

Abstract
The role of the multi-resistance protein 2 (Mrp2) in the nephropathy induced by inorganic mercuric mercury (Hg2+) was studied in rats (TR−) and mice (Mrp2−/−), which lack functional Mrp2, and control animals. Animals were exposed to nephrotoxic doses of HgCl2. Forty-eight or 24 hours after exposure, tissues were harvested and analyzed for Hg content and markers of injury. Histological analyses revealed that the proximal tubular segments affected pathologically by Hg2+ were significantly different between Mrp2-deficient animals and controls. In the absence of Mrp2, cellular injury localized almost exclusively in proximal tubular segments in the subcapsular (S1) to midcortical regions (early S2) of the kidney. In control animals, cellular death occurred mainly in the proximal tubular segments in the inner cortex (late S2) and outer stripe of the outer medulla (S3). These differences in renal pathology indicate that axial heterogeneity exists along the proximal tubule with respect to how mercuric ions are handled. Total renal and hepatic accumulation of mercury was also greater in animals lacking Mrp2 than in controls, indicating that Mrp2 normally plays a significant role in eliminating mercuric ions from within proximal tubular cells and hepatocytes. Analyses of plasma creatinine, BUN, and renal expression of Kim-1 and Ngal tend to support the severity of the nephropathies detected histologically. Collectively, our findings indicate that a fraction of mercuric ions is normally secreted by Mrp2 in early portions of proximal tubules into the lumen and then is absorbed downstream in straight portions, where mercuric species typically induce toxic effects.
Various forms of mercury continue to be added to the pool of environmental mercury (ATSDR, 1999). Following exposure of humans to mercury, mercuric ions accumulate preferentially in the kidney (Bridges and Zalups, 2005, 2010; Zalups, 2000). Specifically, mercuric ions accumulate almost exclusively along the S1, S2, and S3 segments of the proximal tubule (Rodier and Kates, 1988; Rodier et al., 1988; Zalups, 1991, 2000). The uptake of mercuric ions by these cells can be attributed largely to transport of thiol S-conjugates of Hg2+ by carriers present in luminal and basolateral membranes of these cells (Bridges and Zalups, 2010; Zalups, 2000).
In virtually all mammalian species, exposure to inorganic mercury (Hg2+) can result in acute renal pathological changes (Zalups, 2000; Zalups and Diamond, 1987; Zalups et al., 1991). The pathological changes associated with the nephropathy induced by Hg2+ consist primarily of degeneration and death of the epithelial cells lining the pars recta (straight segment) of the proximal tubule. In mildly to moderately severe forms, cellular injury and death occur primarily in S2 segments present at the cortico-medullary junction and S3 segments present in the outer stripe of the outer medulla (OSOM) (Bridges and Zalups, 2010; Zalups, 1997, 2000; Zalups and Diamond, 1987; Zalups et al., 1991). When the nephropathy is severe, other segments of the nephron can be affected adversely, particularly the convoluted (S1) segments of the proximal tubules. Following exposure to relatively high, but not lethal, doses of Hg2+, the severity of acute proximal tubular necrosis (ATN) can be great enough to induce acute renal failure (ARF) (Gstraunthaler et al., 1983; Houser et al., 1992; Sherwood et al., 1974; Zager, 1983). Interestingly, the amount of Hg2+ required to induce proximal tubular injury varies significantly among mammalian species. Even among rodents, there can be great differences in the dose required to manifest histological evidence of even the mildest forms of Hg2+-induced nephropathy. In rats, for example, as little as a 1.5-μmol • kg−1 dose of HgCl2, administered intravenously (iv) can induce a relatively mild nephropathy characterized by cellular degeneration and death in the pars recta segments of proximal tubules at the cortico-medullary junction and in the OSOM (Gritzka and Trump, 1968; Zalups, 1997, 2000; Zalups and Barfuss, 1996; Zalups and Diamond, 1987; Zalups et al., 1991). By contrast, we have discovered that intraperitoneal (ip) administration of an 18.5-μmol • kg−1 dose of HgCl2 is required to induce a nephropathy in mice that is comparable in severity to that in rats exposed to the 1.5-μmol • kg−1 dose (unpublished findings). The reason for the differences in the nephrotoxic effects of various doses of Hg2+ among species of mammals is not known at present.
Despite numerous studies on mechanisms by which mercuric species gain entry into various epithelial cells in the body (Bridges and Zalups, 2010; Zalups, 2000), it is only recently that mechanisms participating in the export and elimination of mercuric ions by proximal tubular cells have been studied and identified. A series of recent studies show that the multidrug resistance-associated protein 2 (Mrp2), which is localized exclusively in the luminal plasma membrane of proximal tubular epithelial cells (Schaub et al., 1999), is capable of transporting certain mercuric species from the cytoplasm into the lumen of proximal tubular segments (Bridges and Zalups, 2010; Bridges et al., 2008a,b, 2011; Zalups and Bridges, 2009). Overall, these findings indicate that the absence of functional Mrp2 protein in proximal tubular epithelial cells results in enhanced accumulation and retention of mercuric ions in the renal cortex and OSOM.
Considering that the proximal tubular epithelial cells in rats or mice lacking a functional form of Mrp2 are responsible for the enhanced renal accumulation and retention of Hg2+, we hypothesize that, in the absence of Mrp2, the nature and/or severity of the acute nephropathy induced by Hg2+ may be altered significantly. Accordingly, the present study was designed to test the hypothesis that the absence of functional Mrp2 protein in proximal tubular epithelial cells enhances the severity of and/or alters the tubular site of injury in the nephropathy induced by various nephrotoxic doses of HgCl2.
MATERIALS AND METHODS
Animals
Breeder pairs of TR− rats were obtained from the laboratory of Dr. Kim Brouwer at the University of North Carolina. TR− rats possess a spontaneous mutation which results in a non-functional Mrp2 protein (Mayer et al., 1995). Wistar rats were used as corresponding control animals and were obtained from Harlan Laboratories (Indianapolis, IN). Breeder pairs of Mrp2−/− mice (Chu et al., 2006) were obtained from Taconic (Germantown, NY). Friend Virus B (FVB) mice, which were used as control mice, were obtained from Charles River Laboratories (Wilmington, MA). All animals were mated in the Mercer University School of Medicine animal facility and were provided a commercial laboratory diet (Tekland 6% rodent diet, Harlan Laboratories) and water ad libitum throughout all aspects of experimentation. All procedures involving animals were reviewed and approved by the Mercer University Institutional Animal Care and Use Committee. Animals were handled in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Manufacture of 203Hg
The protocol for generating the radioisotope of Hg, 203Hg2+, has been described previously (Belanger et al., 2001; Bridges et al., 2004). Briefly, 3 mg of mercuric oxide (enriched with the stable isotope of Hg, 202Hg) was sealed in quartz tubing and was irradiated by neutron activation for 4 weeks at the Missouri University Research Reactor facility. After irradiation, the mercuric oxide containing 203Hg2+ was dissolved in 1 N hydrochloric acid (HCl). The radioactivity of the solution was determined using an Ion Chamber survey meter (Fluke Biomedical, Everett, WA). The specific activities of the [203Hg] ranged from 12 to 16 mCi/mg.
