicide, is known to result in multiple organ damage and failure (Cristovao et al., 2009). Epidemiological studies have suggested that prolonged exposure of paraquat could increase the risk of developing Parkinson's disease in human subjects (Dagda et al., 2013; Liou et al., 1997). Paraquat, reminiscent of the dopaminergic neurotoxin MPP+, may deplete dopaminergic neurons in the midbrain, en route to apoptotic cell death (McCormack et al., 2002; Niso-Santano et al., 2006, 2011; Peng et al., 2004). A number of cellular mechanisms have been proposed for paraquat-induced cell injury including inhibition of mitochondrial oxidative phosphorylation and interruption of mitochondrial respiration chain, resulting in impaired energy metabolism, proteasomal dysfunction, and oxidative stress (Alexi et al., 2000; Gonzalez-Polo et al., 2004; McCormack et al., 2005; Niso-Santano et al., 2010; Tawara et al., 1996). Recent studies suggest that paraquat compromises myocardial survival and contractile function, en route to cardiopulmonary failure (Chan et al., 2007; Ge et al., 2010; Koo et al., 2002; Li et al., 2007). It has shown that paraquat could induce autophagy as evidenced by elevated autophagic markers such as Beclin-1, LC3 II, p62 degradation, and mammalian target of rapamycin (mTOR) dephosphorylation (Gonzalez-Polo et al., 2007; Niso-Santano et al., 2011). However, a number of neurological studies suggested that paraquat possibly promotes protein aggregation and blocks autophagy due to impaired basal autophagy (Garcia-Garcia et al., 2013; Manning-Bog et al., 2002,2003). Nonetheless, whether and how autophagy is involved in paraquat-induced myocardial dysfunction still remains unclear.
The AMP-activated protein kinase (AMPK), a master regulator of several metabolic processes, acts as an autophagy inducer by directly phosphorylating TSC2 to activate the Rheb-GTPase, leading to suppressed mTOR complex 1 (mTORC1) and autophagy activation (Gottlieb, 2012; Inoki et al., 2003; Tripathi et al., 2013; Wullschleger et al., 2006). AMPK can also directly phosphorylate and activate unc-51-like kinase 1 (ULK1) to turn on autophagy independent of mTORC1 (Egan et al., 2011; Kim et al., 2011; Lee et al., 2010; Tripathi et al., 2013). However, activated ULK1 may participate in a negative feedback mechanism to suppress AMPK activity (Loffler et al., 2011). To this end, this study took advantage of a transgenic mouse model with overexpression of mutant AMPKα2 subunit to examine the impact of AMPK deficiency on paraquat-induced myocardial dysfunction and the underlying mechanisms with a special focus on autophagy.
MATERIALS AND METHODS
Experimental animals and paraquat treatment
All animal experimental procedures carried out here were approved by the Animal Use and Care Committees at the University of Wyoming (Laramie, WY). Adult muscle-specific AMPK kinase dead (KD) transgenic mice that express a KD rat α2 isoform (K45R mutation) and their wild-type (WT) littermates C57BL/6 were used as described (Mu et al., 2001; Russell et al., 2004). AMPK KD mice were genotyped using polymerase chain reaction (PCR) technique. The PCR primers used were: GGT CGA CGG TAT CGA TAA GCT TGA TAT C (forward) and GAA GGA ACC CGT TGG AGG ACT GGA GGC GAG G (reverse) (Mu et al., 2001; Turdi et al., 2011). All mice were housed in a temperature-controlled room (22.8 ± 2.0°C, 45–50% humidity) under a 12 h/12 h light/dark and allowed access to food and tap water ad libitum. For acute paraquat challenge, 4-month-old mice of both sex were delivered a single intraperitoneal injection of paraquat (45 mg/kg, ∼100 μl, paraquat dichloride, Sigma-Aldrich, St. Louis, MO) or the vehicle saline, and were examined 48 h later (Day and Crapo, 1996). Body weight was recorded before and after treatment of paraquat or vehicle.
Echocardiographic assessment
Cardiac geometry and function were evaluated in anesthetized (ketamine 80 mg/kg and xylazine 12 mg/kg, i.p.) mice using the two-dimensional guided M-mode echocardiography (Philips SONOS 5500) equipped with a 15–6 MHz linear transducer. Left ventricular (LV) anterior and posterior wall dimensions during diastole and systole were recorded from three consecutive cycles in M mode using the method adopted by the American Society of Echocardiography. Fractional shortening was calculated from LV end-diastolic (LVEDD) and end-systolic (LVESD) diameters using the equation (LVEDD − LVESD)/LVEDD*100. Echocardiographic LV mass was estimated by [(LVEDD + septal wall thickness + posterior wall thickness)3 − LVEDD3] × 1.055, where 1.055 (mg/mm3) is the density of myocardium. Heart rates were averaged over 10 consecutive cycles (Ren et al., 2008).
Cardiomyocyte isolation
After ketamine/xylazine sedation, hearts were rapidly removed from anesthetized 4-month-old adult mice of both sex and were mounted onto a temperature-controlled (37°C) Langendorff system. After perfusing with a modified Tyrode solution (Ca2+ free) for 2 min, the heart was digested for 16–20 min with a Ca2+-free KHB buffer containing Liberase Blendzyme 4 (Hoffmann-La Roche Inc., Indianapolis, IN). The modified Tyrode solution (pH 7.4) contained the following (in mM): NaCl 135, KCl 4.0, MgCl2 1.0, HEPES 10, NaH2PO4 0.33, glucose 10, and butanedione monoxime 10, and the solution was gassed with 5% CO2–95% O2. The digested heart was then removed from the cannula and left ventricle was cut into small pieces in the modified Tyrode's solution. Tissue pieces were gently agitated and pellet of cells was resuspended. Extracellular Ca2+ was added incrementally back to 1.20 mM over a period of 30 min. A yield of at least 60–70% viable rod-shaped cardiomyocytes with clear sarcomere striations was achieved. Only rod-shaped cardiomyocytes with clear edges were selected for mechanical and intracellular Ca2+ studies (Li et al., 2007).
Cell shortening/relengthening
Mechanical properties of myocytes were assessed using a SoftEdge Myocam system (IonOptix Corporation, Milton, MA). IonOptix SoftEdge software was used to capture changes in cardiomyocyte length during shortening and relengthening. In brief, cardiomyocytes were placed in a Warner chamber mounted on the stage of an inverted microscope (Olympus IX-70) and superfused (∼1 ml/min at 25°C) with a buffer containing (in mM) 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES at pH 7.4. Myocytes were field stimulated with supra-threshold voltage at a frequency of 0.5 Hz (unless otherwise stated), 3 ms duration, using a pair of platinum wires placed on opposite sides of the chamber connected to a FHC stimulator (Brunswick, NE). The myocyte being studied was displayed on the computer monitor using an IonOptix MyoCam camera. IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. Cell shortening and relengthening were assessed using the following indices: peak shortening (PS)—the amplitude myocytes shortened on electrical stimulation, which is indicative of peak ventricular contractility; time-to-PS (TPS)—the duration of myocyte shortening, which is indicative of contraction duration; time-to-90% relengthening (TR90)—the duration to reach 90% relengthening, which represents cardiomyocyte relaxation duration (90% rather than 100% relengthening was used to avoid noisy signal at baseline concentration); and maximal velocities of shortening (+ dL/dt) and relengthening (− dL/dt)—maximal slope (derivative) of shortening and relengthening phases, which are indicatives of maximal velocities of ventricular pressure rise/fall (Ren et al., 2008).
To assess the role of AMPK and autophagy in paraquat-induced cardiomyocyte contractile response, cardiomyocytes from adult WT mice were treated with paraquat (100 μM) at 37°C for 3 h (Ge et al., 2010) in the presence or absence of the AMPK inhibitor compound C (5 μM) for 4 h (Guo and Ren, 2012), the mTOR inhibitor rapamycin (5 μM) for 4 h (Yuan et al., 2009), the lysosomal inhibitor bafilomycin A1 (100 μM) for 2 h (Xu et al., 2013b), or the autophagy inhibitor 3-methyladenine (3-MA, 10 mM) for 4 h (Guo and Ren, 2012).
Intracellular Ca2+ transients
A cohort of myocytes was loaded with fura-2/AM (0.5 μM) for 10 min and fluorescence intensity was recorded with a dual-excitation fluorescence photomultiplier system (Ionoptix). Myocytes were placed onto an Olympus IX-70 inverted microscope and imaged through a Fluor × 40 oil objective. Cells were exposed to light emitted by a 75W lamp and passed through either a 360 or a 380 nm filter, while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm and qualitative change in fura-2 fluorescence intensity (FFI) was inferred from FFI ratio at the two wavelengths (360/380). Fluorescence decay time (both single and bi-exponential decay rates) was measured as an indication of intracellular Ca2+ clearing rate.
Measurement of mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential was measured using JC-1 (Invitrogen, T-3168) staining as described (Xu et al., 2013a). Cardiomyocytes (∼50 cells/mouse, 3 mice/group) that were isolated from WT and KD mice with or without paraquat treatment were seeded on gelatin-coated culture chamber slides and stained with JC-1 (5 μmol/l) at 37°C for 10 min. Cells then were washed with 1× PBS. Fluorescence of each sample was read at an excitation wavelength of 490 nm an emission wavelengths of 530 and 590 nm by a spectrofluorimeter (Spectra MaxGeminiXS, Spectra Max, Atlanta, GA). The aggregate form of JC-1 that yields high concentration of red fluorescence at ∼590 nm indicates a healthy mitochondrial with normal membrane potential, whereas the monomer form of JC-1 which emitting low concentration of green fluorescence at ∼530 nm. The fluorescence intensity results were evaluated by the ratio of 590-to-530-nm emission.
Western blot analysis
Heart tissues from study mice were homogenized and sonicated in a lysis buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 1mM EDTA, 1 mM EGTA, 1% Triton, 0.1% sodium dodecyl sulfate, and a protease inhibitor cocktail. For in vitro study, isolated cardiomyocytes from WT mice treated with paraquat or rapamycin were sonicated in the lysis buffer as described above. Myocardial protein samples were incubated with anti-AMPK, anti-phosphorylated AMPK (pAMPK, Thr172), anti-LC3B, anti-Beclin1, anti-TSC2, anti-phosphorylated TSC2 (pTSC2, Ser939), anti-mTOR, anti-phosphorylated mTOR (pmTOR, Ser2448), anti-S6K, anti-phosphorylated-S6K (pS6K, Ser792), anti-ULK1, anti-phosphorylated ULK1 (pULK1, Ser777 and pULK1, Ser757), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; loading control) (1:1000; Rabbit; Cell Signaling Technology, Danvers, MA), and anti-p62 (1:1000; Guinea Pig; Enzo Life Sciences, Plymouth Meeting, PA) antibodies. Protein samples from isolated cardiomyocytes were incubated with anti-Akt, anti-phosphorylated Akt (pAkt, Ser473) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; loading control) (1:1000; Rabbit; Cell Signaling Technology) antibodies. Horseradish peroxidase-coupled secondary antibodies were used for membrane incubation. After immunoblotting, the films were scanned and detected with a Bio-Rad calibrated densitometer and the intensity of immunoblot bands was normalized with corresponding band intensity of GAPDH (Ren et al., 2009).
Data analysis
Data are mean ± SEM. Statistical significance (p < 0.05) for each variable was estimated by a one-way analysis of variance followed by Tukey's test for the post hoc analysis.
RESULTS
General Features and Echocardiographic Characteristics of WT and AMPK-KD Mice with or without Paraquat Treatment
Paraquat challenge overtly reduced body weight in WT mice compared with the vehicle injected counterparts. AMPK deficiency did not affect body weight. With the paraquat treatment, KD mice did not exhibit any significant decrease in body weight (Supplementary table 1S). Neither AMPK deficiency nor paraquat treatment overtly affected LV wall thickness and LVEDD. Paraquat treatment significantly reduced heart rate and fractional shortening whereas increased LVESD and LV mass/body weight ratio. Although AMPK deficiency did not exert any significant effect on these echocardiographic indices measured in the absence of paraquat treatment, it mitigated or significantly attenuated paraquat-induced change in these echocardiographic parameters (Fig. 1).
FIG. 1.