Administration of Hg2+
Male Wistar and TR− rats weighing 250–275 g were used in the present study. According to our published protocol (Bridges et al., 2008a; Zalups, 1993), groups of four randomly selected Wistar and TR− rats were injected (iv) with a 1.5 or 2.25 μmol dose of HgCl2 • kg−1 • 2 ml−1 (0.9% w/v aqueous NaCl). The injection solutions were designed to deliver approximately 1 μCi [203Hg] per rat. The 1.5-μmol • kg−1 dose was chosen because it has been demonstrated previously that the nephropathy induced by this dose in normal rats is of relatively mild severity (Zalups, 1997; Zalups and Diamond, 1987). The 2.25 μmol/kg dose was chosen because it induces a moderately severe nephropathy in Wistar rats within 48 h (unpublished findings). In addition, groups of four Wistar and TR− rats, not treated with HgCl2, were used to obtain control kidneys for histology.
Groups of four randomly selected male, FVB and Mrp2−/− mice, weighing 25–30 g, were injected ip with 18.5, 19.0, or 19.5 μmol HgCl2 • kg−1 • 8 ml−1 (0.9% NaCl). As in the experiments with rats, the injection solutions were designed to deliver 1 μCi [203Hg] per animal. The 18.5-μmol • kg−1 dose was chosen because it induces a mild form of the nephropathy induced by HgCl2 in FVB mice (unpublished findings). By contrast, the 19.0 and 19.5 μmol/kg doses were chosen, respectively, because they induce moderate to moderately severe forms of the nephropathy, respectively, in FVB mice (unpublished findings). Groups of four FVB and Mrp2−/− mice that were not treated with HgCl2 were also used to obtain control kidneys for histology.
After injecting the animals with the designated dose of HgCl2, the animals were placed individually in plastic metabolic cages. Forty-eight hours after exposure to HgCl2, all the rats were anesthetized with an intraperitoneal injection of ketamine (70 mg • kg−1) and xylazine (30 mg • kg−1). In a similar manner, mice were anesthetized with ketamine and xylazine 24 h after exposure to HgCl2. Once a state of deep anesthesia had been attained (determined by lack of corneal and tail reflexes), tissues and organs were harvested for determination of mercury content.
Collection of tissues, organs, urine, and feces and quantification of content of Hg2+
After inducing a state of deep anesthesia in each animal, a midline incision extending from the xiphoid process to pubis was made through the skin and underlying musculature with a scalpel in each rat or mouse. A 3-ml sample of blood was then obtained from the inferior vena cava. One milliliter of whole blood was used for estimation of [203Hg] content. Five-hundred microliters were placed in a plasma-separation tube designed to separate plasma from the cellular components of blood. The total volume of blood for the rat was estimated to be 6% of body weight (Lee and Blaufox, 1985).
Subsequently, the kidneys of each rat or mouse were removed, weighed, and cut in half along a transverse plane. A 3-mm transverse slice of the left kidney was use to isolate relatively pure samples of cortex (which contain S1 and S2 proximal tubular segments of both cortical and juxtamedullary nephrons) and OSOM (which contain exclusively the S3 segments of proximal tubules of cortical and juxtamedullary nephrons). Each zone of the kidney was weighed and placed in a separate polystyrene tube for estimation of [203Hg] content.
In addition, a small slice of left kidney from each rat or mouse was frozen in liquid nitrogen for future isolation of RNA. One half of the right kidney (from each animal) was fixed with 50% glutaraldehyde and 40% formaldehyde for 48 h at 4°C for histopathological analyses. The remaining half was used for estimation of [203Hg] content. The liver of each rat or mouse was then excised, weighed, and a 1-g section was removed for determination of the hepatic content of Hg2+.
Urine and feces were collected from each rat during two consecutive 24-h periods following injection with HgCl2. The total volume of urine excreted during each of the two 24-h periods was determined for all animals. A 1-ml sample of urine was obtained from each 24-h collection after the sample was mixed thoroughly by vortexing. Each 1-ml sample was weighed and placed in a polystyrene tube for estimation of [203Hg] content. Samples of urine and feces were not obtained from the mice used in this study.
The content of [203Hg] in each sample of tissue, urine, or feces was determined using standard isotopic methods after counting in a Wallac Wizard 3 automated gamma counter (Perkin Elmer, Boston, MA).
Real-time PCR analyses of the renal expression of Kim-1 and Ngal
Kidneys from mice and rats were isolated, cut into sections, and frozen at the time of animal euthanasia. At the time of RNA isolation, frozen kidney sections were pulverized with a mortar and pestle. TRIzol Reagent (Life Technologies, Grand Island, NY) was added to the ground kidney and RNA was extracted according to the manufacturer's protocol.
Reverse transcription of 1 μg of RNA was carried out using reverse transcriptase and random hexamers (Life Technologies). For real-time PCR analyses, 2 μl of the reverse transcriptase reaction was utilized. Kidney injury molecule (Kim-1) and neutrophil gelatinase-associated lipocalin (Ngal) were chosen for analysis because they have been shown to be reliable markers of renal injury (Prozialeck et al., 2007; Singer et al., 2013; Sinha et al., 2013). Analysis of kidney injury molecule-1 (Kim-1) and neutrophil gelatinase-associated lipocalin (Ngal) was performed with an ABI Prism 7000 detection system using the following gene expression assays: Rat Kim-1 (Rn00597701_m1); Rat Ngal (Rn00590612_m1); Mouse Kim-1 (Mm00506686_m1); Mouse Ngal (Mm01324470_m1) (Life Technologies). Glyceraldehyde 3-phosphate dehydrogenase (Gapdh; rat, Rn01775763_g1; mouse, Mm99999915_g1) was used as a reference gene.
Histopathological analyses
Kidneys were fixed in 40% formaldehyde, 50% glutaraldehyde in 96.7 mM NaH2PO4, and 67.5 mM NaOH for 48 h at 4°C. Following fixation, kidneys were washed twice with normal saline and placed in 70% ethanol. Tissues were processed in a Tissue-Tek VIP processor using the following sequence: 95% ethanol for 30 min (twice), 100% ethanol for 30 min (twice), and 100% xylene (twice). Samples were subsequently embedded in POLY/Fin paraffin (Fisher) and 5-μm sections were cut using a Leitz 1512 microtome and mounted on glass slides. Sections were stained with hematoxylin and eosin and were viewed using an Olympus IX70 microscope. The entire midtransverse, cross-section of each slice of kidney was evaluated for cellular and tubular pathology at both a qualitative and semi-quantitative level. Images were captured with a Jenoptix Progress C12 digital camera.
Measurement of creatinine and blood urea nitrogen in plasma
Plasma creatinine and blood urea nitrogen (BUN) levels were measured to assess overall renal function (i.e., glomerular filtration rate). Following separation of plasma from cellular components of blood, samples of plasma were stored at −20°C. For determination of plasma creatinine, 30 μl of plasma was utilized and the concentration of creatinine was assessed using the QuantiChrome creatinine assay (BioAssay). Similarly, using a 5-μl sample of plasma, the concentration of BUN was determined using the QuantiChrome urea assay (BioAssay).
Western blot analyses of Mrp2
Western blot analyses utilized kidney sections from male Wistar rats that were not exposed to HgCl2. Animals were anesthetized with ketamine (70 mg • kg−1) and xylazine (30 mg • kg-1) and both kidneys were removed. A 3-mm slice was obtained from each kidney and the cortex, OSOM, inner stripe of the outer medulla (ISOM), and inner medulla were isolated. Each section was placed in an individual tube and frozen immediately in liquid nitrogen. At the time of protein extraction, tissue sections were pulverized in liquid nitrogen using a mortar and pestle. Following pulverization, RIPA buffer (Sigma, St Louis, MO), protease inhibitor cocktail, and phosphatase inhibitor cocktail were added to the powdered tissue. Samples were mixed and incubated on ice for 45 min. Samples were homogenized, centrifuged, and the supernatant was collected for analysis. Protein concentrations were determined using Bradford's method and the concentration of each sample was adjusted so that 20 μg of protein in Laemmli buffer with β-mercaptoethanol was loaded into each well of a 7.5% Tris-HCl gel (Bio-Rad). The proteins were transferred to a PDVF membrane (Bio-Rad, Hercules, CA) using a Criterion blotter. The membrane was incubated in blocking buffer (Bio-Rad) for 1 h, followed by incubation with mouse, anti-rat Mrp2 antibody (1:500; Abcam) or mouse, anti-rat β-actin antibody (1:1000; Abcam). The membrane was washed and subsequently incubated with goat, anti-mouse IgG (1:3000; Bio-Rad) and StrepTactin-AP (1:5000, Bio-Rad). After washing, the membrane was incubated in alkaline phosphatase substrate solution (Bio-Rad) and exposed to X-ray film for 1 min. Band intensity was analyzed using Image J software.
Data analysis
Data for each parameter assessed from the groups of four rats or three mice are expressed as mean ± standard error. Corresponding sets of data for each parameter assessed were analyzed first with the Kolmogorov-Smirnov test for normality and Levene's test for homogeneity of variances. Following these tests, data were analyzed using a 2 × 1 (rats) or 3 x1 (mice), one-way analysis of variance (ANOVA) to assess differences among relevant means. When statistically significant F-values were obtained with ANOVA, the data were analyzed using Tukey's post hoc multiple comparison test. A p-value of <0.05 was considered statistically and biologically significant.
RESULTS
Histopathological Assessment of the Nephropathy Induced by Hg2+
Data from TR− and Wistar rats
Representative sections of kidneys from Wistar and TR− rats that were not exposed to HgCl2 are shown in Figure 1. No evidence of glomerular or tubular pathology was present. Markedly different histological features of the nephropathy induced by Hg2+ were observed between Wistar (control) and TR− (Mrp2-deficient) rats 48 h following exposure (iv) to either the 1.5 or 2.25 μmol • kg−1 dose of HgCl2. The most obvious difference in the nephropathy between the two strains of rats was the localization of tubular pathology.
FIG. 1.
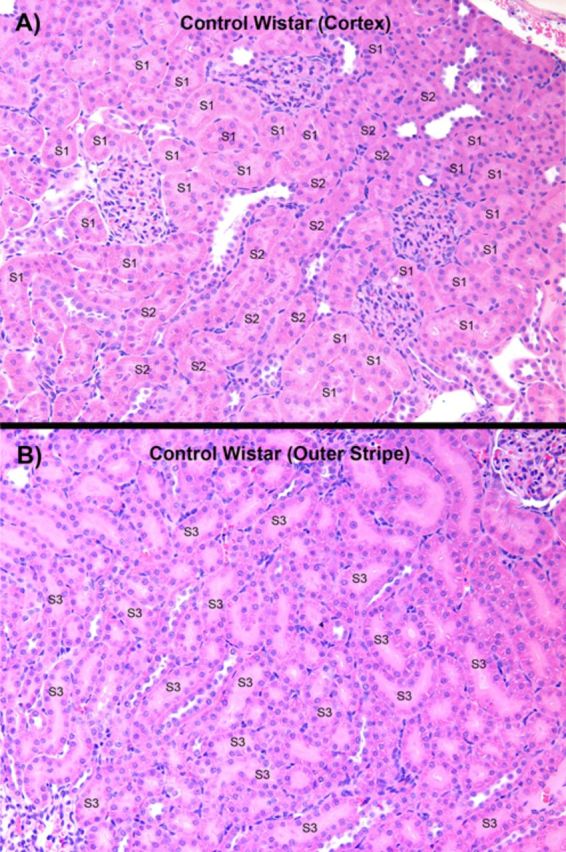
A representative low-power, light microscopic field of the outer renal cortex (A) and the OSOM of a control Wistar rat not treated with HgCl2. In both micrographs, examples of the segments of the proximal tubule (i.e., S1, S2, and S3) present in the two zones have been identified. In panel B, all of the proximal tubules in the OSOM are S3 segments of proximal tubules of cortical and juxtamedullary nephrons, although only some have been identified.
Microscopic examination of the histological sections obtained from the kidneys of TR− rats exposed to either dose of HgCl2 revealed very surprising findings at low-power (approximately 40–100×). Renal cellular injury and death were localized exclusively in proximal tubular segments in the cortex; primarily those segments (S1 and early S2) present in the subcapsular to midcortical region (Fig. 2A). The extent of tubular damage was more severe in the TR− rats administered the 2.25-μmol • kg−1 dose of HgCl2 than in the TR− rats administered the lower dose of HgCl2. The current study is the first to document this pattern of localization of cellular injury and death along the length of the proximal tubule following exposure to Hg2+. It should be stressed that proximal tubular pathology was virtually absent in the OSOM of the TR− rats.
FIG. 2.
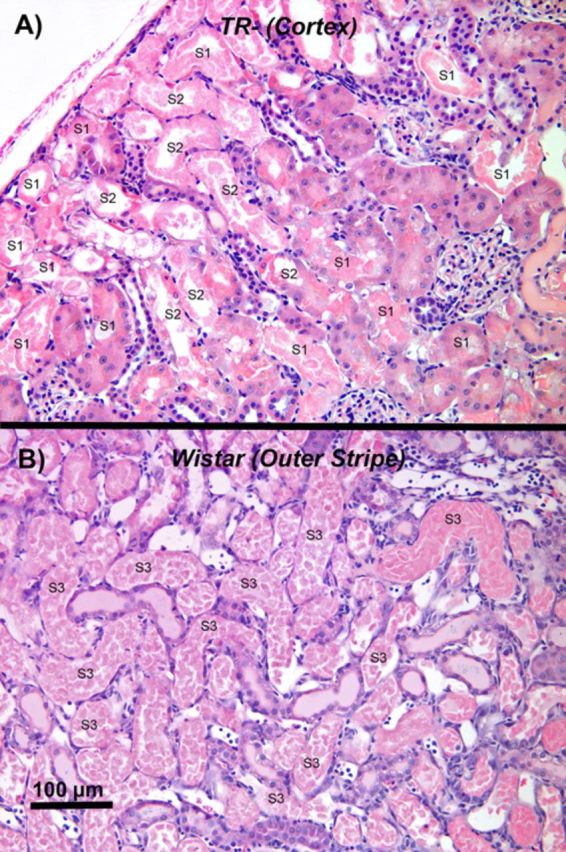
A representative low-power, light microscopic field of the subcapsular and outer renal cortex of a TR− rat (panel A) and the OSOM of a Wistar rat (panel B) 48 h after being exposed to a 2.25-μmol • kg dose of HgCl2. In both micrographs, examples of the segments of the proximal tubule (i.e., S1, S2, and S3) present in the two zones have been identified. Note that a significant degree of epithelial cell death occurred in outer cortical and subcapsular S1 and S2 segments of proximal convoluted tubules in the TR− rat (A). In the most severely damaged tubular segments, only strongly eosin-stained remnants of dead proximal tubular epithelial cells (lacking the presence of a nucleus) remain. In addition, a homogenous, proteinaceous-like material (possibly Tamm's-Horsfall protein) can be seen in the lumen of some proximal and distal tubular segments. Although not seen in the field, proximal tubular cell death was essentially absent in the OSOM of TR− rats. In contrast, extensive cellular injury and death in kidneys of Wistar rats (B) were detected primarily along the terminal portions of the pars recta of proximal tubules at the cortico-medullary junction (late S2 segments) and the OSOM (S3 segments). Also note that the luminal compartments of many proximal tubular segments are filled with strongly eosin-stained, dead epithelial cells, which lack an intact nucleus. Also note that in many of the affected proximal tubular segments the only remnant of tubular structure remaining is an intact basal lamina. Homogenous protein-containing material can also be seen in some of the distal tubular segments in the microscopic field. In panel B, all of the proximal tubules in the OSOM are S3 segments of the proximal tubules, although only some have been identified.