Echocardiographic properties of WT and KD mice with or without treatment of paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (A) Representative M-mode echocardiographic images from WT and KD mice treated with paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (B) Heart rate (beat per minute); (C) LV wall thickness; (D) LV end diastolic diameter (EDD); (E) LV end systolic diameter (ESD); (F) fractional shortening (%); (G) calculated LV mass. Mean ± SEM, n = 6–8 mice per group; *p < 0.05 versus WT group, #p < 0.05 versus WT-paraquat group.
Effect of Paraquat Exposure and AMPK Deficiency on Cardiomyocyte Contractile Properties
Neither paraquat treatment nor AMPK deficiency affected cell length. However, cardiomyocytes from paraquat-treated WT mice displayed significantly reduced PS and ± dL/dt associated with unchanged TPS and TR90. AMPK deficiency significantly abrogated paraquat-induced cardiomyocyte mechanical dysfunctions without eliciting any obvious effect by itself (Fig. 2).
FIG. 2.

Cardiomyocyte contractile properties in WT and KD transgenic mice treated with or without paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (A) Representative traces from cardiomyocytes isolated from WT mice treated with paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (B) Representative traces from cardiomyocytes isolated from KD mice treated with paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (C) Resting cell length; (D) peak shortening (normalized to cell length); (E) maximal velocity of shortening (+ dL/dt); (F) maximal velocity of relengthening (− dL/dt); (G) time-to-peak shortening (TPS); (H) time-to-90% relengthening (TR90). Mean ± SEM, n = 114–141 cells per group; *p < 0.05 versus WT group, #p < 0.05 versus WT-paraquat group.
Cardiomyocyte Intracellular Ca2+ Transient Properties
To explore the possible mechanism of action behind AMPK deficiency- and paraquat-induced cardiac responses, intracellular Ca2+ handling was evaluated using fura-2 fluorescence. Our data indicated that paraquat significantly suppressed the resting and peak intracellular Ca2+ levels (but not the rise in intracellular Ca2+, ΔFFI), whereas it facilitated intracellular Ca2+ clearance. These changes of intracellular Ca2+ properties in response to paraquat were attenuated by AMPK deficiency. AMPK deficiency itself did not affect the electrically stimulated rise in intracellular Ca2+ (ΔFFI) (Fig. 3).
FIG. 3.

Intracellular Ca2+ and frequency response in cardiomyocytes from WT and KD transgenic mice with or without paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (A) Resting fura-2 fluorescence intensity (FFI). (B) Electrically stimulated rise in FFI (ΔFFI). (C) Peak fura-2 fluorescence intensity (peak FFI). (D) Single exponential intracellular Ca2+ decay. Mean ± SEM, n = 79–117 cells per group; *p < 0.05 versus WT group, #p < 0.05 versus WT-paraquat group.
AMPK Deficiency Attenuates Paraquat-Induced Mitochondrial Dysfunction
As reported previously (Garcia-Garcia et al., 2013; Gonzalez-Polo et al., 2004, 2007; Niso-Santano et al., 2011), paraquat challenge triggered mitochondrial injury. To evaluate the impact of AMPK deficiency on paraquat-induced mitochondrial injury, if any, mitochondrial function was assessed using JC-1 staining to monitor the mitochondrial membrane potential (ΔΨm) in cardiomyocytes. Our results showed a significant reduction in the red, green fluorescence ratio in cardiomyocytes from paraquat-treated WT mice, the effect of which was abrogated by AMPK deficiency. AMPK deficiency itself did not exert any significant effect on ΔΨm (Fig. 4).
FIG. 4.
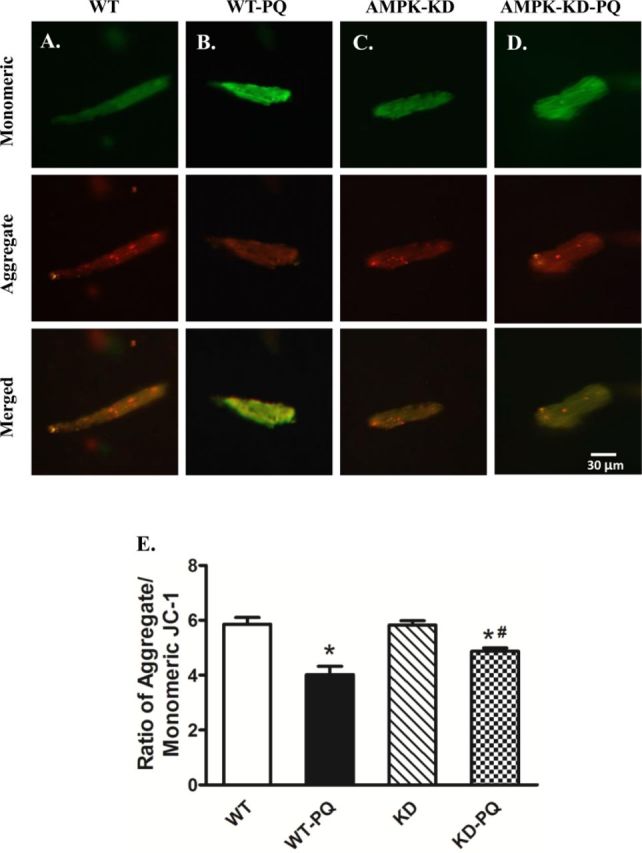
JC-1 staining of cardiomyocytes (A through D, scale bar = 30 μM) from WT and KD mice treated with or without paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (E) Quantitative analysis of the red/green fluorescence ratio (≈50 cardiomyocytes from three mice per group); *p < 0.05 versus WT group, #p < 0.05 versus WT-paraquat group.
Effect of Paraquat Exposure and AMPK Deficiency on Autophagy Markers
To explore the potential role of autophagy in paraquat-induced cardiac damage, levels of the autophagy markers Beclin-1, LC3I/II, and the cargo receptor p62 were evaluated by Western blot using myocardium from WT and KD mice following paraquat treatment. Our data suggested that paraquat treatment significantly elevated expression levels of Beclin1, LC3I, LC3II as well as the LC3II-to-LC3I ratio in WT mice. The expression level of p62 was significantly reduced in myocardium from paraquat-treated WT mice. Although AMPK deficiency itself did not affect the expression of these autophagy markers, it obliterated paraquat-induced changes in autophagy (Fig. 5).