In contrast to the findings from TR− rats, the nephropathy in corresponding Wistar rats was characterized mainly by cellular injury and death in pars recta segments of the proximal tubule situated at the cortico-medullary junction and the OSOM (Fig. 2B). However, following exposure to the 2.25-μmol • kg−1 dose of HgCl2, evidence of cellular injury and death were evident throughout the length of the pars recta of proximal tubules. In some cases, cellular death appeared to have not only occurred along the entire length of the S3 segments but also in most of the S2 segments in medullary rays.
It was difficult to discern differences in the overall level of severity of proximal tubular injury and death between the Wistar and TR− rats, especially at the 2.25-μmol • kg−1 dose of HgCl2. The difference in localization of tubular damage between Wistar and TR− rats was a major confounding factor in attempting to quantify histologically any differences in severity of the nephropathy induced by the 2.25-μmol • kg−1 dose of HgCl2. The nephropathy induced by the lowest dose of HgCl2 tended to be slightly more severe in the TR− rats than in the Wistar rats.
Additional features of the nephropathy induced by Hg2+ in both the Wistar and TR− rats included cellular debris and a homogeneous, strongly eosinophilic, proteinaceous material filling the luminal compartment of both proximal and distal segments of the nephron in the cortex and medulla and in cortical and medullary segments of collecting ducts. In addition, numerous inflammatory leukocytes were also present in numerous peritubular capillaries.
Data from Mrp2−/− and FVB mice
Sections of kidneys from FVB and Mrp2−/− mice not exposed to HgCl2 were similar to that of rats shown in Figure 1. No glomerular or tubular pathology was detected in these sections. In rats exposed to HgCl2, differences in the histological presentation of the nephropathy induced by Hg2+ were also observed between Mrp2−/− mice (also referred to as Mrp2-knock out (KO) mice) and FVB mice. Cellular injury and necrosis in the Mrp2−/− mice were localized predominantly along the proximal tubular segments in the subcapsular to midcortical regions of the kidneys (Fig. 3A). All three doses of HgCl2 induced cellular death in cortical proximal tubules. Moreover, the severity and magnitude of cellular death detected histologically was on average greater in the Mrp2−/− mice than in the FVB control mice. Twenty-four hours after the administration of the largest dose (19.5 μmol Hg2+ • kg−1) of HgCl2, the level of proximal tubular cellular injury and death detected histologically in the mid-to-outer cortex varied from moderately to very severe.
FIG. 3.
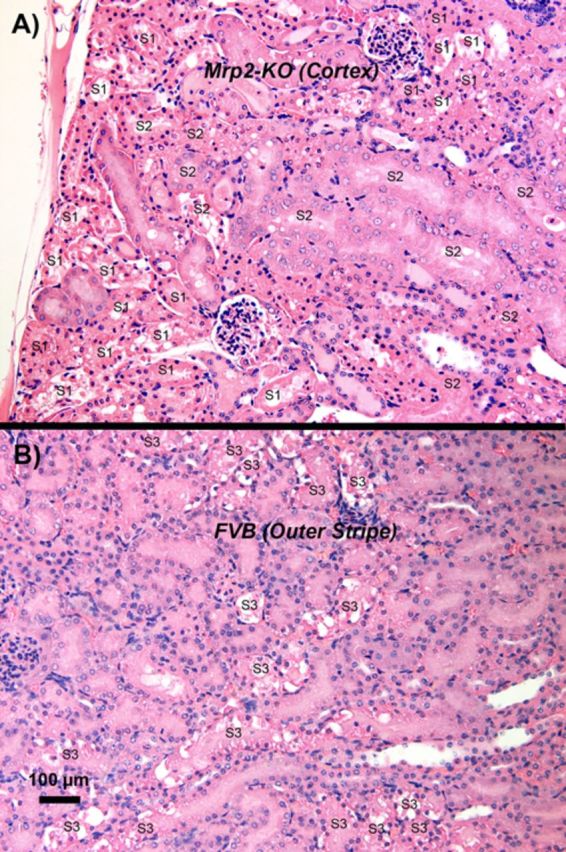
A representative low-power, light microscopic field, of the subcapsular and outer renal cortex of a Mrp2−/− (Mrp2-KO) mouse (panel A) and the OSOM of a FVB mouse (panel B) 24 h after receiving a 19.5-μmol • kg-1 dose of HgCl2. In both micrographs, examples of the segments of the proximal tubule (i.e., S1, S2, and S3) present in the two zones have been identified. A significant degree of epithelial cell death occurred in outer cortical and subcapsular S1 and S2 segments of proximal convoluted tubules in the Mrp2−/− mouse (A). In addition, a homogenous, proteinaceous-like material (possibly Tamm's-Horsfall protein) can be seen in the lumen of some distal tubular segments. Although not shown in the image, proximal tubular cell death was essentially absent in the OSOM of Mrp2−/− mice. In contrast, kidneys of FVB mice (B) exhibited extensive cellular injury and death in the terminal portions of the pars recta of proximal tubules at the cortico-medullary junction (late S2) and OSOM (S3 segments). Many proximal tubular lumina are filled with eosinophilic epithelial cells, which represent denuded proximal tubular epithelial cells at various stages after death. In panel B, all of the proximal tubules in the OSOM are S3 segments of the proximal tubules, although only some have been identified.
All three ip doses of HgCl2 induced cellular injury and death in proximal tubular cells in the FVB mice. However, unlike the localization of cellular injury and death detected histologically in the kidneys of the Mrp2−/− mice, proximal tubular cellular injury and death was predominantly localized along terminal portions of the pars recta (late portions of S2 segments and S3 segments) in the kidneys of the FVB mice. After administration of the largest (19.5-μmol • kg−1) dose of HgCl2, the level of cellular injury and death along the terminal portions of the pars recta of proximal tubule varied from mild to modestly severe in the FVB mice (Fig. 3B). Thus, severity of proximal tubular damage was more extensive and severe in Mrp2−/− mice than in the FVB control mice, notwithstanding the difference in localization of proximal tubular pathology.