FIG. 5.

Western blot analysis of autophagy markers in heart from WT and KD mice treated with or without paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (A) Representative gel blots depicting LC3 I, LC3 II, p62, Beclin-2, and GAPDH (loading control) using specific antibodies. (B) LC3 I expression; (C) LC3 II expression; (D) LC3 II-to-LC3 I ratio; (E) Beclin-1 expression; and (F) p62 expression. Mean ± SEM, n = 4–5; *p < 0.05 versus WT group, #p < 0.05 versus WT-paraquat group. Gel density of all groups was normalized with the respective gel density of WT group.
Effect of Paraquat Exposure and AMPK Deficiency on AMPK-TSC-mTOR Signaling
To further consolidate the cell signaling mechanisms of AMPK deficiency in paraquat-induced autophagy, AMPK-related autophagic regulatory signaling molecules including TSC2, mTOR, S6K, and ULK1 were examined. Our data revealed reduced pan protein expression of AMPK in AMPK KD mice, validating the murine model (Figs. 6A and B). AMPK phosphorylation was significantly upregulated following paraquat treatment in WT mice, the effect of which was mitigated by AMPK deficiency. The AMPK downstream target, TSC2, was also examined. Our data revealed that paraquat dramatically increased myocardial TSC2 phosphorylation/activation (Figs. 6E–G). Meanwhile, phosphorylation of mTOR and S6K, as well as phosphorylation of ULK1 at Ser757, was significantly dampened in the heart from paraquat-treated WT mice. However, phosphorylation of ULK1 at Ser777, a downstream target regulated by AMPK, was elevated following paraquat treatment. Although AMPK deficiency itself failed to alter either pan or phosphorylated levels of these signaling molecules, it effectively attenuated paraquat-induced changes in these molecules (Fig. 7). Neither paraquat treatment nor AMPK deficiency showed any notable effect on pan expression of TSC2, mTOR, S6K, and ULK1 (Figs. 6 and 7).
FIG. 6.

Western blot analysis of AMPK-TSC signaling proteins from WT and KD mice treated with or without paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (A) Representative gel blots depicting pan and phosphorylated AMPK, TSC2, and GAPDH (loading control) using specific antibodies; (B) AMPKα; (C) phospho-AMPKα (pAMPK, Thr172); (D) pAMPKα-to-AMPKα ratio; (E) pan TSC2; (F) phospho-TSC2 (pTSC2, Ser939); (G) pTSC2-to-TSC2 ratio. Mean ± SEM, n = 4–5; *p < 0.05 versus WT group, #p < 0.05 versus WT-paraquat group. Gel density of all groups was normalized with the respective gel density of WT group.
FIG. 7.

Western blot analysis of mTORC1-related autophage signaling proteins from WT and KD mice treated with or without paraquat (45 mg/kg, i.p.) or vehicle for 48 h. (A) Representative gel blots depicting mTOR, phospho-mTOR (pmTOR, Ser2448), S6K, phospho-S6K (pS6K, Ser792), ULK1, phosphoULK1 (pULK1, Ser757 and pULK1, Ser777), and α-tubulin (loading control) using specific antibodies; (B) pan mTOR; (C) pmTOR; (D) pmTOR-to-mTOR ratio; (E) pan S6K; (F) pS6K; (G) pS6K-to-S6K ratio; (H) pan ULK1; (I) pULK1 Ser757; (J) pULK1757-to-ULK1 ratio; (K) pULK1 Ser777; (L) pULK1777-to-ULK1 ratio. Mean ± SEM, n = 4–5, *p < 0.05 versus WT group, #p < 0.05 versus WT-paraquat group. Gel density of all groups was normalized with the respective gel density of WT group.
Effect of AMPK Inhibitor Compound C, Rapamycin, Lysosomal Inhibitor Bafilomycin A1, and Autophagy Inhibitor 3-MA on Paraquat-Induced Cardiomyocyte Contractile Defects
To better elucidate a cause-effect relationship for AMPK and autophagy in paraquat-induced cardiac contractile response, freshly isolated cardiomyocytes from WT mice were exposed to paraquat in the presence or the absence of the AMPK inhibitor compound C, the mTOR inhibitor rapamycin, the lysosomal inhibitor bafilomycin, and the autophagy inhibitor 3-MA. Our data revealed that paraquat significantly depressed PS, maximal velocity of shortening/relengthening, and prolonged duration of relengthening without influencing resting cell length and duration of shortening. Although compound C and 3-MA themselves did not affect cardiomyocyte mechanical properties in the absence of paraquat exposure, they ablated or significantly attenuated paraquat-induced cardiomyocyte mechanical anomalies. However, the mTOR inhibitor rapamycin and lysosomal inhibitor bafilomycin exacerbated paraquat-induced cardiomyocyte contractile dysfunction. Combination of compound C with rapamycin further accentuated paraquat-induced contractile dysfunction in cardiomyocytes. Nonetheless, combination of compound C and bafilomycin mitigated the paraquat-induced cardiomyocyte mechanical anomalies (Fig. 8). Furthermore, levels of pan and phosphorylated (Ser473) Akt were measured in isolated cardiomyocytes treated with paraquat or rapamycin. Our results revealed that treatment of either paraquat or rapamycin did not affect pan or phosphorylated level of Akt (Supplementary fig. 1S).
FIG. 8.

Effect of compound C, rapamycin, bafilomycin A1, and 3-MA on paraquat-induced cardiomyocyte contractile defects. Freshly isolated cardiomyocytes from WT mice were incubated with paraquat (100 μM) in the presence or absence of the AMPK inhibitor compound C (5 μM), rapamycin (5 μM), lysosomal inhibitor bafilomycin A1 (100 nM), and 3-MA (10 mM) for 3 h. (A) Resting cell length; (B) peak shortening (normalized to resting cell length); (C) maximal velocity of shortening (+ dL/dt); (D) maximal velocity of relengthening (− dL/dt); (E) time-to-peak shortening (TPS); (F) time-to-90% relengthening (TR90). Mean ± SEM, n = 64–157 cells from four mice per group; *p < 0.05 versus control group, #p < 0.05 versus paraquat group, ‡p < 0.05 versus paraquat compound C group.
DISCUSSION
The salient findings from our study suggested that AMPK deficiency significantly attenuated paraquat-induced damages in myocardial geometry and function, intracellular Ca2+ homeostasis, as well as mitochondrial injury (JC-1 fluorescence). Our data further revealed activation of autophagy in response to paraquat exposure, the effect of which was reversed by AMPK deficiency. More importantly, our findings provided compelling evidence for a permissive role of the AMPK-TSC2-mTOR-ULK1 signaling in paraquat-elicited autophagy response, which may help to interpret paraquat-induced myocardial dysfunction. In addition, inhibition of AMPK and autophagy (compound C and 3-MA) protects against paraquat-induced cardiomyocyte contractile dysfunction. However, induction of autophagy by inhibiting mTOR (rapamycin) and impairment of autophagy flux by inhibiting lysosomal function (bafilomycin) exacerbated paraquat-induced cardiomyocyte contractile dysfunction. These observations are in favor of the notion that paraquat induces myocardial dysfunction through activation of autophagy via over-stimulation of AMPK and inhibition of mTOR.
The toxicity of paraquat has been intensively studied since 1960s (Bus et al., 1976; Clark et al., 1966). Mice have been used for a long time as a model to study the toxicity of paraquat (Day and Crapo, 1996; Day et al., 1995; Giri et al., 1981). Bus and colleagues reported a median lethal dose of paraquat for intraperitoneal injection at 30 mg/kg in mice (Bus et al., 1976). Day and coworkers challenged mice with paraquat at 0, 35, 45, or 55 mg/kg for 48 h. In the 55 mg/kg treatment group, two out of five mice died prior to 48 h. All mice from other groups survived after 48 h of treatment (Day et al., 1995). Moreover, Giri and colleagues found significantly elevated pulmonary vascular permeability at 24 h and 48 h following paraquat treatment at 50 mg/kg (Giri et al., 1981). Day and Crapo treated mice with 45 mg/kg paraquat intraperitoneal injection and studied the lung injury 48 h later (Day and Crapo, 1996). Based on these studies of paraquat in mice, we selected the dosage of 45 mg/kg i.p. for 48 h. Following the treatment, animal body weight was significantly decreased, whereas there was little significant change in body weight in KD mice. Our finding of weight loss is concordant with the previous report on paraquat toxicity in mice (Prasad et al., 2009).
Our echocardiographic data showed that paraquat treatment significantly increased LVESD and LV mass-to-body weight ratio, as well as decreased fractional shortening. These findings are in line with the previous observations of myocardial dysfunction induced by acute paraquat treatment (Ge et al., 2010; Li et al., 2007). In addition, data from our study revealed that paraquat treatment triggered cardiomyocyte contractile dysfunction, as evidenced by reduced PS and maximal velocity of shortening/relengthening. Furthermore, intracellular Ca2+ homeostasis was disrupted in response to paraquat treatment, including depressed basal and peak intracellular Ca2+ levels and prolonged intracellular Ca2+ clearance. AMPK deficiency itself failed to elicit any notable change in cardiac geometry, cardiomyocyte contractile function, and intracellular Ca2+ handling properties, consistent with our previous reports (Guo and Ren, 2012; Turdi et al., 2011). This finding suggests that AMPK deficiency may not be innately harmful to cardiac function (Guo and Ren, 2012; Musi et al., 2005; Xing et al., 2003). Nonetheless, overexpression of negative AMPKα2 subunit significantly ameliorated paraquat-induced cardiac remodeling, cardiomyocyte contractile dysfunction, and intracellular Ca2+ mishandling. These data indicate that AMPK may participate in the maintenance of intracellular Ca2+ homeostasis in cardiac pathological responses although the underlying mechanism remains unclear.
Our results further revealed that AMPK-TSC2-mTORC1-ULK1 signaling cascade played an essential role in paraquat-induced myocardial dysfunction. A robust increase in AMPKα phosphorylation was observed following paraquat treatment, which was reversed by AMPK deficiency. As the direct downstream target of AMPK and upstream regulator of mTORC1, TSC2 phosphorylation was also significantly elevated following paraquat exposure. It has been shown that mTORC1 is negatively regulated by TSC1/TSC2, whereas phosphorylation of TSC (aka, TSC1/TSC2 activation) inhibits mTORC1 en route to autophagy activation (Gottlieb, 2012; Xu et al., 2014). In our hands, mTORC1 activity was significantly reduced in the paraquat-challenged mouse heart, a good indication for activated autophagy. AMPK deficiency was demonstrated to interrupt the inhibition of TSC2 on mTORC1 signaling, leading to phosphorylation of mTORC1 (Tripathi et al., 2013). Accumulating studies have suggested that mTORC1 inhibition is an adaptive response during cardiac stress (Sciarretta et al., 2014). It is consistent with our findings that in vitro treatment of rapamycin, a specific mTOR inhibitor, exacerbated paraquat-induced cardiomyocyte contractile dysfunction. Previous studies have shown that during acute treatment, rapamycin has a high specificity to inhibit mTORC1. However, chronic exposure of rapamycin may also inhibit mTORC2 (Lamming et al., 2013). Moreover, mTORC2 is capable of further activating Akt through phosphorylation at Ser473 and therefore regulate Akt/mTORC1 signaling in a positive feedback loop (Efeyan and Sabatini, 2010; Fumarola et al., 2014). For our short-term acute treatment of rapamycin in isolated cardiomyocytes, we did not observe any change in pan and phosphorylated (Ser473) Akt in paraquat and rapamycin-treated groups. More importantly, autophagic protein ULK1/2, an essential regulator in the initiation process of autophagy, can be phosphorylated by either mTORC1 or AMPK (Ganley et al., 2009; Sciarretta et al., 2014). On one hand, our results revealed that ULK1 phosphorylation at Ser757 was significantly reduced following acute paraquat exposure, which is negatively regulated by AMPK (Bach et al., 2011). On the other hand, we found that paraquat-induced distinct ULK1 phosphorylation at Ser777 was in concert with elevated AMPK phosphorylation, which was reversed by AMPK deficiency. It has been suggested that phosphorylation of ULK1 at Ser757 by active mTORC1 might prevent ULK1 from interacting with AMPK (Bach et al., 2011). Dephosphorylation of ULK1 at Ser757 is thought to be enzymatically active and important for recruiting downstream autophagic proteins and subsequent autophagosome formation (Chan et al., 2009; Guo and Ren, 2012). ULK1 can be regulated by multiple phosphorylation and dephosphorylation processes (Bach et al., 2011). Studies have shown that AMPK can induce autophagy directly through phosphorylation of ULK1 at Ser317 and Ser777, though other phosphorylation sites such as Ser555 and Ser467 may also participate in this process (Egan et al., 2011; Kim et al., 2011). A schematic diagram is provided to summarize the paraquat-induced autophagic and contractile responses through AMPK and TSC2, and subsequent inhibition of mTORC1 and phosphorylation of ULK1 (Fig. 9).