As in the kidneys of the two strains of rats studied, cellular debris and proteinaceous material were seen filling the luminal compartment of both proximal and distal segments of the nephron in the cortex and medulla and in cortical and medullary segments of collecting ducts. The number of collecting ducts and distal nephron segments containing eosinophilic debris or proteinaceous casts tended to be greater in the transverse sections of kidneys from Mrp2−/− mice than from FVB mice. Numerous inflammatory leukocytes were also observed in peritubular capillaries.
An important point needs to be stressed at this juncture; the dose-effect relationship between dose and the severity of the nephropathy induced by HgCl2 were greatly different between the rats and mice used in the present study. Sensitivity to the nephrotoxic effects of HgCl2 tended to be, on average, more than 10-fold greater in rats than in mice. Despite the differences in dose of HgCl2 required to induce renal pathological events in the rats and mice used, the histopathological characteristics of the nephropathy induced by Hg2+ were very similar in the Wistar rats and FVB mice. More specifically, the primary target affected by the toxic effects of Hg2+ was the pars recta of proximal tubules, particularly those portions present in the S2 segments in the inner and midcortex and the S3 segments in the OSOM.
Influence of Hg2+ on Serum Creatinine and BUN
Serum creatinine in Wistar and TR− rats (Fig. 4A) exposed to the 1.5-μmol • kg−1 dose of HgCl2 was similar to normal values reported previously (Amini et al., 2012; Moeini et al., 2013; Palm and Lundblad, 2005). By contrast, serum creatinine was elevated significantly in both strains of rats when they were exposed to 2.25 μmol HgCl2• kg−1.
FIG. 4.

Plasma concentration of creatinine and BUN in Wistar and TR− rats 48 h after being exposed to 1.5 or 2.25 μmol HgCl2 • kg−1. * = significantly different (p < 0.05) from the mean for rats of the same strain that received the 1.5-μmol • kg−1 dose of HgCl2. + = significantly different (p < 0.05) from the mean for the corresponding group of Wistar rats that received the same dose of HgCl2.
BUN was also normal in Wistar and TR− rats following exposure to 1.5 μmol • kg−1 HgCl2 (Fig. 4B). Exposure to the 2.25-μmol • kg−1 dose of HgCl2 caused BUN to increase significantly in both strains of rats.
Influence of Hg2+ on Renal Expression of Kim-1 and Ngal
In the two groups of rats exposed to 1.5 μmol • kg−1 HgCl2, the renal expression of Kim-1 (Fig. 5A) and Ngal (Fig. 5B) was significantly greater in TR− rats than in Wistar rats. In contrast, differences were not detected between TR− and Wistar rats exposed to 2.25 μmol • kg−1 HgCl2. On the other hand, renal expression of Kim-1 and Ngal was increased significantly in both strains of rats when the dose of HgCl2 was increased from 1.5 μmol • kg−1 to 2.25 μmol • kg−1.
FIG. 5.

Real-time PCR assessment of the renal expression of Kim-1 and Ngal in Wistar and TR− rats 48 h after exposure to 1.5 or 2.25 μmol HgCl2 • kg−1. * = significantly different (p < 0.05) from the mean for rats of the same strain that received the 1.5-μmol • kg−1 dose of HgCl2. + = significantly different (p < 0.05) from the mean for the corresponding group of Wistar rats that received the same dose of HgCl2.
Disposition of Hg2+ in the Total Renal Mass and Urine
Data from TR− and Wistar rats
The content of Hg2+ in the total renal mass of TR− rats exposed to either dose of HgCl2 was significantly greater than that of corresponding Wistar rats (Fig. 6A). In Wistar and TR− rats exposed to 2.25 μmol • kg−1 HgCl2, the percent of the administered dose in the total renal mass was less than that in the corresponding strain of rats exposed to the 1.5-μmol • kg−1 dose.
FIG. 6.

Renal content of Hg2+ (A) and the total amount of Hg2+ excreted in urine (B) 48 h after Wistar and TR− rats were exposed to 1.5 or 2.25 μmol HgCl2 • kg−1. * = significantly different (p < 0.05) from the mean for rats of the same strain that received the 1.5-μmol • kg−1 dose of HgCl2. + = significantly different (p < 0.05) from the mean for the corresponding group of Wistar rats that received the same dose of HgCl2.
Excretion of Hg2+ in urine was significantly greater in Wistar rats that in TR− rats during the initial 48 h after exposure to either dose of HgCl2 (Fig. 6B). Wistar rats excreted less of the dose of Hg2+ after the 2.25-μmol • kg−1 dose than after the 1.5-μmol • kg−1 dose. By contrast, the total amount of Hg2+ excreted in urine by TR− rats was significantly greater after exposure to the 2.25-μmol • kg−1 dose of HgCl2 than after the 1.5-μmol • kg−1 dose.
Data from Mrp2−/− and FVB mice
The content of Hg2+ in the total renal mass of Mrp2−/− mice was approximately fourfold to fivefold greater than that in the total renal mass of corresponding FVB mice following the administration any of the three doses of HgCl2 (Fig. 7). No significant differences in the content of Hg2+ in the total renal mass were detected among the three groups of FVB mice or Mrp2−/− mice.
FIG. 7.

Content of Hg2+ in the combined renal mass of Mrp2−/− and FVB mice 24 h after exposure (ip) to 18.5, 19, or 19.5 μmol HgCl2 • kg−1. + = significantly different (p < 0.05) from the mean for the corresponding group of FVB mice that received the same dose of HgCl2.
Disposition of Hg2+ in the Renal Cortex and OSOM
Data from TR− and Wistar rats
The concentration of Hg2+ in the renal cortex was significantly greater in the groups of TR− rats than in the corresponding groups of Wistar rats after exposure to either dose of HgCl2 (Fig. 8A). Interestingly, the renal cortical concentration of Hg2+ in the group of TR− rats treated with the 1.5-μmol • kg−1 dose of HgCl2 was significantly greater than that in the group of TR− rats exposed to the 2.25-μmol • kg−1 dose.
FIG. 8.

Concentration of Hg2+ in the renal cortex (A) and OSOM (B) of Wistar and TR− rats 48 h after exposure to 1.5 or 2.25 μmol HgCl2 • kg−1. * = significantly different (p < 0.05) from the mean for rats of the same strain that received the 1.5-μmol • kg−1 dose of HgCl2. + = significantly different (p < 0.05) from the mean for the corresponding group of Wistar rats that received the same dose of HgCl2.
No significant differences in the concentration of Hg2+ in the OSOM were detected between the two groups of TR− rats (Fig. 8B). However, the concentration of Hg2+ in the OSOM was significantly lower in Wistar rats exposed to 2.25 μmol • kg−1 HgCl2 than in the Wistar rats exposed to 1.5 μmol HgCl2 • kg−1.
Data from Mrp2−/− and FVB mice
The cortical concentration of Hg2+ was significantly greater in Mrp2−/− mice than in corresponding FVB mice following all doses of HgCl2 (Fig. 9A). No significant differences were detected among the three corresponding groups of FVB mice or the three groups of Mrp2−/− mice.
FIG. 9.

Concentration of Hg2+ in the renal cortex (A) and OSOM (B) of Mrp2−/− mice and FVB mice 24 h after exposure to 18.5, 19, or 19.5 μmol HgCl2 • kg−1. + = significantly different (p < 0.05) from the mean for the corresponding group of FVB mice that received the same dose of HgCl2.
The concentration of Hg2+ in the OSOM was significantly greater in groups of Mrp2−/− mice than in corresponding groups of FVB mice (Fig. 9B). The differences in the concentration of Hg2+ in the OSOM between the FVB mice and Mrp2−/− mice were not as great as the differences in the cortical concentration of Hg2+ between the same groups of animals.
Disposition of Hg2+ in the Liver and Feces
The hepatic burden of Hg2+ was significantly greater in the TR− rats than in the Wistar rats following exposure to either dose of HgCl2 (Fig. 10A). Within each strain, however, the hepatic burden of Hg2+ was significantly greater following exposure to the 2.25-μmol • kg−1 dose than after exposure to the 1.5-μmol • kg−1 dose.
FIG. 10.

Hepatic content of Hg2+ (A) and the total amount of Hg2+ excreted in the feces (B) 48 h after Wistar and TR− rats were exposed to 1.5 or 2.25 μmol HgCl2 • kg−1. * = significantly different (p < 0.05) from the mean for rats of the same strain that received the 1.5-μmol • kg−1 dose of HgCl2. + = significantly different (p < 0.05) from the mean for the corresponding group of Wistar rats that received the same dose of HgCl2.
The amount of Hg2+ excreted in the feces was not significantly different between the strains of rats 48 h after being exposed to the 1.5-μmol • kg−1 dose of HgCl2 (Fig. 10B). Exposure to the 2.25-μmol kg−1 dose led to significant reductions in the percent of the dose excreted in feces in both strains of rats, with the Wistar rats excreting the least.
Content of Mrp2 Protein in Renal Zones
Protein from samples of renal cortex, OSOM, ISOM, and inner medulla from the kidneys of normal Wistar rats (not treated with HgCl2) was utilized for Western blotting analyses. The analyses demonstrate that Mrp2 protein is localized only in the renal cortex and OSOM (Fig. 11). The relative densities of the bands for Mrp2 were 3.24 ± 0.17 in the cortex and 1.00 ± 0.08 for the OSOM.
FIG. 11.
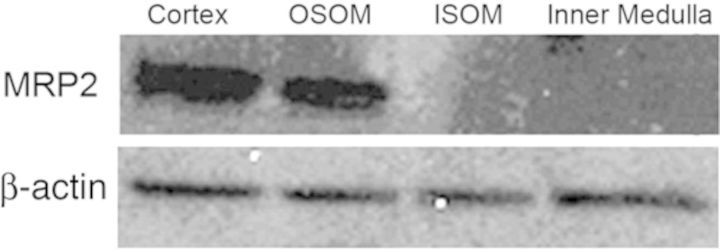
Western blot for Mrp2 and β-actin in samples of renal cortex, OSOM, ISOM, and inner medulla obtained from several Wistar rats (not treated with HgCl2).
DISCUSSION
Mrp2 is an ATP-binding cassette protein found in the apical plasma membrane of various transporting epithelial cells. The primary role of this protein is to participate in the cellular elimination of xenobiotics and cellular metabolites (some in the form of phase II drug metabolism conjugates) into excretory compartments such as bile, feces, and urine (Schaub et al., 1999). Given the localization and function of Mrp2, we hypothesized that it plays an important role in the handling of mercuric species. Findings from our initial studies using TR− rats and Mrp2−/− mice indicate that the absence of Mrp2 in the luminal membrane of proximal tubular epithelial cells and the sinusoidal membrane of hepatocytes leads to enhanced accumulation/retention of mercuric species in these cells. On the whole, the findings strongly support the hypothesis that Mrp2 plays an important role in the hepatocellular and proximal tubular elimination of some species of Hg2+ and organic mercury (Bridges et al., 2008a,b, 2011; Bridges and Zalups, 2010; Zalups and Bridges, 2009).
The purpose of the present study was to extend our previous findings by studying and characterizing the nephrotoxic effects of Hg2+ in TR− rats and Mrp2−/− mice in order to test the hypothesis that the absence of Mrp2 in proximal tubular cells leads to significant alterations in the nephropathy induced by Hg2+. Following exposure to any form of mercury, mercuric ions accumulate primarily in the kidneys, specifically the three segments (S1, S2, and S3) of the proximal tubule. Despite the fact that mercuric ions accumulate along the entire length of the proximal tubule, the nephrotoxic effects of mercury in normal rats and mice are manifested primarily in the epithelial cells lining the pars recta (or straight) segments (Bridges and Zalups, 2010; Ganote et al., 1974; Gritzka and Trump, 1968; McDowell et al., 1976; Zalups, 2000; Zalups and Diamond, 1987; Zalups et al., 1991). In the present study, histological evidence confirmed that in control rats and mice exposed to a mild or modestly severe nephrotoxic dose of Hg2+ the typical pattern of Hg2+-induced nephropathy was present. Specifically, the terminal portions of S2 segments at the cortico-medullary junction and the inner cortex and the S3 segments in the OSOM, which make up much of the pars recta of the proximal tubule, were the primary sites where the Hg2+-induced nephropathy was manifested. Interestingly, this pattern of nephrotoxicity differed significantly in corresponding TR− rats and Mrp2−/− mice. Unlike in control animals, the nephropathy in TR− rats and Mrp2−/− mice was characterized by cellular injury and death in S1 and S2 segments of proximal tubules in the subcapsular and midcortical regions of the kidneys. Perhaps most intriguingly, the S3 segments of proximal tubules, which are normally affected by the toxic effects of Hg2+, did not display any significant cellular or tubular pathology (at least with the doses of HgCl2 used in the present study). The greatest differences in tubular injury and cellular death were most evident between the two strains of mice studied, with the most severe level of proximal tubular damage in the Mrp2−/− mice.
We propose that, in the absence of Mrp2, mercuric ions within cells of S1 and early S2 segments are unable to be exported into the tubular lumen. The retention of mercuric ions within these cells can lead to intoxicating events and subsequent cellular injury and necrosis. Interestingly, in animals lacking Mrp2, there was virtually no intoxication or cellular injury in epithelial cells lining the S3 segments in the OSOM, which is likely due to the inability of mercuric ions to access these segments of the nephron.
The dose-dependent level of renal tubular damage rats is supported by our measurements of the levels of BUN, plasma creatinine, and the renal expression of Kim-1 and Ngal. Renal proximal tubular expression of Kim-1 and Ngal are reliable biomarkers of renal proximal tubular injury and impairment (Bridges et al., 2014; Prozialeck et al., 2007; Singer et al., 2013; Sinha et al., 2013). Because BUN and plasma creatinine were elevated significantly in both Wistar and TR− rats exposed to the largest dose of HgCl2 (2.25 μmol HgCl2 • kg−1), glomerular filtration rate was likely compromised significantly in these animals, indicating some level of ARF. Interestingly, when rats were exposed to the lowest dose of Hg2+, renal expression of Kim-1 and Ngal was significantly greater in TR− rats than in Wistar rats. These data correlate with the present and previous (Bridges et al., 2008a,b) observations that the renal cortical accumulation of Hg2+ in the TR− rats was greater than that in Wistar rats. The absence of a functional form of Mrp2 and the consequent increase in the cellular burden of Hg2+ likely overwhelmed the protective mechanisms in the epithelial cells in the S1 and early S2 segments of the proximal tubule, which resulted in cellular injury and death. Surprisingly, the proximal tubular injury induced by the lowest dose of Hg2+ did not affect the levels of plasma creatinine or BUN in either strain of rat, indicating that despite the induction of ATN, the degree of injury was not sufficient to induced ARF.