FIG. 9.

Schematic diagram depicting possible mechanism(s) involved in paraquat- and AMPK deficiency (KD) induced changes in cardiac intracellular Ca2+ homeostasis and contractile function. Paraquat administration triggers activation of AMPK signaling. Activation of AMPK activates TSC complex (through phosphorylation) to enhance its inhibition on mTOR signaling and reduced phosphorylation of ULK1 at Ser757, leading to excessive autophagy and autophagy flux, which leads to cardiac dysfunction. AMPK can alternatively activate ULK1 at Ser777 to upregulate autophagy. AMPK deficiency suppresses autophagy via inhibition of AMPK/TSC/mTOR/ULK1757 and AMPK/ULK1777 signaling cascade en route to cardioprotection. Arrows indicate activation whereas the lines with a “T” ending denote inhibition. TSC, tuber sclerosis complex.
Our in vivo data revealed, for the first time, that AMPK deficiency or inhibition ameliorates paraquat-induced excessive autophagy in the heart manifested as increased expression level of both Beclin-1 and LC3 (LC3I and LC3II) and decreased autophagosome cargo protein p62 along with contractile anomalies. These findings indicate that autophagy may play a causal role in paraquat-elicited cardiac damage possibly by over-activating autophagy, which was supported by in vitro findings that autophagy inhibition using 3-MA reversed mimicked paraquat-induced cardiomyocyte dysfunction. Moreover, in vitro treatment of rapamycin, which inhibits mTOR and activates autophagy, exacerbated cardiomyocyte dysfunction following the paraquat exposure, the effects of which were unaffected by AMPK inhibition. Similar effects were also observed in the cardiomyocyte contractile properties with the in vitro treatment of lysosomal inhibitor bafilomycin that blocks the autophagosome removal and resultant interruption of autophagic flux. However, AMPK inhibition effectively reversed these unfavorable effects. These findings suggest that autophagy activation is the key cause effect of paraquat-induced myocardial dysfunction; AMPK is a major regulator of paraquat-induced autophagy; impaired autophagic flux can also lead to cardiomyocyte contractile anomalies. It is possible that under AMPK deficiency or inhibition, autophagy is inactivated through either elevated mTORC1 activity (en route to elevated ULK1 Ser757 phosphorylation) or suppressed ULK1 Ser777 phosphorylation. These findings supported a crucial role of autophagy in paraquat-induced cardiac contractile dysfunction with over-activated AMPK, which suggests that autophagy induction may be detrimental in this process.
Perhaps, the most intriguing finding from our study is that autophagic flux plays an important role in cardiac response to pathological stress. Autophagic flux is the entire process of autophagy including the delivery of autophagosomal cargo to lysosome, degradation of autophagolysosome, as well as the release of the degradation product back into the cytosol. In responding to perturbations in the extracellular environment, cells have the ability to adjust autophagic flux to meet intracellular metabolic demands (Garcia-Garcia et al., 2013). A number of studies have shown that paraquat toxicity robustly increases the number of autophagic vacuoles or cause accumulation of autophagosomes (Garcia-Garcia et al., 2013; Gonzalez-Polo et al., 2007; Niso-Santano et al., 2010). Accumulation of autophagosomes can result from either increased autophagic flux or impaired autophagolysosome clearance (Garcia-Garcia et al., 2013). Paraquat has been reported to facilitate autophagy with improved autophagic flux (Gonzalez-Polo et al., 2007; Niso-Santano et al., 2010), whereas others show that paraquat impairs autophagic flux and the resulting protein accumulation (Garcia-Garcia et al., 2013; Wills et al., 2012). Here, we demonstrated that paraquat stimulates autophagic flux; meanwhile, our in vitro studies with treatment of bafilomycin, mimicking impaired autophagic flux, revealed that blocked autophagy can exacerbate the cardiomyocyte contractile dysfunction.
In conclusion, findings from our present study provided convincing evidence for the first time that AMPK deficiency helps to ameliorate paraquat-induced myocardial contractile dysfunction. Our data revealed a pivotal role of autophagy in cardiac stress response to environmental toxin paraquat, which may be possibly via AMPK-TSC2-mTORC1-ULK1-mediated regulation of autophagy. This is supported by the finding that inhibition of AMPK or autophagy protected against paraquat-induced cardiac anomalies. Although it is still premature to discern the precise mechanism underneath AMPK-mediated autophagy following paraquat exposure, our current investigation should shed some light toward better understanding of the role of autophagy in paraquat toxicity in heart damage. Further study is warranted to elucidate the therapeutic value of AMPK and AMPK-related autophagic signaling molecule in the management of paraquat-induced myopathic anomalies.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institute of Health/National Center for Research Resources (5P20RR016474); National Institute of Health/National Institute of General Medical Sciences (8P20GM103432).