As mentioned above, the renal accumulation of Hg2+, especially in the renal cortex, was significantly greater in the TR− rats and Mrp2−/− mice than in corresponding control animals. Moreover, the urinary excretion of Hg2+ in the TR− rats was significantly less than that in corresponding Wistar rats. Overall, the renal and urinary dispositional data from both the rats and mice confirm that the lack of Mrp2 contributes to the enhanced accumulation of Hg2+ in the epithelial cells of the proximal tubule secondary to diminished secretion of Hg2+ into the lumen, particularly in the S1 and S2 segments in the renal cortex. Additionally, diminished secretion of mercuric species from proximal tubular cells leads to decreased urinary excretion of mercury Hg2+. It should be kept in mind though, that at higher doses of HgCl2, some fraction of the Hg2+ excreted in the urine may come from dead proximal tubular epithelial cells carried out into the final urine.
To better understand the role of Mrp2 in the disposition and nephrotoxicology of Hg2+, we evaluated the relative distribution of Mrp2 protein in the zones of the kidney. Mrp2 has been localized in the apical plasma membrane of cortical proximal tubular segments (Schaub et al., 1999), but it is not clear from the literature if other renal zones have not been assessed for the presence of this protein. Because the proximal tubule extends from the cortex through the OSOM, we tested the hypothesis that Mrp2 is present in proximal tubular cells lining the entire proximal tubule. Indeed, our findings show that Mrp2 protein is localized in both the renal cortex and OSOM. The amount of Mrp2 protein in the cortex appears to be approximately threefold greater than that in the OSOM, suggesting that Mrp2 is more abundant in the cells of early proximal tubular segments. This distribution of Mrp2 indirectly correlates with the dispositional and toxicological findings in the TR− rats and Mrp2−/− mice.
Our findings have led us to suggest that under normal conditions, epithelial cells of S1 segments, which possess Mrp2, are capable of secreting intracellular mercuric ions into the tubular lumen. Moreover, we propose that the secreted mercuric ions are in the form of a thiol (likely glutathione) S-conjugate formed intracellularly. Indeed, Mrp2 has been shown to utilize glutathione as a co-factor for certain substrates (Deeley et al., 2006). Once in the lumen the secreted species are carried within the lumen to more distal portions of the proximal tubule, where they are absorbed by epithelial cells in the terminal S2 and S3 segments. Absorption of these mercuric species can then lead to cellular injury and/or death in these segments.
In addition to providing important information regarding the role of Mrp2 in the renal handling of mercuric species, the data from the current study also support a role for Mrp2 in the hepatobiliary transport of mercuric species. Following exposure to either dose of Hg2+, hepatic accumulation of Hg2+ in TR− rats was enhanced whereas fecal excretion of Hg2+ was significantly lower than that of control rats. Enhanced hepatic accumulation and decreased fecal excretion of Hg2+ have also been documented in TR− rats after being exposed to a non-nephrotoxic, 0.5-μmol • kg−1 dose of HgCl2 (Bridges et al., 2008a,b, 2011). Collectively, these data provide strong evidence supporting a role for Mrp2 in the hepatobiliary elimination of mercuric species.
In conclusion, we demonstrate and describe a unique Hg2+-induced nephropathy that is evident in Mrp2-deficient animals. The features of this nephropathy provide evidence for axial heterogeneity in the metabolic pathway that is involved in handling certain mercuric species by proximal tubular epithelial cells. Due to the broad substrate selectively of Mrp2, it is possible that the pathway described above may also pertain to the proximal tubular handling of other toxic, divalent metals, or perhaps certain organic electrophiles.
FUNDING
National Institutes of Health (NIH) (National Institute of Environmental Health Sciences) [ES019991 to C.C.B.]; NIH (National Institute of General Medicine) [GM41935 to K.B.].
REFERENCES
- Amini F. G., Rafieian-Kopaei M., Nematbakhsh M., Baradaran A., Nasri H. Ameliorative effects of metformin on renal histologic and biochemical alterations of gentamicin-induced renal toxicity in Wistar rats. J. Res. Med. Sci. 2012;17:621–625. [PMC free article] [PubMed] [Google Scholar]
- ATSDR. Atlanta: United States Public Health Service/Agency for Toxic Substance Registry; 1999. Toxicological profile for mercury; pp. 1–617. [Google Scholar]
- Belanger M., Westin A., Barfuss D. W. Some health physics aspects of working with 203Hg in university research. Health Phys. 2001;80:S28–S30. [PubMed] [Google Scholar]
- Bridges C. C., Bauch C., Verrey F., Zalups R. K. Mercuric conjugates of cysteine are transported by the amino acid transporter system b(0,+): Implications of molecular mimicry. J. Am. Soc. Nephrol. 2004;15:663–673. doi: 10.1097/01.ASN.0000113553.62380.F5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. C., Joshee L., Zalups R. K. MRP2 and the DMPS- and DMSA-mediated elimination of mercury in TR(-) and control rats exposed to thiol S-conjugates of inorganic mercury. Toxicol. Sci. 2008a;105:211–220. doi: 10.1093/toxsci/kfn107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. C., Joshee L., Zalups R. K. Multidrug resistance proteins and the renal elimination of inorganic mercury mediated by 2,3-dimercaptopropane-1-sulfonic acid and meso-2,3-dimercaptosuccinic acid. J. Pharmacol. Exp. Ther. 2008b;324:383–390. doi: 10.1124/jpet.107.130708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. C., Joshee L., Zalups R. K. MRP2 and the handling of mercuric ions in rats exposed acutely to inorganic and organic species of mercury. Toxicol. Appl. Pharmacol. 2011;251:50–58. doi: 10.1016/j.taap.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. C., Joshee L., Zalups R. K. Aging and the disposition and toxicity of mercury in rats. Exp. Gerontol. 2014;53:31–39. doi: 10.1016/j.exger.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. C., Zalups R. K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005;204:274–308. doi: 10.1016/j.taap.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges C. C., Zalups R. K. Transport of inorganic mercury and methylmercury in target tissues and organs. J. Toxicol. Environ. Health B Crit. Rev. 2010;13:385–410. doi: 10.1080/10937401003673750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X. Y., Strauss J. R., Mariano M. A., Li J., Newton D. J., Cai X., Wang R. W., Yabut J., Hartley D. P., Evans D. C., et al. Characterization of mice lacking the multidrug resistance protein MRP2 (ABCC2) J. Pharmacol. Exp. Ther. 2006;317:579–589. doi: 10.1124/jpet.105.098665. [DOI] [PubMed] [Google Scholar]