Supplementary Material
REFERENCES
- Alexi T., Borlongan C. V., Faull R. L., Williams C. E., Clark R. G., Gluckman P. D., Hughes P. E. Neuroprotective strategies for basal ganglia degeneration: Parkinson's and Huntington's diseases. Prog. Neurobiol. 2000;60:409–470. doi: 10.1016/s0301-0082(99)00032-5. [DOI] [PubMed] [Google Scholar]
- Bach M., Larance M., James D. E., Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011;440:283–291. doi: 10.1042/BJ20101894. [DOI] [PubMed] [Google Scholar]
- Bus J. S., Aust S. D., Gibson J. E. Paraquat toxicity: Proposed mechanism of action involving lipid peroxidation. Environ. Health Perspect. 1976;16:139–146. doi: 10.1289/ehp.7616139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y. C., Chang S. C., Hsuan S. L., Chien M. S., Lee W. C., Kang J. J., Wang S. C., Liao J. W. Cardiovascular effects of herbicides and formulated adjuvants on isolated rat aorta and heart. Toxicology In Vitro. 2007;21:595–603. doi: 10.1016/j.tiv.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Chan E. Y., Longatti A., McKnight N. C., Tooze S. A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. G., McElligott T. F., Hurst E. W. The toxicity of paraquat. Br. J. Ind. Med. 1966;23:126–132. doi: 10.1136/oem.23.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristovao A. C., Choi D. H., Baltazar G., Beal M. F., Kim Y. S. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid. Redox Signal. 2009;11:2105–2118. doi: 10.1089/ars.2009.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda R. K., Das Banerjee T., Janda E. How Parkinsonian toxins dysregulate the autophagy machinery. Int. J. Mol. Sci. 2013;14:22163–22189. doi: 10.3390/ijms141122163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day B. J., Crapo J. D. A metalloporphyrin superoxide dismutase mimetic protects against paraquat-induced lung injury in vivo. Toxicol. Appl. Pharmacol. 1996;140:94–100. doi: 10.1006/taap.1996.0201. [DOI] [PubMed] [Google Scholar]
- Day B. J., Hatch A., Carstens D. D., Crapo J. D. An easy and economical method to prepare cells for cytologic analyses. J. Pharmacol. Toxicol. Methods. 1995;34:57–62. doi: 10.1016/1056-8719(95)00029-h. [DOI] [PubMed] [Google Scholar]
- Efeyan A., Sabatini D. M. mTOR and cancer: Many loops in one pathway. Curr. Opin. Cell Biol. 2010;22:169–176. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D. F., Shackelford D. B., Mihaylova M. M., Gelino S., Kohnz R. A., Mair W., Vasquez D. S., Joshi A., Gwinn D. M., Taylor R., et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumarola C., Bonelli M. A., Petronini P. G., Alfieri R. R. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem. Pharmacol. 2014;90:197–207. doi: 10.1016/j.bcp.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Ganley I. G., Lam du H., Wang J., Ding X., Chen S., Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia A., Anandhan A., Burns M., Chen H., Zhou Y., Franco R. Impairment of Atg5-dependent autophagic flux promotes paraquat- and MPP(+)-induced apoptosis but not rotenone or 6-hydroxydopamine toxicity. Toxicol. Sci. 2013;136:166–182. doi: 10.1093/toxsci/kft188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W., Zhang Y., Han X., Ren J. Cardiac-specific overexpression of catalase attenuates paraquat-induced myocardial geometric and contractile alteration: Role of ER stress. Free Radic. Biol. Med. 2010;49:2068–2077. doi: 10.1016/j.freeradbiomed.2010.10.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S. N., Hollinger M. A., Schiedt M. J. The effects of paraquat and superoxide dismutase on pulmonary vascular permeability and edema in mice. Arch. Environ. Health. 1981;36:149–154. doi: 10.1080/00039896.1981.10667619. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo R. A., Niso-Santano M., Ortiz-Ortiz M. A., Gomez-Martin A., Moran J. M., Garcia-Rubio L., Francisco-Morcillo J., Zaragoza C., Soler G., Fuentes J. M. Inhibition of paraquat-induced autophagy accelerates the apoptotic cell death in neuroblastoma SH-SY5Y cells. Toxicol. Sci. 2007;97:448–458. doi: 10.1093/toxsci/kfm040. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo R. A., Rodriguez-Martin A., Moran J. M., Niso M., Soler G., Fuentes J. M. Paraquat-induced apoptotic cell death in cerebellar granule cells. Brain Res. 2004;1011:170–176. doi: 10.1016/j.brainres.2004.02.078. [DOI] [PubMed] [Google Scholar]
- Gottlieb R. Autophagy in Health and Disease. London: Academic Press; 2012. [Google Scholar]
- Guo R., Ren J. Deficiency in AMPK attenuates ethanol-induced cardiac contractile dysfunction through inhibition of autophagosome formation. Cardiovasc. Res. 2012;94:480–491. doi: 10.1093/cvr/cvs127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K., Zhu T., Guan K. L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Kassiotis C., Ballal K., Wellnitz K., Vela D., Gong M., Salazar R., Frazier O. H., Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120(Suppl. 11):S191–S197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J., Emr S. D. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo J. R., Kim J. C., Yoon J. W., Kim G. H., Jeon R. W., Kim H. J., Chae D. W., Noh J. W. Failure of continuous venovenous hemofiltration to prevent death in paraquat poisoning. Am. J. kidney Dis. 2002;39:55–59. doi: 10.1053/ajkd.2002.29880. [DOI] [PubMed] [Google Scholar]
- Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T., Ohsumi Y., Tokuhisa T., Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lamming D. W., Ye L., Sabatini D. M., Baur J. A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Invest. 2013;123:980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. W., Park S., Takahashi Y., Wang H. G. The association of AMPK with ULK1 regulates autophagy. PloS One. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Klionsky D. J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Liou H. H., Tsai M. C., Chen C. J., Jeng J. S., Chang Y. C., Chen S. Y., Chen R. C. Environmental risk factors and Parkinson's disease: A case-control study in Taiwan. Neurology. 1997;48:1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- Li Q., Yang X., Sreejayan N., Ren J. Insulin-like growth factor I deficiency prolongs survival and antagonizes paraquat-induced cardiomyocyte dysfunction: Role of oxidative stress. Rejuvenation Res. 2007;10:501–512. doi: 10.1089/rej.2007.0552. [DOI] [PubMed] [Google Scholar]