- Deeley R. G., Westlake C., Cole S. P. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006;86:849–899. doi: 10.1152/physrev.00035.2005. [DOI] [PubMed] [Google Scholar]
- Ganote C. E., Reimer K. A., Jennings R. B. Acute mercuric chloride nephrotoxicity. An electron microscopic and metabolic study. Lab. Invest. 1974;31:633–647. [PubMed] [Google Scholar]
- Gritzka T. L., Trump B. F. Renal tubular lesions caused by mercuric chloride. Electron microscopic observations: Degeneration of the pars recta. Am. J. Pathol. 1968;52:1225–1277. [PMC free article] [PubMed] [Google Scholar]
- Gstraunthaler G., Pfaller W., Kotanko P. Glutathione depletion and in vitro lipid peroxidation in mercury or maleate induced acute renal failure. Biochem. Pharmacol. 1983;32:2969–2972. doi: 10.1016/0006-2952(83)90404-5. [DOI] [PubMed] [Google Scholar]
- Houser M. T., Milner L. S., Kolbeck P. C., Wei S. H., Stohs S. J. Glutathione monoethyl ester moderates mercuric chloride-induced acute renal failure. Nephron. 1992;61:449–455. doi: 10.1159/000186965. [DOI] [PubMed] [Google Scholar]
- Lee H. B., Blaufox M. D. Blood volume in the rat. J. Nucl. Med. 1985;26:72–76. [PubMed] [Google Scholar]
- Mayer R., Kartenbeck J., Buchler M., Jedlitschky G., Leier I., Keppler D. Expression of the MRP gene-encoded conjugate export pump in liver and its selective absence from the canalicular membrane in transport-deficient mutant hepatocytes. J. Cell Biol. 1995;131:137–150. doi: 10.1083/jcb.131.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell E. M., Nagle R. B., Zalme R. C., McNeil J. S., Flamenbaum W., Trump B. F. Studies on the pathophysiology of acute renal failure. I. Correlation of ultrastructure and function in the proximal tubule of the rat following administration of mercuric chloride. Virchows Archiv. B Cell Pathol. 1976;22:173–196. doi: 10.1007/BF02889215. [DOI] [PubMed] [Google Scholar]
- Moeini M., Nematbakhsh M., Fazilati M., Talebi A., Pilehvarian A. A., Azarkish F., Eshraghi-Jazi F., Pezeshki Z. Protective role of recombinant human erythropoietin in kidney and lung injury following renal bilateral ischemia-reperfusion in rat model. Int. J. Prev. Med. 2013;4:648–655. [PMC free article] [PubMed] [Google Scholar]
- Palm M., Lundblad A. Creatinine concentration in plasma from dog, rat, and mouse: A comparison of 3 different methods. Vet. Clin. Pathol. 2005;34:232–236. doi: 10.1111/j.1939-165x.2005.tb00046.x. [DOI] [PubMed] [Google Scholar]
- Prozialeck W. C., Vaidya V. S., Liu J., Waalkes M. P., Edwards J. R., Lamar P. C., Bernard A. M., Dumont X., Bonventre J. V. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007;72:985–993. doi: 10.1038/sj.ki.5002467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier P. M., Kates B. Histological localization of methylmercury in mouse brain and kidney by emulsion autoradiography of 203Hg. Toxicol. Appl. Pharmacol. 1988;92:224–234. doi: 10.1016/0041-008x(88)90382-1. [DOI] [PubMed] [Google Scholar]
- Rodier P. M., Kates B., Simons R. Mercury localization in mouse kidney over time: Autoradiography versus silver staining. Toxicol. Appl. Pharmacol. 1988;92:235–245. doi: 10.1016/0041-008x(88)90383-3. [DOI] [PubMed] [Google Scholar]
- Schaub T. P., Kartenbeck J., Konig J., Spring H., Dorsam J., Staehler G., Storkel S., Thon W. F., Keppler D. Expression of the MRP2 gene-encoded conjugate export pump in human kidney proximal tubules and in renal cell carcinoma. J. Am. Soc. Nephrol. 1999;10:1159–1169. doi: 10.1681/ASN.V1061159. [DOI] [PubMed] [Google Scholar]
- Sherwood T., Lavender J. P., Russell S. B. Mercury-induced renal vascular shut-down: Observations in experimental acute renal failure. Euro. J. Clin. Invest. 1974;4:1–8. doi: 10.1111/j.1365-2362.1974.tb00364.x. [DOI] [PubMed] [Google Scholar]
- Singer E., Marko L., Paragas N., Barasch J., Dragun D., Muller D. N., Budde K., Schmidt-Ott K. M. Neutrophil gelatinase-associated lipocalin: Pathophysiology and clinical applications. Acta Physiol. 2013;207:663–672. doi: 10.1111/apha.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha V., Vence L. M., Salahudeen A. K. Urinary tubular protein-based biomarkers in the rodent model of cisplatin nephrotoxicity: A comparative analysis of serum creatinine, renal histology, and urinary KIM-1, NGAL, and NAG in the initiation, maintenance, and recovery phases of acute kidney injury. J. Invest. Med. 2013;61:564–568. doi: 10.2310/JIM.0b013e31828233a8. [DOI] [PubMed] [Google Scholar]
- Zager R. A. Glomerular filtration rate and brush border debris excretion after mercuric chloride and ischemic acute renal failure: Mannitol versus furosemide diuresis. Nephron. 1983;33:196–201. doi: 10.1159/000182942. [DOI] [PubMed] [Google Scholar]
- Zalups R. K. Autometallographic localization of inorganic mercury in the kidneys of rats: Effect of unilateral nephrectomy and compensatory renal growth. Exp. Mol. Pathol. 1991;54:10–21. doi: 10.1016/0014-4800(91)90039-z. [DOI] [PubMed] [Google Scholar]
- Zalups R. K. Influence of 2,3-dimercaptopropane-1-sulfonate (DMPS) and meso-2,3-dimercaptosuccinic acid (DMSA) on the renal disposition of mercury in normal and uninephrectomized rats exposed to inorganic mercury. J. Pharmacol. Exp. Ther. 1993;267:791–800. [PubMed] [Google Scholar]
- Zalups R. K. Reductions in renal mass and the nephropathy induced by mercury. Toxicol. Appl. Pharmacol. 1997;143:366–379. doi: 10.1006/taap.1996.8084. [DOI] [PubMed] [Google Scholar]
- Zalups R. K. Molecular interactions with mercury in the kidney. Pharmacol. Rev. 2000;52:113–143. [PubMed] [Google Scholar]
- Zalups R. K., Barfuss D. W. Nephrotoxicity of inorganic mercury co-administrated with L-cysteine. Toxicol. 1996;109:15–29. doi: 10.1016/0300-483x(95)03297-s. [DOI] [PubMed] [Google Scholar]
- Zalups R. K., Bridges C. C. MRP2 involvement in renal proximal tubular elimination of methylmercury mediated by DMPS or DMSA. Toxicol. Appl. Pharmacol. 2009;235:10–17. doi: 10.1016/j.taap.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Zalups R. K., Diamond G. L. Mercuric chloride-induced nephrotoxicity in the rat following unilateral nephrectomy and compensatory renal growth. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1987;53:336–346. doi: 10.1007/BF02890261. [DOI] [PubMed] [Google Scholar]
- Zalups R. K., Gelein R. M., Cernichiari E. DMPS as a rescue agent for the nephropathy induced by mercuric chloride. J. Pharmacol. Exp. Ther. 1991;256:1–10. [PubMed] [Google Scholar]