- Loffler A. S., Alers S., Dieterle A. M., Keppeler H., Franz-Wachtel M., Kundu M., Campbell D. G., Wesselborg S., Alessi D. R., Stork B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- Manning-Bog A. B., McCormack A. L., Li J., Uversky V. N., Fink A. L., Di Monte D. A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- Manning-Bog A. B., McCormack A. L., Purisai M. G., Bolin L. M., Di Monte D. A. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J. Neurosci. 2003;23:3095–3099. doi: 10.1523/JNEUROSCI.23-08-03095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack A. L., Atienza J. G., Johnston L. C., Andersen J. K., Vu S., Di Monte D. A. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J. Neurochem. 2005;93:1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x. [DOI] [PubMed] [Google Scholar]
- McCormack A. L., Thiruchelvam M., Manning-Bog A. B., Thiffault C., Langston J. W., Cory-Slechta D. A., Di Monte D. A. Environmental risk factors and Parkinson's disease: Selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- Musi N., Hirshman M. F., Arad M., Xing Y., Fujii N., Pomerleau J., Ahmad F., Berul C. I., Seidman J. G., Tian R., et al. Functional role of AMP-activated protein kinase in the heart during exercise. FEBS Lett. 2005;579:2045–2050. doi: 10.1016/j.febslet.2005.02.052. [DOI] [PubMed] [Google Scholar]
- Mu J., Brozinick J. T., Jr, Valladares O., Bucan M., Birnbaum M. J. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Nemchenko A., Chiong M., Turer A., Lavandero S., Hill J. A. Autophagy as a therapeutic target in cardiovascular disease. J. Mol. Cell. Cardiol. 2011;51:584–593. doi: 10.1016/j.yjmcc.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niso-Santano M., Gonzalez-Polo R. A., Bravo-San Pedro J. M., Gomez-Sanchez R., Lastres-Becker I., Ortiz-Ortiz M. A., Soler G., Moran J. M., Cuadrado A., Fuentes J. M., et al. Activation of apoptosis signal-regulating kinase 1 is a key factor in paraquat-induced cell death: Modulation by the Nrf2/Trx axis. Free Radic. Biol. Med. 2010;48:1370–1381. doi: 10.1016/j.freeradbiomed.2010.02.024. [DOI] [PubMed] [Google Scholar]
- Niso-Santano M., Moran J. M., Garcia-Rubio L., Gomez-Martin A., Gonzalez-Polo R. A., Soler G., Fuentes J. M. Low concentrations of paraquat induces early activation of extracellular signal-regulated kinase 1/2, protein kinase B, and c-Jun N-terminal kinase 1/2 pathways: Role of c-Jun N-terminal kinase in paraquat-induced cell death. Toxicol. Sci. 2006;92:507–515. doi: 10.1093/toxsci/kfl013. [DOI] [PubMed] [Google Scholar]
- Niso-Santano M., Bravo-San Pedro J. M., Gomez-Sanchez R., Climent V., Soler G., Fuentes J. M., Gonzalez-Polo R. A. ASK1 overexpression accelerates paraquat-induced autophagy via endoplasmic reticulum stress. Toxicol. Sci. 2011;119:156–168. doi: 10.1093/toxsci/kfq313. [DOI] [PubMed] [Google Scholar]
- Peng J., Mao X. O., Stevenson F. F., Hsu M., Andersen J. K. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J. Biol. Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- Prasad K., Tarasewicz E., Mathew J., Strickland P. A., Buckley B., Richardson J. R., Richfield E. K. Toxicokinetics and toxicodynamics of paraquat accumulation in mouse brain. Exp. Neurol. 2009;215:358–367. doi: 10.1016/j.expneurol.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Babcock S. A., Li Q., Huff A. F., Li S. Y., Doser T. A. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicol. Lett. 2009;187:149–156. doi: 10.1016/j.toxlet.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Privratsky J. R., Yang X., Dong F., Carlson E. C. Metallothionein alleviates glutathione depletion-induced oxidative cardiomyopathy in murine hearts. Crit. Care Med. 2008;36:2106–2116. doi: 10.1097/CCM.0b013e31817bf925. [DOI] [PubMed] [Google Scholar]
- Russell R. R. III, Li J., Coven D. L., Pypaert M., Zechner C., Palmeri M., Giordano F. J., Mu J., Birnbaum M. J., Young L. H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta S., Volpe M., Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014;114:549–564. doi: 10.1161/CIRCRESAHA.114.302022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawara T., Fukushima T., Hojo N., Isobe A., Shiwaku K., Setogawa T., Yamane Y. Effects of paraquat on mitochondrial electron transport system and catecholamine contents in rat brain. Arch. Toxicol. 1996;70:585–589. doi: 10.1007/s002040050316. [DOI] [PubMed] [Google Scholar]
- Tripathi D. N., Chowdhury R., Trudel L. J., Tee A. R., Slack R. S., Walker C. L., Wogan G. N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E2950–E2957. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turdi S., Kandadi M. R., Zhao J., Huff A. F., Du M., Ren J. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J. Mol. Cell. Cardiol. 2011;50:712–722. doi: 10.1016/j.yjmcc.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills J., Credle J., Oaks A. W., Duka V., Lee J. H., Jones J., Sidhu A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PloS One. 2012;7:e30745. doi: 10.1371/journal.pone.0030745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S., Loewith R., Hall M. N. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xie M., Morales C. R., Lavandero S., Hill J. A. Tuning flux: Autophagy as a target of heart disease therapy. Curr. Opin. Cardiol. 2011;26:216–222. doi: 10.1097/HCO.0b013e328345980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y., Musi N., Fujii N., Zou L., Luptak I., Hirshman M. F., Goodyear L. J., Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J. Biol. Chem. 2003;278:28372–28377. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- Xu X., Bucala R., Ren J. Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J. Am. Heart Assoc. 2013a;2:e000439. doi: 10.1161/JAHA.113.000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Hua Y., Nair S., Zhang Y., Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J. Mol. Cell Biol. 2013b;5:61–63. doi: 10.1093/jmcb/mjs055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Roe N. D., Weiser-Evans M. C., Ren J. Inhibition of mammalian target of rapamycin with rapamycin reverses hypertrophic cardiomyopathy in mice with cardiomyocyte-specific knockout of PTEN. Hypertension. 2014;63:729–739. doi: 10.1161/HYPERTENSIONAHA.113.02526. [DOI] [PubMed] [Google Scholar]
- Yuan H., Perry C. N., Huang C., Iwai-Kanai E., Carreira R. S., Glembotski C. C., Gottlieb R. A. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am. J. Physiol. Heart Circ. Physiol. 2009;296:H470–H479. doi: 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.